Biological Evaluation and Molecular Docking Studies of Dimethylpyridine Derivatives



Abstract
1. Introduction
2. Results and Discussion
2.1. Chemistry
2.2. Biological Evaluation
2.2.1. Anti-COX Activity
2.2.2. Antiradical Activity
2.2.3. Cytotoxicity
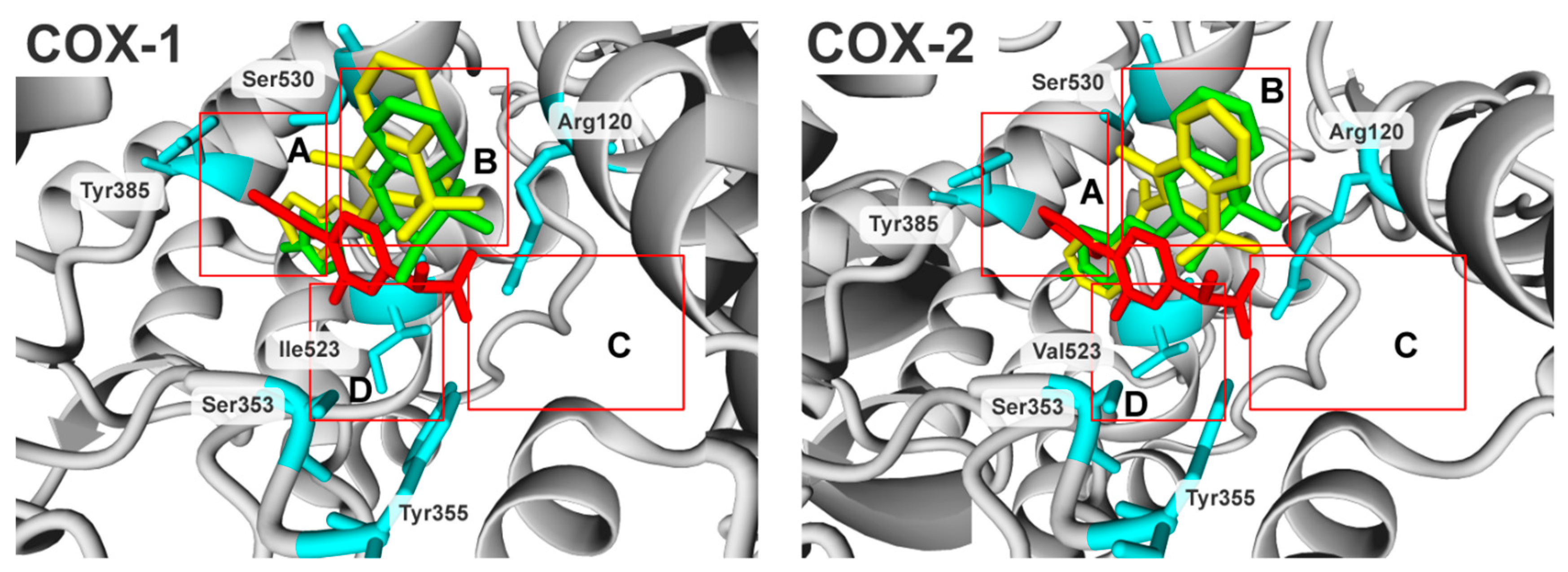
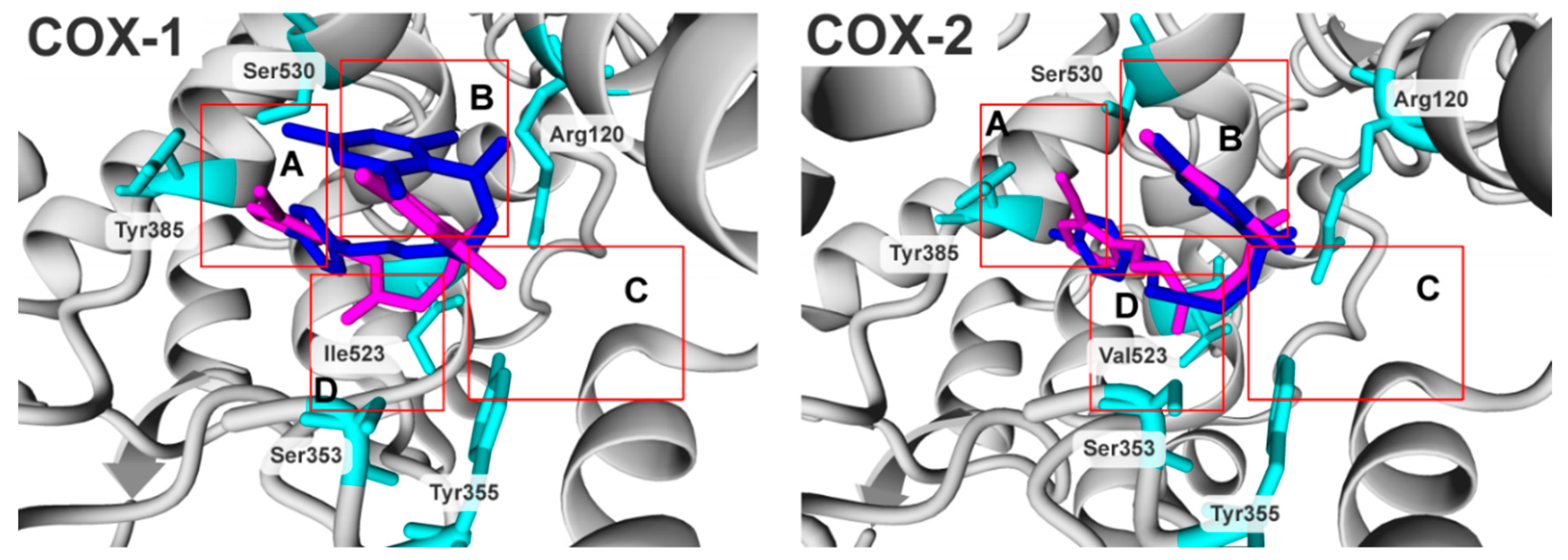
2.3. Molecular Modelling Studies
3. Materials and Methods
3.1. Chemicals
3.2. Cells
3.3. Methods
3.3.1. COX Colorimetric Inhibitor Screening Assay Kit (Cayman Chemical Company, Ann Arbor, MI, USA)
3.3.2. Cell and Culture Conditions
3.3.3. Determination of Cell Density/Cell Proliferation
3.3.4. Evaluation of Intracellular ROS Level
3.3.5. Statistical Analysis
3.3.6. Molecular Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kam, P.C.A.; See, A.U.-L. Cyclo-Oxygenase Isoenzymes: Physiological and Pharmacological Role. Anaesthesia 2000, 55, 442–449. [Google Scholar] [CrossRef] [PubMed]
- Süleyman, H.; Demircan, B.; Karagöz, Y. Anti-Inflammatory and Side Effects of Cyclooxygenase Inhibitors. Pharmacol. Rep. 2007, 59, 247–258. [Google Scholar] [PubMed]
- Hsu, A.L.; Ching, T.T.; Wang, D.S.; Song, X.; Rangnekar, V.M.; Chen, C.S. The Cyclooxygenase-2 Inhibitor Celecoxib Induces Apoptosisby Blocking Akt Activationin Human ProstateCancer Cells Independently of Bcl-2. J. Biol. Chem. 2000, 275, 11397–11403. [Google Scholar] [CrossRef]
- Song, X.; Lin, H.-P.; Johnson, A.J.; Tseng, P.-H.; Yang, Y.-T.; Kulp, S.K.; Chen, C.-S. Cyclooxygenase-2, Playeror Spectatorin Cyclooxygenase-2 Inhibitor-Induced Apoptosisin Prostate Cancer Cells. J. Natl. Cancer Inst. 2002, 94, 585–591. [Google Scholar] [CrossRef]
- Liu, X.H.; Kirschenbaum, A.; Yao, S.; Lee, R.; Holland, J.F.; Levine, A.C. Inhibition of Cyclooxygenase-2 Suppresses Angiogenesis and the Growth of Prostate Cancer in Vivo. J. Urol. 2000, 164, 820–825. [Google Scholar] [CrossRef]
- Narayanan, B.A.; Condon, M.S.; Bosland, M.C.; Narayanan, N.K.; Reddy, B.S. Suppression of N-Methyl-N-Nitrosourea/Testosterone-Induced Rat Prostate Cancer Growth by Celecoxib. Clin. Cancer Res. 2003, 9, 3503–3513. [Google Scholar] [PubMed]
