Biological Evaluation and Molecular Dynamics Simulation of Chalcone Derivatives as Epidermal Growth Factor-Tyrosine Kinase Inhibitors

 , , , and
, , , and
Abstract

1. Introduction
2. Results and Discussion
2.1. Cytotoxicity Effect Against the Wild Type (A431 and A549) and Mutant EGFR (H1650 and H1975) Cancer Cell Lines
2.2. EGFR-TKI Activity by Chalcones
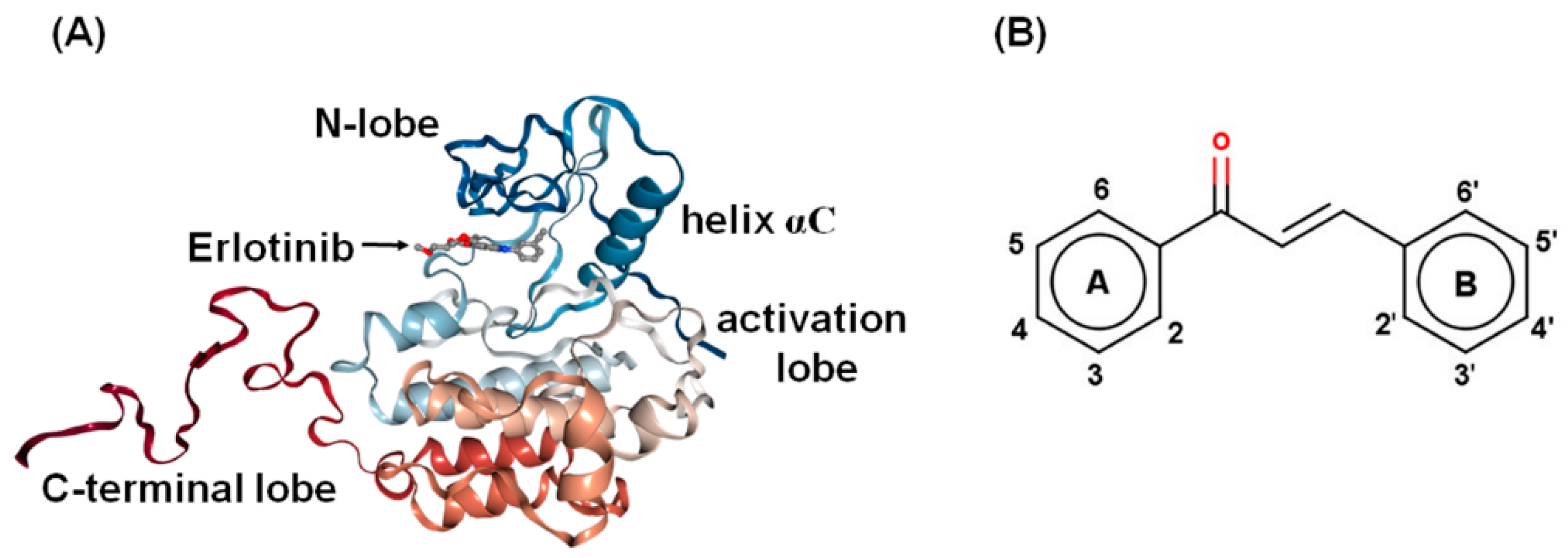
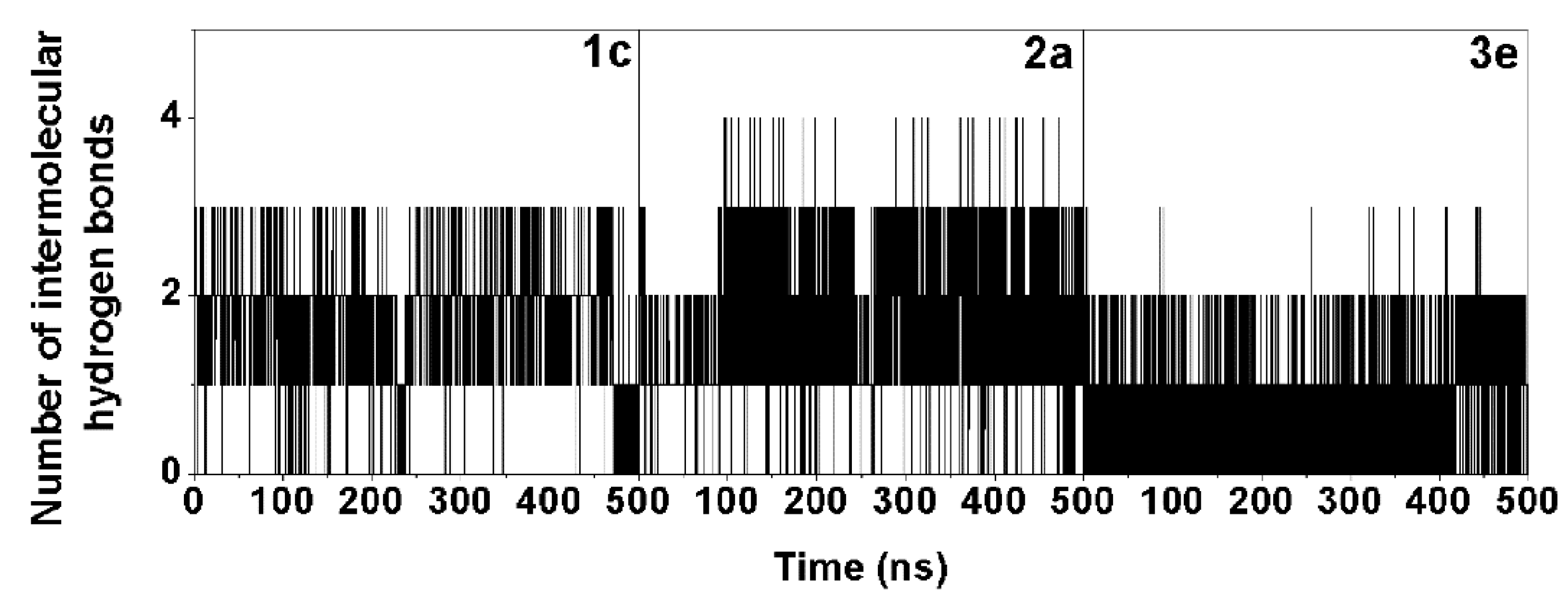
2.3. Molecular Binding and Interaction of Potent Chalcones
2.4. Physicochemical Properties of the Potent Chalcones
3. Materials and Methods
3.1. Materials and Measurements
3.2. Cell Culture and Cell Viability Assay (MTT Assay)
3.3. Enrichment of the ICD of rEGFR from Transfected Hela Cells
3.4. EGFR-TKI Assay
3.5. Molecular Dynamics Simulation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Cancer. 2018. Available online: http://www.who.int/en/news-room/fact-sheets/detail/cancer (accessed on 12 September 2018).
- Xu, G.; McLeod, H.L. Strategies for enzyme/prodrug cancer therapy. Clin. Cancer Res. 2001, 7, 3314–3324. [Google Scholar] [PubMed]
- Woodburn, J. The epidermal growth factor receptor and its inhibition in cancer therapy. Pharmacol. Ther. 1999, 82, 241–250. [Google Scholar] [CrossRef]
- Ogiso, H.; Ishitani, R.; Nureki, O.; Fukai, S.; Yamanaka, M.; Kim, J.-H.; Saito, K.; Sakamoto, A.; Inoue, M.; Shirouzu, M. Crystal structure of the complex of human epidermal growth factor and receptor extracellular domains. Cell 2002, 110, 775–787. [Google Scholar] [CrossRef]
- Okita, R.; Maeda, A.; Shimizu, K.; Nojima, Y.; Saisho, S.; Nakata, M. PD-L1 overexpression is partially regulated by EGFR/HER2 signaling and associated with poor prognosis in patients with non-small-cell lung cancer. Cancer Immunol. Immunother. 2017, 66, 865–876. [Google Scholar] [CrossRef] [PubMed]
- Forcella, M.; Oldani, M.; Epistolio, S.; Freguia, S.; Monti, E.; Fusi, P.; Frattini, M. Non-small cell lung cancer (NSCLC), EGFR downstream pathway activation and TKI targeted therapies sensitivity: Effect of the plasma membrane-associated NEU3. PLoS ONE 2017, 12, e0187289. [Google Scholar] [CrossRef] [PubMed]
- Doyle, H.A.; Koski, R.A.; Bonafé, N.; Bruck, R.A.; Tagliatela, S.M.; Gee, R.J.; Mamula, M.J. Epidermal growth factor receptor peptide vaccination induces cross-reactive immunity to human EGFR, HER2, and HER3. Cancer Immunol. Immunother. 2018, 67, 1559–1569. [Google Scholar] [CrossRef]
