
A Smart Epoxy Composite Based on Phase Change Microcapsules: Preparation, Microstructure, Thermal and Dynamic Mechanical Performances



Abstract
1. Introduction
2. Results and Discussion
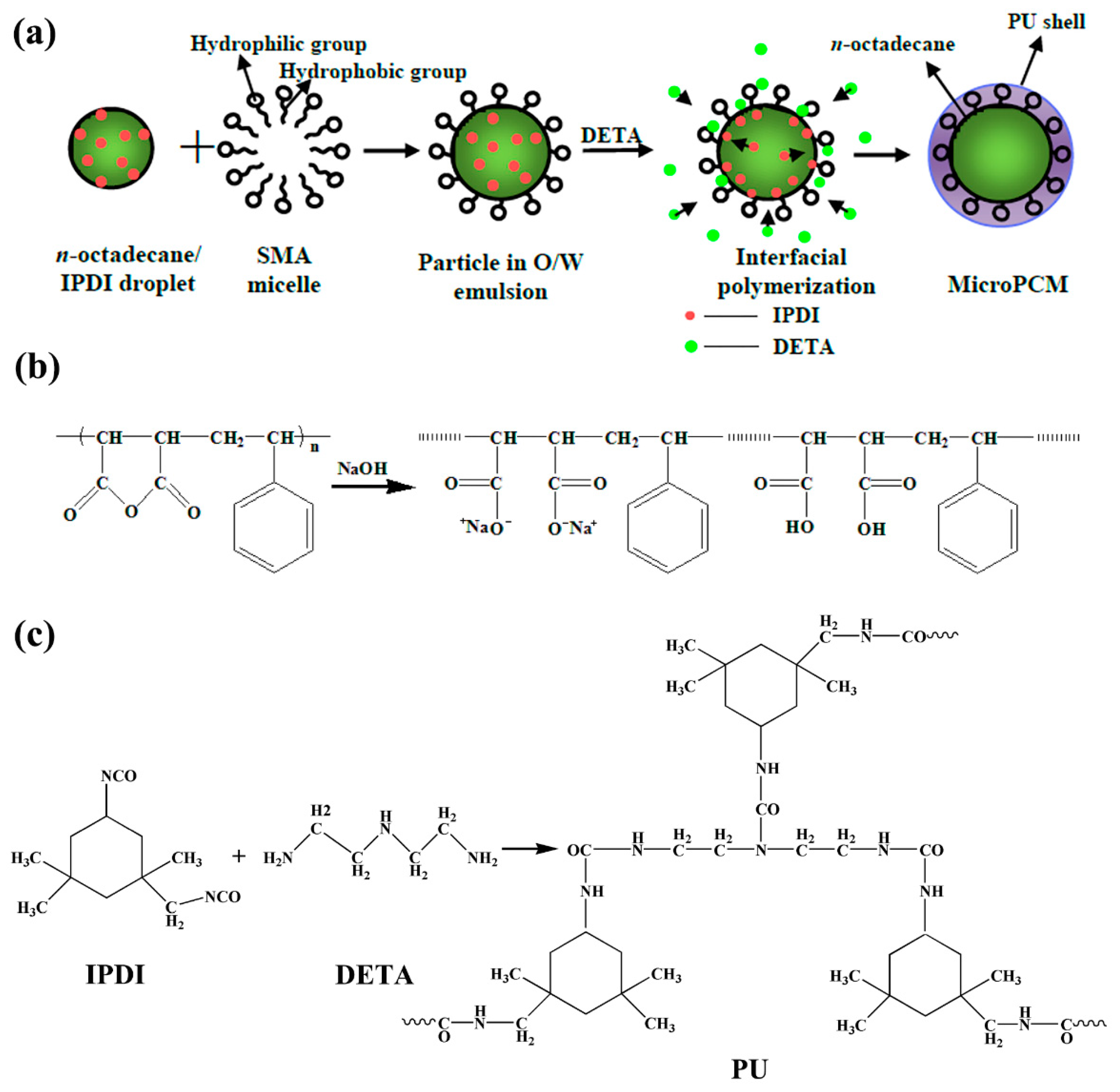
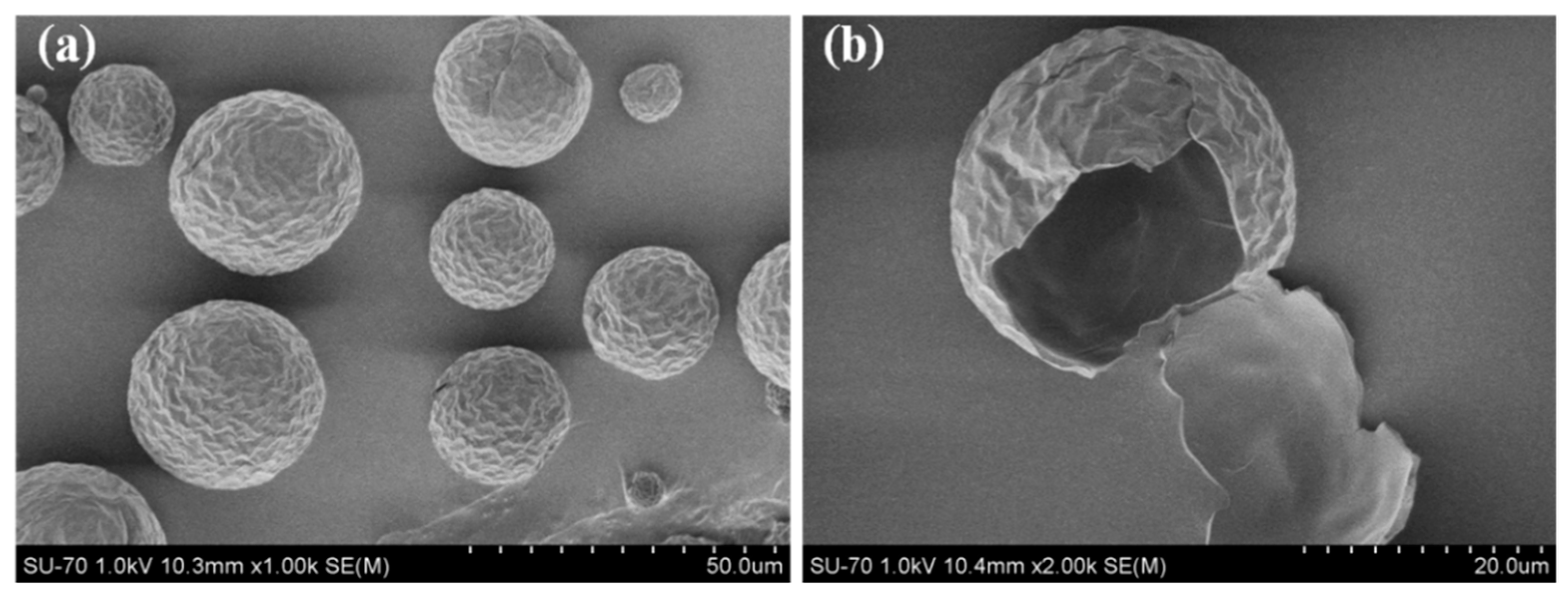
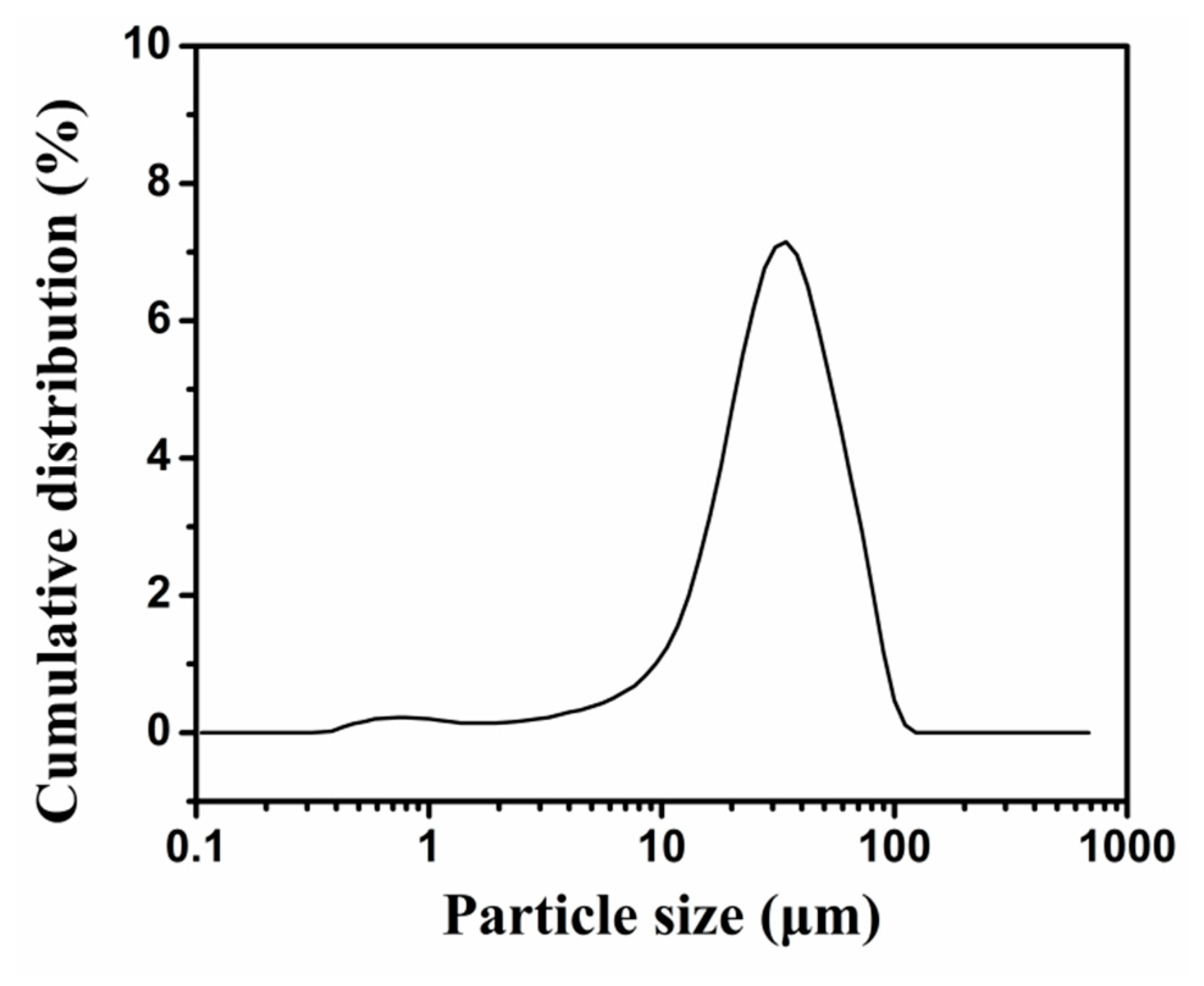