- Vital-Reyes, V.; Rodríguez-Burford, C.; Chhieng, D.C.; Oelschlager, D.K.; Reyes-Fuentes, A.; Barnes, M.; Grizzle, W.E. Celecoxib Inhibits Cellular Growth, Decreases Ki-67 Expression and Modifies Apoptosisin Ovarian Cancer Cell Lines. Arch. Med. Res. 2006, 37, 689–695. [Google Scholar] [CrossRef] [PubMed]
- Parada, B.; Sereno, J.; Reis, F.; Teixeira-Lemos, E.; Garrido, P.; Pinto, A.F.; Cunha, M.F.; Pinto, R.; Mota, A.; Figueiredo, A.; et al. Anti-Inflammatory, Anti-Proliferativeand Antioxidant Profiles of Selective Cyclooxygenase-2 Inhibitionas Chemoprevention for RatBladder Carcinogenesis. Cancer Biol. Ther. 2009, 8, 1615–1622. [Google Scholar] [CrossRef] [PubMed]
- Sereno, J.; Parada, B.; Reis, F.; Cunha, F.X.; Teixeira-Lemos, E.; Garrido, P.; Pinto, R.; Rocha-Pereira, P.; Neto, P.; Ruivo, J.; et al. Preventive but Not Curative Efficacy of Celecoxibon Bladder Carcinogenesisina Rat Model. Mediat. Inflamm. 2010, 2010, 380937. [Google Scholar] [CrossRef] [PubMed]
- Grubbs, C.J.; Lubet, R.A.; Koki, A.T.; Leahy, K.M.; Masferrer, J.L.; Steele, V.E.; Kelloff, G.J.; Hill, D.L.; Seibert, K. Celecoxib Inhibits N-Butyl-N-(4-Hydroxybutyl)-Nitrosamine-Induced Urinary Bladder Cancersin Male B6D2F1 Miceand Female Fischer-344 Rats. Cancer Res. 2000, 60, 5599–5602. [Google Scholar] [PubMed]
- Reddy, B.S.; Hirose, Y.; Lubet, R.; Steele, V.; Kelloff, G.; Paulson, S.; Seibert, K.; Rao, C.V. Chemoprevention of Colon Cancer by Specific Cyclooxygenase-2 Inhibitor, Celecoxib, Administeredduring Different Stages of Carcinogenesis. Cancer Res. 2000, 60, 293–297. [Google Scholar]
- Federico, A.; Morgillo, F.; Tuccillo, C.; Ciardiello, F.; Loguercio, C. Chronic Inflammation and Oxidative Stressin Human Carcinogenesis. Int. J. Cancer 2007, 121, 2381–2386. [Google Scholar] [CrossRef]
- Fosslien, E. Molecular Pathology of Cyclooxygenase-2 in Neoplasia. Ann. Clin. Lab. Sci. 2000, 30, 3–21. [Google Scholar] [PubMed]
- Subbaramaiah, K.; Dannenberg, A.J. Cyclooxygenase 2: A Molecular Target for Cancer Prevention and Treatment. Trends Pharmacol. Sci. 2003, 24, 96–102. [Google Scholar] [CrossRef]