- Liu, W.-J.; Liu, X.-J.; Xu, J.; Li, L.; Li, Y.; Zhang, S.-H.; Wang, J.-L.; Miao, Q.-F.; Zhen, Y.-S. EGFR-targeting, β-defensin-tailored fusion protein exhibits high therapeutic efficacy against EGFR-expressed human carcinoma via mitochondria-mediated apoptosis. Acta Pharmacol. Sin. 2018, 39, 1777. [Google Scholar] [CrossRef] [PubMed]
- Maemondo, M.; Inoue, A.; Kobayashi, K.; Sugawara, S.; Oizumi, S.; Isobe, H.; Gemma, A.; Harada, M.; Yoshizawa, H.; Kinoshita, I. Gefitinib or chemotherapy for non–small-cell lung cancer with mutated EGFR. N. Engl. J. Med. 2010, 362, 2380–2388. [Google Scholar] [CrossRef] [PubMed]
- Ganjoo, K.N.; Wakelee, H. Review of erlotinib in the treatment of advanced non-small cell lung cancer. Biol. Targets Ther. 2007, 1, 335. [Google Scholar]
- Geyer, C.E.; Forster, J.; Lindquist, D.; Chan, S.; Romieu, C.G.; Pienkowski, T.; Jagiello-Gruszfeld, A.; Crown, J.; Chan, A.; Kaufman, B. Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N. Engl. J. Med. 2006, 355, 2733–2743. [Google Scholar] [CrossRef]
- Li, D.; Ambrogio, L.; Shimamura, T.; Kubo, S.; Takahashi, M.; Chirieac, L.; Padera, R.; Shapiro, G.; Baum, A.; Himmelsbach, F. BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene 2008, 27, 4702. [Google Scholar] [CrossRef] [PubMed]
- Kalous, O.; Conklin, D.; Desai, A.J.; O’Brien, N.A.; Ginther, C.; Anderson, L.; Cohen, D.J.; Britten, C.D.; Taylor, I.; Christensen, J.G. Dacomitinib (PF-00299804), an irreversible Pan-HER inhibitor, inhibits proliferation of HER2-amplified breast cancer cell lines resistant to trastuzumab and lapatinib. Mol. Cancer Ther. 2012, 11, 1978–1987. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Srivastava, A.K.; Pandey, O. A Review on Chalcones Synthesis and their Biological Activity. PharmaTutor 2018, 6, 22–39. [Google Scholar] [CrossRef]
- Singh, P.; Anand, A.; Kumar, V. Recent developments in biological activities of chalcones: A mini review. Eur. J. Med. Chem. 2014, 85, 758–777. [Google Scholar] [CrossRef]
- Rao, C.M.M.P.; Yejella, R.P.; Rehman, R.S.A.; Basha, S.H. Molecular docking based screening of novel designed chalcone series of compounds for their anti-cancer activity targeting EGFR kinase domain. Bioinformation 2015, 11, 322. [Google Scholar] [CrossRef]
- Nowakowska, Z. A review of anti-infective and anti-inflammatory chalcones. Eur. J. Med. Chem. 2007, 42, 125–137. [Google Scholar] [CrossRef]
- Lee, S.H.; Seo, G.S.; Kim, J.Y.; Jin, X.Y.; Kim, H.-D.; Sohn, D.H. Heme oxygenase 1 mediates anti-inflammatory effects of 2′, 4′, 6′-tris (methoxymethoxy) chalcone. Eur. J. Pharmacol. 2006, 532, 178–186. [Google Scholar] [CrossRef]