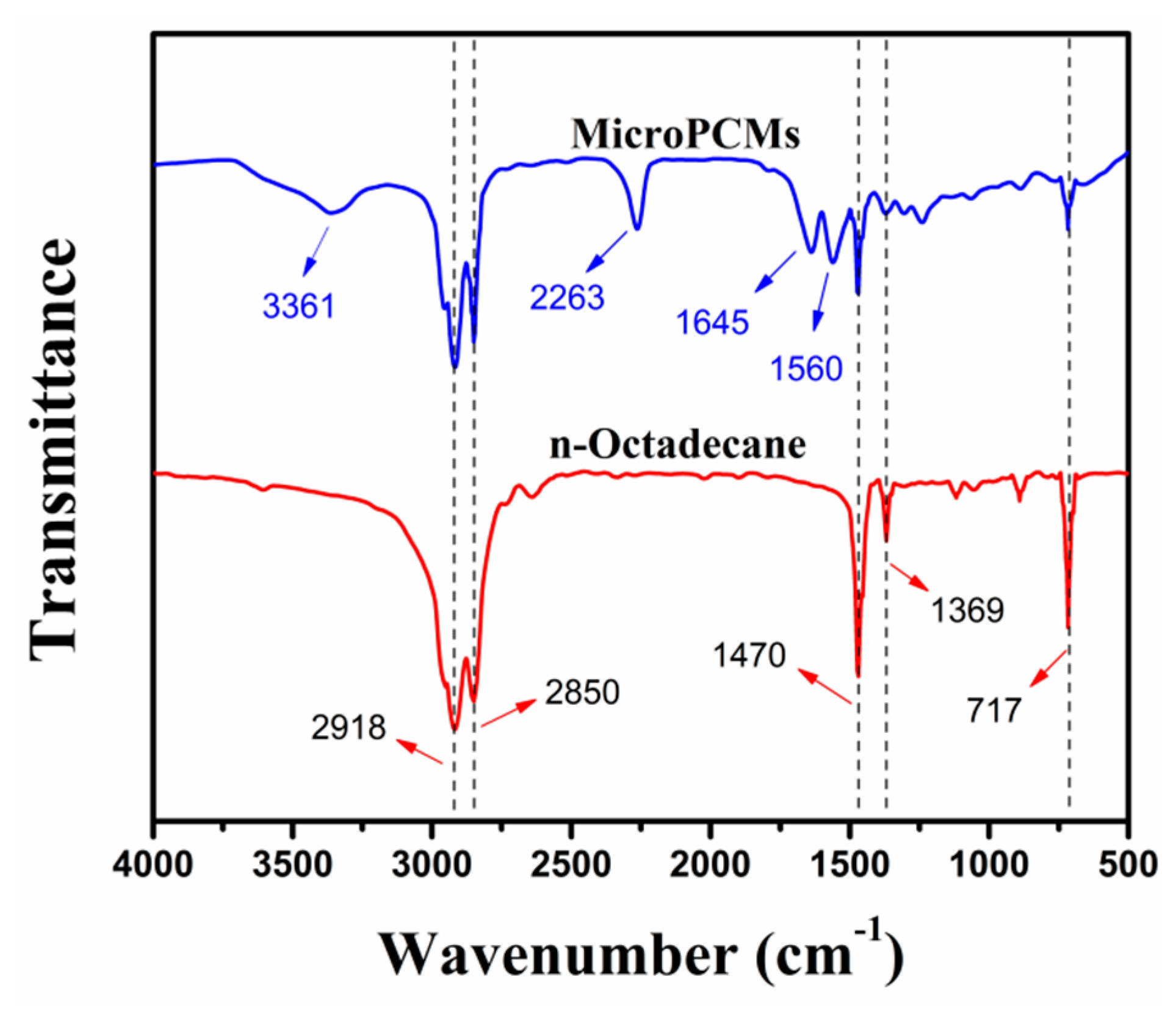
2.1. Synthesis and Structure of PU-Shelled n-Octadecane MicroPCMs