- Bezerra-Netto, H.J.C.; Lacerda, D.I.; Miranda, A.L.P.; Alves, H.M.; Barreiro, E.J.; Fraga, C.A.M. Design and Synthesis of 3,4-Methylenedioxy-6-Nitrophenoxyacetylhydrazone Derivatives Obtainedfrom Natural Safrole: New Lead-Agents with Analgesic and Antipyretic Properties. Bioorg. Med. Chem. 2006, 14, 7924–7935. [Google Scholar] [CrossRef] [PubMed]
- Lima, P.C.; Lima, L.M.; daSilva, K.C.M.; Léda, P.H.O.; deMiranda, A.L.P.; Fraga, C.A.M.; Barreiro, E.J. Synthesis and Analgesic Activity of Novel N-Acylarylhydrazonesand Isosters, Derivedfrom Natural Safrole. Eur. J. Med. Chem. 2000, 35, 187–203. [Google Scholar] [CrossRef]
- Gaston, M.A.; Dias, L.R.; Freitas, A.C.; Miranda, A.P.; Barrerio, E.J. Synthesis and Analgesic Properties of New4-Arylhydrazone 1-HPyrazole[3,4-b] Pyridine Derivatives. Pharm. Acta Helv. 1996, 71, 213–219. [Google Scholar] [CrossRef]
- Mahy, J.P.; Gaspard, S.; Mansuy, D. Phenylhydrazonesas New Good Substrates for the Dioxygenase and Peroxidase Reactions of Prostaglandin Synthase: Form ationofIron(III)-Sigma-Phenyl Complexes. Biochemistry 1993, 32, 4014–4021. [Google Scholar] [CrossRef] [PubMed]
- Silva, G.A.; Costa, L.M.M.; Brito, F.C.F.; Miranda, A.L.P.; Barreiro, E.J.; Fraga, C.A.M. New Class of Potent Antinociceptive and Antiplatelet10 H-Phenothiazine-1-Acylhydrazone Derivatives. Bioorg. Med. Chem. 2004, 12, 3149–3158. [Google Scholar] [CrossRef] [PubMed]
- Świątek, P.; Saczko, J.; Rembiałkowska, N.; Kulbacka, J. Synthesis of New Hydrazone Derivatives and Evaluation of Their Efficacy as Proliferation Inhibitorsin Human Cancer Cells. Med. Chem. 2019. accepted for publication. [Google Scholar] [CrossRef]
- Wong, S.L.; Lau, C.W.; Wong, W.T.; Xu, A.; Au, C.L.; Ng, C.F.; Ng, S.S.M.; Gollasch, M.; Yao, X.; Huang, Y. Pivotal Role of Protein Kinase Cδin Angiotensin II–Induced Endothelial Cyclooxygenase-2Expression. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1169–1176. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.T.; Tian, X.Y.; Chen, Y.; Leung, F.P.; Liu, L.; Lee, H.K.; Ng, C.F.; Xu, A.; Yao, X.; Vanhoutte, P.M.; et al. Bone Morphogenic Protein-4 Impairs Endothelial Function Through Oxidative Stress–Dependent Cyclooxygenase-2 Upregulation. Circ. Res. 2010, 107, 984–991. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Zhang, H.; Liu, J.; Lau, C.W.; Liu, P.; Chen, Z.Y.; Lee, H.K.; Tipoe, G.L.; Ho, H.M.; Yao, X.; et al. Cyclooxygenase-2-Dependent Oxidative Stress Mediates Palmitate-Induced Impairment of Endothelium-Dependent Relaxationsin Mouse Arteries. Biochem. Pharmacol. 2014, 91, 474–482. [Google Scholar] [CrossRef]
- Świątek, P.; Strzelecka, M.; Urniaz, R.; Gębczak, K.; Gębarowski, T.; Gąsiorowski, K.; Malinka, W. Synthesis, COX-1/2Inhibition Activities and Molecular Docking Study of Isothiazolopyridine Derivatives. Bioorg. Med. Chem. 2017, 25, 316–326. [Google Scholar] [CrossRef] [PubMed]
- Gautam, R.; Jachak, S.M.; Kumar, V.; Mohan, C.G. Synthesis, Biological Evaluation and Molecular Docking Studies of Stellatin Derivativesas Cyclooxygenase(COX-1, COX-2)Inhibitorsand Anti-Inflammatory Agents. Bioorg. Med. Chem. Lett. 2011, 21, 1612–1616. [Google Scholar] [CrossRef] [PubMed]
- Lanzo, C.A.; Sutin, J.; Rowlinson, S.; Talley, J.; Marnett, L.J. Fluorescence Quenching Analysis of the Association and Dissociation of a Diarylheterocycleto Cyclooxygenase-1 and Cyclooxygenase-2: Dynamic Basis of Cyclooxygenase-2 Selectivity. Biochemistry 2000, 39, 6228–6234. [Google Scholar] [CrossRef]
- Rowlinson, S.W.; Kiefer, J.R.; Prusakiewicz, J.J.; Pawlitz, J.L.; Kozak, K.R.; Kalgutkar, A.S.; Stallings, W.C.; Kurumbail, R.G.; Marnett, L.J. A Novel Mechanism of Cyclooxygenase-2 Inhibition Involving Interactionswith Ser-530 and Tyr-385. J. Biol. Chem. 2003, 278, 45763–45769. [Google Scholar] [CrossRef] [PubMed]
- Prusakiewicz, J.J.; Duggan, K.C.; Rouzer, C.A.; Marnett, L.J. Differential Sensitivity and Mechanism of Inhibition of COX-2 Oxygenation of Arachidonic Acidand2-Arachidonoylglycerol by Ibuprofen and Mefenamic Acid. Biochemistry 2009, 48, 7353–7355. [Google Scholar] [CrossRef] [PubMed]