- Yang, H.-M.; Shin, H.-R.; Cho, S.-H.; Bang, S.-C.; Song, G.-Y.; Ju, J.-H.; Kim, M.-K.; Lee, S.-H.; Ryu, J.-C.; Kim, Y. Structural requirement of chalcones for the inhibitory activity of interleukin-5. Bioorg. Med. Chem. 2007, 15, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Nowakowska, Z.; Kędzia, B.; Schroeder, G. Synthesis, physicochemical properties and antimicrobial evaluation of new (E)-chalcones. Eur. J. Med. Chem. 2008, 43, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, Z.N.; Praveen, S.; Musthafa, T.M.; Ahmad, A.; Khan, A.U. Thermal solvent-free synthesis of chromonyl chalcones, pyrazolines and their in vitro antibacterial, antifungal activities. J. Enzyme Inhib. Med. Chem. 2012, 27, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, J.C.; Bariwal, J.B.; Upadhyay, K.D.; Naliapara, Y.T.; Joshi, S.K.; Pannecouque, C.C.; De Clercq, E.; Shah, A.K. Improved and rapid synthesis of new coumarinyl chalcone derivatives and their antiviral activity. Tetrahedron Lett. 2007, 48, 8472–8474. [Google Scholar] [CrossRef]
- Gacche, R.; Dhole, N.; Kamble, S.; Bandgar, B. In-vitro evaluation of selected chalcones for antioxidant activity. J. Enzyme Inhib. Med. Chem. 2008, 23, 28–31. [Google Scholar] [CrossRef] [PubMed]
- Bonesi, M.; Loizzo, M.R.; Statti, G.A.; Michel, S.; Tillequin, F.; Menichini, F. The synthesis and Angiotensin Converting Enzyme (ACE) inhibitory activity of chalcones and their pyrazole derivatives. Bioorg. Med. Chem. Lett. 2010, 20, 1990–1993. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, S.S.; Singh, A.K.; Meena, S.; Lohani, M.; Singh, A.; Arya, R.K.; Cheruvu, S.H.; Sarkar, J.; Gayen, J.R.; Datta, D. Synthesis of novel β-carboline based chalcones with high cytotoxic activity against breast cancer cells. Bioorg. Med. Chem. Lett. 2014, 24, 2820–2824. [Google Scholar] [CrossRef]
- Kolundžija, B.; Marković, V.; Stanojković, T.; Joksović, L.; Matić, I.; Todorović, N.; Nikolić, M.; Joksović, M.D. Novel anthraquinone based chalcone analogues containing an imine fragment: Synthesis, cytotoxicity and anti-angiogenic activity. Bioorg. Med. Chem. Lett. 2014, 24, 65–71. [Google Scholar] [CrossRef]
- Wan, M.; Xu, L.; Hua, L.; Li, A.; Li, S.; Lu, W.; Pang, Y.; Cao, C.; Liu, X.; Jiao, P. Synthesis and evaluation of novel isoxazolyl chalcones as potential anticancer agents. Bioorg. Chem. 2014, 54, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Jain, U.K.; Bhatia, R.K.; Rao, A.R.; Singh, R.; Saxena, A.K.; Sehar, I. Design and development of halogenated chalcone derivatives as potential anticancer agents. Trop. J. Pharm. Res. 2014, 13, 73–80. [Google Scholar] [CrossRef]