2.2. Microstructure of MicroPCM/Epoxy Composites
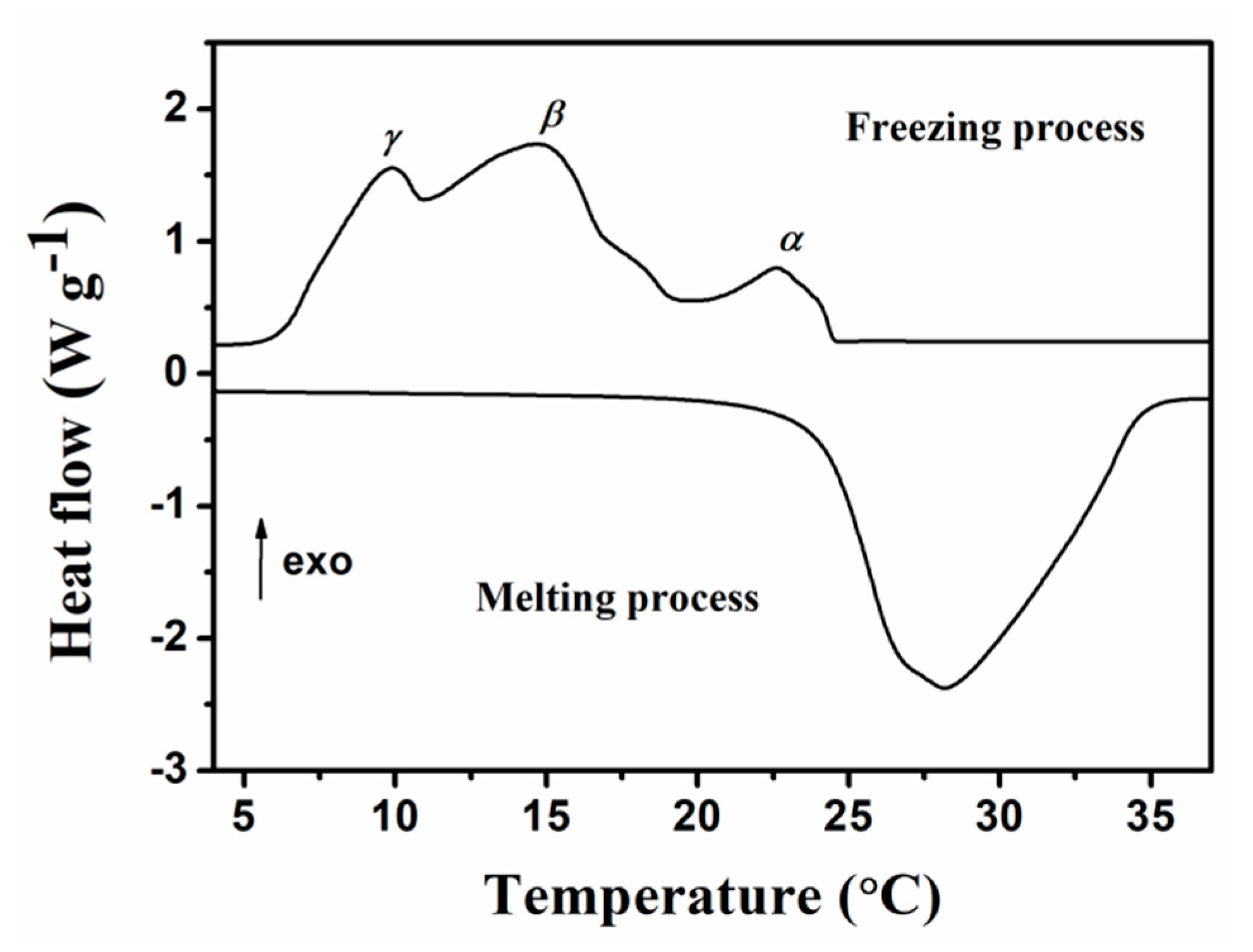