- Gierse, J.K.; McDonald, J.J.; Hauser, S.D.; Rangwala, S.H.; Koboldt, C.M.; Seibert, K. A Single Amino Acid Difference between Cyclooxygenase-1(COX-1) and -2(COX-2) Reverses the Selectivity of COX-2 SpecificInhibitors. J. Biol. Chem. 1996, 271, 15810–15814. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Hermanson, D.J.; Banerjee, S.; Ghebreselasie, K.; Clayton, G.M.; Garavito, R.M.; Marnett, L.J. Oxicams Bindina Novel Modeto the Cyclooxygenase Active Siteviaa Two-Water-Mediated H-Bonding Network. J. Biol. Chem. 2014, 289, 6799–6808. [Google Scholar] [CrossRef] [PubMed]
- Hood, W.F.; Gierse, J.K.; Isakson, P.C.; Kiefer, J.R.; Kurumbail, R.G.; Seibert, K.; Monahan, J.B. Characterization of Celecoxib and Valdecoxib Binding to Cyclooxygenase. Mol. Pharmacol. 2003, 63, 870–877. [Google Scholar] [CrossRef]
- Vichai, V.; Kirtikara, K. SulforhodamineB Colorimetric Assay for Cytotoxicity Screening. Nat. Protoc. 2006, 1, 1112–1116. [Google Scholar] [CrossRef] [PubMed]
- Eruslanov, E.; Kusmartsev, S. Identification of ROS Using Oxidized DCFDA and Flow-Cytometry. Methods Inmolecularbiol. 2010, 594, 57–72. [Google Scholar] [CrossRef]
- Roy, S.; Khanna, S.; Nallu, K.; Hunt, T.K.; Sen, C.K. Dermal Wound Healing Is Subject to Redox Control. Mol. Ther. 2006, 13, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Loo, A.E.K.; Halliwell, B. Effects of Hydrogen Peroxideina Keratinocyte-Fibroblast Co-Culture Model of Wound Healing. Biochem. Biophys. Res. Commun. 2012, 423, 253–258. [Google Scholar] [CrossRef]
- Gogoi, D.; Bezbaruah, R.L.; Bordoloi, M.; Sarmah, R.; Bora, T.C. Insights from the Docking Analysis of Biologically Active Compounds from Plant Litsea Genusas Potential COX-2 Inhibitors. Bioinformation 2012, 8, 812–815. [Google Scholar] [CrossRef] [PubMed]
- Palkar, M.B.; Singhai, A.S.; Ronad, P.M.; Vishwanathswamy, A.H.M.; Boreddy, T.S.; Veerapur, V.P.; Shaikh, M.S.; Rane, R.A.; Karpoormath, R. Synthesis, Pharmacological Screening and in Silico Studies of New Class of Diclofenac Analoguesasa Promising Anti-Inflammatory Agents. Bioorg. Med. Chem. 2014, 22, 2855–2866. [Google Scholar] [CrossRef]