- Mizuno, C.S.; Paul, S.; Suh, N.; Rimando, A.M. Synthesis and biological evaluation of retinoid-chalcones as inhibitors of colon cancer cell growth. Bioorg. Med. Chem. Lett. 2010, 20, 7385–7387. [Google Scholar] [CrossRef] [PubMed]
- Vasconcelos, A.; Campos, V.F.; Nedel, F.; Seixas, F.K.; Dellagostin, O.A.; Smith, K.R.; Pereira, C.M.P.; Stefanello, F.M.; Collares, T.; Barschak, A.G. Cytotoxic and apoptotic effects of chalcone derivatives of 2-acetyl thiophene on human colon adenocarcinoma cells. Cell Biochem. Funct. 2013, 31, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Jandial, D.D.; Blair, C.A.; Zhang, S.; Krill, L.S.; Zhang, Y.-B.; Zi, X. Molecular Targeted Approaches to Cancer Therapy and Prevention Using Chalcones. Curr. Cancer Drug Targets 2014, 14, 181–200. [Google Scholar] [CrossRef] [PubMed]
- Mahapatra, D.K.; Bharti, S.K.; Asati, V. Anti-cancer chalcones: Structural and molecular target perspectives. J. Enzyme Inhib. Med. Chem. 2015, 98, 69–114. [Google Scholar] [CrossRef] [PubMed]
- Gaur, R.; Mishra, L. Synthesis and characterization of Ru (II)–DMSO–Cl–chalcone complexes: DNA binding, nuclease, and topoisomerase II inhibitory activity. Inorg. Chem. 2012, 51, 3059–3070. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-H.; Lee, E.; Baek, K.H.; Kwon, H.B.; Woo, H.; Lee, E.-S.; Kwon, Y.; Na, Y. Chalcones, inhibitors for topoisomerase I and cathepsin B and L, as potential anti-cancer agents. Bioorg. Med. Chem. Lett. 2013, 23, 3320–3324. [Google Scholar] [CrossRef] [PubMed]
- Jeon, K.-H.; Yu, H.-B.; Kwak, S.Y.; Kwon, Y.; Na, Y. Synthesis and topoisomerases inhibitory activity of heteroaromatic chalcones. Bioorg. Med. Chem. 2016, 24, 5921–5928. [Google Scholar] [CrossRef] [PubMed]
- Alswah, M.; Bayoumi, A.H.; Elgamal, K.; Elmorsy, A.; Ihmaid, S.; Ahmed, H.E. Design, Synthesis and Cytotoxic Evaluation of Novel Chalcone Derivatives Bearing Triazolo [4,3-a]-quinoxaline Moieties as Potent Anticancer Agents with Dual EGFR Kinase and Tubulin Polymerization Inhibitory Effects. Molecules 2017, 23, 48. [Google Scholar] [CrossRef]
- Mohamed, M.F.; Hassaneen, H.M.; Abdelhamid, I.A. Cytotoxicity, molecular modeling, cell cycle arrest, and apoptotic induction induced by novel tetrahydro-[1,2,4]triazolo[3,4-a]isoquinoline chalcones. Eur. J. Med. Chem. 2018, 143, 532–541. [Google Scholar] [CrossRef] [PubMed]
- Syam, S.; Abdelwahab, S.I.; Al-Mamary, M.A.; Mohan, S. Synthesis of chalcones with anticancer activities. Molecules 2012, 17, 6179–6195. [Google Scholar] [CrossRef]
- Sangpheak, K.; Mueller, M.; Darai, N.; Wolschann, P.; Suwattanasophon, C.; Ruga, R.; Chavasiri, W.; Seetaha, S.; Choowongkomon, K.; Kungwan, N. Computational screening of chalcones acting against topoisomerase IIα and their cytotoxicity towards cancer cell lines. J. Enzyme Inhib. Med. Chem. 2019, 34, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Ono, M.; Hirata, A.; Kometani, T.; Miyagawa, M.; Ueda, S.-I.; Kinoshita, H.; Fujii, T.; Kuwano, M. Sensitivity to gefitinib (Iressa, ZD1839) in non-small cell lung cancer cell lines correlates with dependence on the epidermal growth factor (EGF) receptor/extracellular signal-regulated kinase 1/2 and EGF receptor/Akt pathway for proliferation. Mol. Cancer Ther. 2004, 3, 465–472. [Google Scholar]
- Zhang, F.; Wang, S.; Yin, L.; Yang, Y.; Guan, Y.; Wang, W.; Xu, H.; Tao, N. Quantification of epidermal growth factor receptor expression level and binding kinetics on cell surfaces by surface plasmon resonance imaging. Anal. Chem. 2015, 87, 9960–9965. [Google Scholar] [CrossRef]
- Acquaviva, J.; Smith, D.L.; Sang, J.; Friedland, J.C.; He, S.; Sequeira, M.; Zhang, C.; Wada, Y.; Proia, D.A. Targeting KRAS-mutant non–small cell lung cancer with the Hsp90 inhibitor ganetespib. Mol. Cancer Ther. 2012, 11, 2633–2643. [Google Scholar] [CrossRef]
- Stamatkin, C.; Ratermann, K.L.; Overley, C.W.; Black, E.P. Inhibition of class IA PI3K enzymes in non-small cell lung cancer cells uncovers functional compensation among isoforms. Cancer Biol. Ther. 2015, 16, 1341–1352. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Fan, X.-X.; Jiang, Z.-B.; Loo, W.T.Y.; Yao, X.-J.; Leung, E.L.-H.; Chow, L.W.C.; Liu, L. Shikonin inhibits gefitinib-resistant non-small cell lung cancer by inhibiting TrxR and activating the EGFR proteasomal degradation pathway. Pharmacol. Res. 2017, 115, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Coco, S.; Truini, A.; Alama, A.; Dal Bello, M.G.; Venè, R.; Garuti, A.; Carminati, E.; Rijavec, E.; Genova, C.; Barletta, G.; et al. Afatinib resistance in non-small cell lung cancer involves the PI3K/AKT and MAPK/ERK signalling pathways and epithelial-to-mesenchymal transition. Target Oncol. 2015, 10, 393–404. [Google Scholar] [CrossRef] [PubMed]
- Hsu, S.-C.; Hung, M.-C. Characterization of a novel tripartite nuclear localization sequence in the EGFR family. J. Biol. Chem. 2007, 282, 10432–10440. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.K.; Nandekar, P.P.; Sangamwar, A.; Pérez-Sánchez, H.; Agarwal, S.M. Structure guided design and binding analysis of EGFR inhibiting analogues of erlotinib and AEE788 using ensemble docking, molecular dynamics and MM-GBSA. RSC Adv. 2016, 6, 65725–65735. [Google Scholar] [CrossRef]