- Urniaż, R.D.; Jóźwiak, K. X-Ray Crystallographic Structuresasa Source of Lig and Alignmentin 3D-QSAR. J. Chem. Inf. Model. 2013, 53, 1406–1414. [Google Scholar] [CrossRef]
- Urniaż, R.D.; Rutkowska, E.E.; Plazinska, A.; Jozwiak, K. FRMSD chiral: A Novel Algorithm to Represent Differences between Positions of Stereoisomers in Complex with Dissymmetric BindingSite. J. Chromatogr. B 2014, 955–956, 110–115. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| IC50 (µM),(SD) | Ratio: COX-2/COX-1 | ||
|---|---|---|---|
| Compound | COX-1 | COX-2 | |
| Meloxicam | 85.8 (12.5) | 71.5 (5.1) | 0.8 |
| Piroxicam | 170.5 (15.7) | 127.6 (11.9) | 0.7 |
| PS18 | 57.3 (1.1)*Δ | 139.5 (15.7) Δ | 2.4 |
| PS19 | 92.9 (4.4) * | NA | - |
| PS33 | 51.8 (0.6) *Δ | 142.9 (22.9) Δ | 2.8 |
| PS34 | NA | NA | - |
| PS35 | NA | NA | - |
| PS36 | NA | 730.7 (202.8) *Δ | - |
| PS38 | NA | NA | - |
| PS39 | NA | NA | - |
| PS40 | 83.6 (13.5) * | NA | - |
| PS41 | NA | NA | - |
| PS42 | 72.8 (33.8) * | 176.7 (14.3) *Δ | 2.4 |
| PS43 | 78.8 (5.5) * | 132.2 (128.1) | 1.7 |
| E/E0 | Without H2O2 | With H2O2 | ||||
|---|---|---|---|---|---|---|
| Compound | Average | Standard Deviation | p | Average | Standard Deviation | p |
| Meloxicam | 0.99 | 0.20 | NS | 1.22 | 0.14 | NS |
| Piroxicam | 1.19 | 0.17 | NS | 1.59 | 0.25 | NS |
| PS18 | 0.87 | 0.08 | NS | 0.70 | 0.06 | 0.01 * |
| PS19 | 0.84 | 0.12 | NS | 0.72 | 0.06 | NS |
| PS33 | 0.86 | 0.09 | NS | 0.80 | 0.16 | NS |
| PS34 | 0.73 | 0.03 | 0.004 * | 0.58 | 0.07 | 0.01 * |
| PS35 | 1.07 | 0.28 | NS | 0.84 | 0.05 | 0.03 * |
| PS36 | 1.05 | 0.29 | NS | 0.75 | 0.06 | 0.02 * |
| PS38 | 1.10 | 0.29 | NS | 0.89 | 0.05 | NS |
| PS39 | 1.06 | 0.24 | NS | 0.79 | 0.05 | 0.02 * |
| PS40 | 1.00 | 0.26 | NS | 0.67 | 0.04 | 0.004 * |
| PS41 | 0.95 | 0.21 | NS | 0.53 | 0.04 | 0.003 * |
| PS42 | 1.00 | 0.24 | NS | 0.59 | 0.01 | 0.0002 * |
| PS43 | 1.14 | 0.28 | NS | 0.77 | 0.02 | 0.003 * |
| IC50 (µM) (SD) | ||||
|---|---|---|---|---|
| Compound | NHDF | V79 | LoVo | A549 |
| Meloxicam | 205.6 (44.2) | 231.8 (33.5) | 124.6 (11.2) | 148.3 (37.9) |