- Liu, B.; Bernard, B.; Wu, J.H. Impact of EGFR point mutations on the sensitivity to gefitinib: Insights from comparative structural analyses and molecular dynamics simulations. Proteins 2006, 65, 331–346. [Google Scholar] [CrossRef]
- Rajith, B.; Chakraborty, C.; NagaSundaram, N.; Ali, S.K.; Zhu, H. Structural signature of the G719S-T790M double mutation in the EGFR kinase domain and its response to inhibitors. Sci. Rep. 2014, 4, 5868. [Google Scholar]
- Martínez-Jiménez, F.; Overington, J.P.; Al-Lazikani, B.; Marti-Renom, M.A. Rational design of non-resistant targeted cancer therapies. Sci. Rep. 2017, 7, 46632. [Google Scholar] [CrossRef] [PubMed]
- Stamos, J.; Sliwkowski, M.X.; Eigenbrot, C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J. Biol. Chem. 2002, 277, 46265–46272. [Google Scholar] [CrossRef]
- Ahmed, M.; Sadek, M.M.; Abouzid, K.A.; Wang, F. In silico design: Extended molecular dynamic simulations of a new series of dually acting inhibitors against EGFR and HER2. J. Mol. Graph. Model. 2013, 44, 220–231. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Nadeem, S.; Sirajuddin, M.; Ahmad, S.; Tirmizi, S.A.; Ali, M.I.; Hameed, A. Synthesis, spectral characterization and in vitro antibacterial evaluation and Petra/Osiris/Molinspiration analyses of new Palladium (II) iodide complexes with thioamides. AJM 2016, 52, 279–288. [Google Scholar] [CrossRef]
- Sander, T.; Freyss, J.; von Korff, M.; Rufener, C. DataWarrior: An Open-Source Program For Chemistry Aware Data Visualization And Analysis. J. Chem. Inf. Model. 2015, 55, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Mahalapbutr, P.; Chusuth, P.; Kungwan, N.; Chavasiri, W.; Wolschann, P.; Rungrotmongkol, T. Molecular recognition of naphthoquinone-containing compounds against human DNA topoisomerase IIα ATPase domain: A molecular modeling study. J. Mol. Liq. 2017, 247, 374–385. [Google Scholar] [CrossRef]
- Panman, W.; Nutho, B.; Chamni, S.; Dokmaisrijan, S.; Kungwan, N.; Rungrotmongkol, T. Computational screening of fatty acid synthase inhibitors against thioesterase domain. J. Biomol. Struct. Dyn. 2017, 36, 4114–4125. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09 Revision E.01; Gaussian Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Case, D.A.; Betz, R.M.; Cerutti, D.S.; Cheatham, T.E., III; Darden, T.A.; Duke, R.E.; Giese, T.J.; Gohlke, H.; Goetz, A.W.; Homeyer, N.; et al. AMBER 2016; University of California: San Francisco, CA, USA, 2016. [Google Scholar]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Olsson, M.H.M.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- York, D.M.; Darden, T.A.; Pedersen, L.G. The effect of long-range electrostatic interactions in simulations of macromolecular crystals: A comparison of the Ewald and truncated list methods. J. Chem. Phys. 1993, 99, 8345–8348. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are currently not available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 value (μM) Against: | IC50 Against EGFR-TK | |||
|---|---|---|---|---|---|
| A431 | A549 | H1650 | H1975 | ||
| 1b | 34.0 ± 7.38 | 50.9 ± 3.8 | - | - | - |
| 1c | 8.0 ± 1.2 | 25.4 ± 1.2 | 13.1 ± 2.8 | 9.2 ± 2.3 | 10.3 μM |
| 2a | 9.9 ± 4.9 | 20.2 ± 1.9 | 10.0 ± 0.7 | 5.1 ± 0.3 | 13.8 μM |
| 2b | 29.5 ± 3.5 | 69.4 ± 7.6 | - | - | - |
| 2c | 24.6 ± 6.0 | >100 | - | - | - |
| 2d | 26.6 ± 6.9 | 25.4 ± 1.7 | - | - | - |
| 3c | 20.7 ± 9.8 | >100 | - | - | - |
| 3e | 10.5 ± 7.4 | >100 | 23.8 ± 2.1 | 14.6 ± 1.1 | 15.4 μM |
| 3f | 18.9 ± 11.1 | >100 | - | - | - |
| 4a | 38.9 ± 5.2 | >100 | - | - | - |
| 4b | 25.0 ± 8.7 | 44.1 ± 9.4 | - | - | - |
| 4c | 26.6 ± 5.5 | 20.2 ± 1.9 | - | - | - |
| 4d | 25.1 ± 4.3 | >100 | - | - | - |
| 4e | 10.0 ± 5.8 | 44.2 ± 5.3 | 22.2 ± 7.4 | 17.8 ± 1.8 | - |
| 4f | 38.8 ± 1.6 | >100 | - | - | - |
| 4g | 21.8 ± 5.3 | >100 | - | - | - |
| 4h | 48.8 ± 3.6 | >100 | - | - | - |
| 4j | 24.0 ± 2.6 | >100 | - | - | - |
| 4k | 14.9 ± 7.6 | >100 | - | - | - |
| 4l | 29.1 ± 4.1 | >100 | - | - | - |
| 4m | 55.0 ± 6.7 | >100 | - | - | - |
| 4n | 22.0 ± 5.3 | >100 | - | - | - |
| 4o | 21.7 ± 6.8 | >100 | - | - | - |
| 4p | 37.5 ± 4.0 | >100 | - | - | - |
| 4q | 25.9 ± 3.8 | >100 | - | - | - |
| 4s | 39.5 ± 7.4 | 25.4 ± 2.2 | - | - | - |
| 4t | 5.0 ± 3.5 | >100 | 9.2 ± 0.8 | 6.7 ± 2.8 | - |
| 4u | >100 | >100 | - | - | - |
| 4v | >100 | >100 | - | - | - |
| 4w | 24.2 ± 4.9 | >100 | - | - | - |
| 4x | 41.5 ± 6.6 | >100 | - | - | - |
| 4y | 41.5 ± 2.0 | 49.4 ± 7.9 | - | - | - |
| 4aa | >100 | 74.4 ± 6.5 | - | - | - |
| 5a | >100 | >100 | - | - | - |
| 6b | 33.4 ± 3.1 | >100 | - | - | - |
| 6e | 40.0 ± 3.9 | >100 | - | - | - |
| Erlotinib | 0.6 ± 0.1 | 18.8 ± 2.4 | - | - | 24.29 nM |
| Afatinib | - | - | 2.4 ± 0.4 | 1.9 ± 0.3 | - |
| ADMET Parameter | 1c | 2a | 3e | Erlotinib | |
|---|---|---|---|---|---|
| Toxicity risk | Mutation a | +++ | +++ | +++ | +++ |
| Tumor a | +++ | +++ | +++ | +++ | |