| Piroxicam | 170.5 (23.0) | 200.0 (32.9) | 122.1 (9.6) | 138.1 (27.8) |
| PS18 | 171.4 (6.9) | 275.0 (19.5) | 135.3 (7.5) | 154.2 (28.8) |
| PS19 | 213.9 (48.4) | 338.5 (151.3) | 115.1 (7.3) | 163.9 (22.9) |
| PS33 | 159.1 (3.6) | 177.9 (30.4) | 130.9 (8.6) | 156.3 (12.6) |
| PS34 | NA | 420.9 (145.6) | NA | NA |
| PS35 | 202.4 (32.6) | 196.2(76.2) | 211.9 (76.4) | NA |
| PS36 | 143.7 (7.9) | 140.6 (26.7) | 99.8 (7.9) | 163.5 (33.7) |
| PS38 | NA | 171.6 (36.2) | NA | 503.8 (171.4) |
| PS39 | 391.0 (144.6) | 146.8 (10.3) | 103.0 (6.6) | 119.7 (12.8) |
| PS40 | NA | 608.2 (287.5) | NA | 147.2 (22.3) |
| PS41 | 335.7 (118.3) | 418.1 (349.6) | 153.9 (12.2) | 220.4 (47.8) |
| PS42 | 132.4 (11.6) | 162.0 (21.7) | 133.2 (24.2) | 139.9 (11.0) |
| PS43 | 177.1 (39.3) | 255.8 (8.2) | 258.5 (183.5) | 221.5 (53.1) |
| Name | COX-1 | Name | COX-2 | ||
|---|---|---|---|---|---|
| pIC50 | MolDock Score | pIC50 | MolDock Score | ||
| PS33 | 4.29 | −143.779 | PS43 | 3.88 | −141.132 |
| PS18 | 4.24 | −132.919 | PS18 | 3.86 | −130.663 |
| PS 42 | 4.14 | −129.366 | PS33 | 3.84 | −130.689 |
| PS43 | 4.10 | −127.159 | PS42 | 3.75 | −129.073 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Świątek, P.; Gębczak, K.; Gębarowski, T.; Urniaz, R. Biological Evaluation and Molecular Docking Studies of Dimethylpyridine Derivatives. Molecules 2019, 24, 1093. https://doi.org/10.3390/molecules24061093
Świątek P, Gębczak K, Gębarowski T, Urniaz R. Biological Evaluation and Molecular Docking Studies of Dimethylpyridine Derivatives. Molecules. 2019; 24(6):1093. https://doi.org/10.3390/molecules24061093
Chicago/Turabian StyleŚwiątek, Piotr, Katarzyna Gębczak, Tomasz Gębarowski, and Rafal Urniaz. 2019. "Biological Evaluation and Molecular Docking Studies of Dimethylpyridine Derivatives" Molecules 24, no. 6: 1093. https://doi.org/10.3390/molecules24061093
APA StyleŚwiątek, P., Gębczak, K., Gębarowski, T., & Urniaz, R. (2019). Biological Evaluation and Molecular Docking Studies of Dimethylpyridine Derivatives. Molecules, 24(6), 1093. https://doi.org/10.3390/molecules24061093