| Irritant a | +++ | +++ | +++ | +++ | |
| Reproduction effective a | +++ | +++ | +++ | +++ | |
| Physicochemical properties | Molecular weight (g/mol) b | 224.25 | 254.28 | 314.33 | 393.44 |
| cLOGP b | 2.96 | 2.88 | 2.75 | 2.79 | |
| TPSA (Å2) b | 37.30 | 46.53 | 65.00 | 74.73 | |
| Solubility class c | Moderately soluble | Moderately soluble | Moderately soluble | Poorly soluble | |
| Drug likeness | Lipinski’s rule of five c | Yes; 0 violation | Yes; 0 violation | Yes; 0 violation | Yes; 0 violation |
| Ghose c | Yes | Yes | Yes | Yes | |
| Veber c | Yes | Yes | Yes | Yes | |
| Egan c | Yes | Yes | Yes | Yes | |
| Muegge c | Yes | Yes | Yes | Yes | |
| Pharmacokinetic | Gastro Intestinal absorption (%) c | High | High | High | High |
| Blood-brain barrier permeant c | Yes | Yes | Yes | Yes | |
| P-gp substrate c | No | No | No | No | |
| CYP1A2 inhibitor c | No | Yes | Yes | Yes | |
| CYP2C19 inhibitor c | Yes | Yes | Yes | Yes | |
| CYP2C9 inhibitor c | Yes | Yes | Yes | Yes | |
| CYP2D6 inhibitor c | No | No | No | Yes | |
| CYP3A4 inhibitor c | No | Yes | Yes | Yes | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sangpheak, K.; Tabtimmai, L.; Seetaha, S.; Rungnim, C.; Chavasiri, W.; Wolschann, P.; Choowongkomon, K.; Rungrotmongkol, T. Biological Evaluation and Molecular Dynamics Simulation of Chalcone Derivatives as Epidermal Growth Factor-Tyrosine Kinase Inhibitors. Molecules 2019, 24, 1092. https://doi.org/10.3390/molecules24061092
Sangpheak K, Tabtimmai L, Seetaha S, Rungnim C, Chavasiri W, Wolschann P, Choowongkomon K, Rungrotmongkol T. Biological Evaluation and Molecular Dynamics Simulation of Chalcone Derivatives as Epidermal Growth Factor-Tyrosine Kinase Inhibitors. Molecules. 2019; 24(6):1092. https://doi.org/10.3390/molecules24061092
Chicago/Turabian StyleSangpheak, Kanyani, Lueacha Tabtimmai, Supaphorn Seetaha, Chompoonut Rungnim, Warinthorn Chavasiri, Peter Wolschann, Kiattawee Choowongkomon, and Thanyada Rungrotmongkol. 2019. "Biological Evaluation and Molecular Dynamics Simulation of Chalcone Derivatives as Epidermal Growth Factor-Tyrosine Kinase Inhibitors" Molecules 24, no. 6: 1092. https://doi.org/10.3390/molecules24061092
APA StyleSangpheak, K., Tabtimmai, L., Seetaha, S., Rungnim, C., Chavasiri, W., Wolschann, P., Choowongkomon, K., & Rungrotmongkol, T. (2019). Biological Evaluation and Molecular Dynamics Simulation of Chalcone Derivatives as Epidermal Growth Factor-Tyrosine Kinase Inhibitors. Molecules, 24(6), 1092. https://doi.org/10.3390/molecules24061092