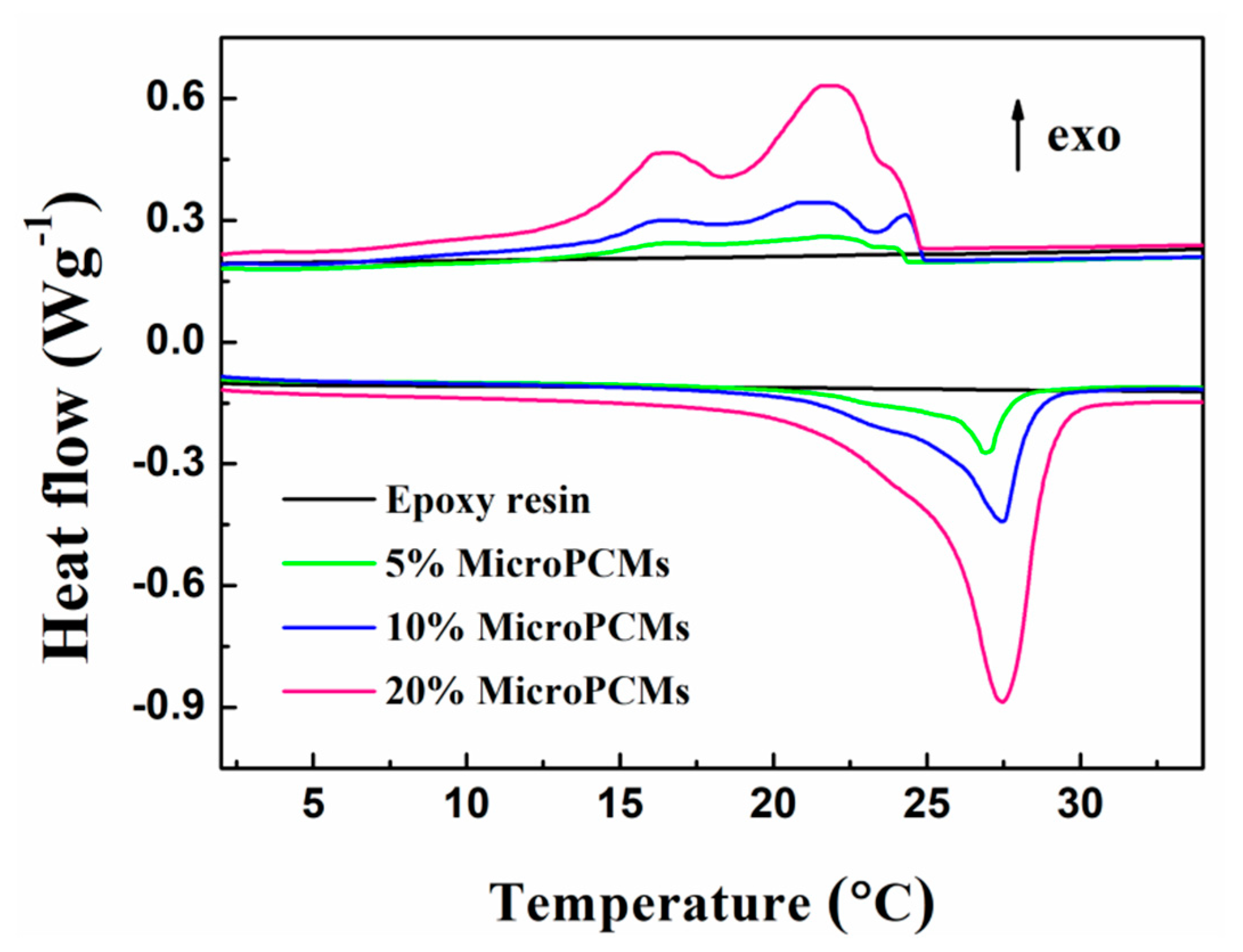
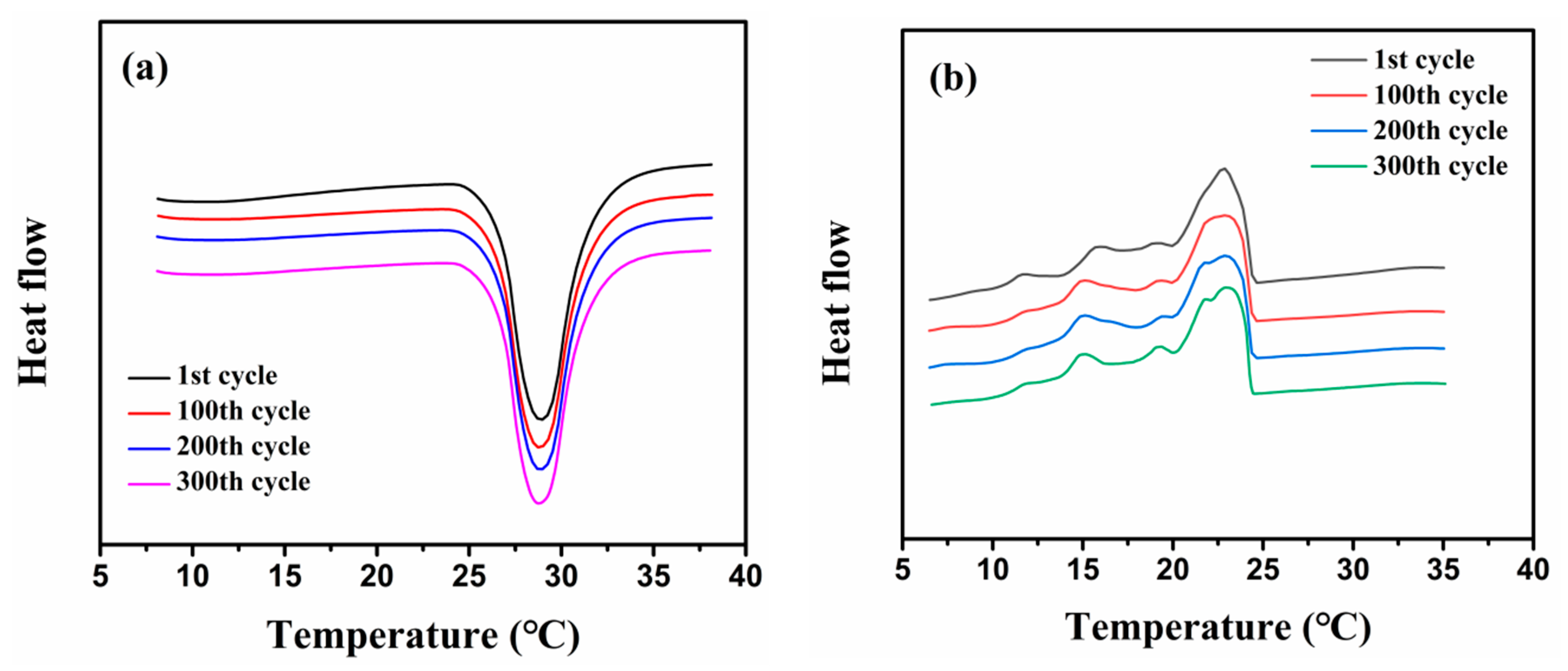
2.3. Phase Change Properties
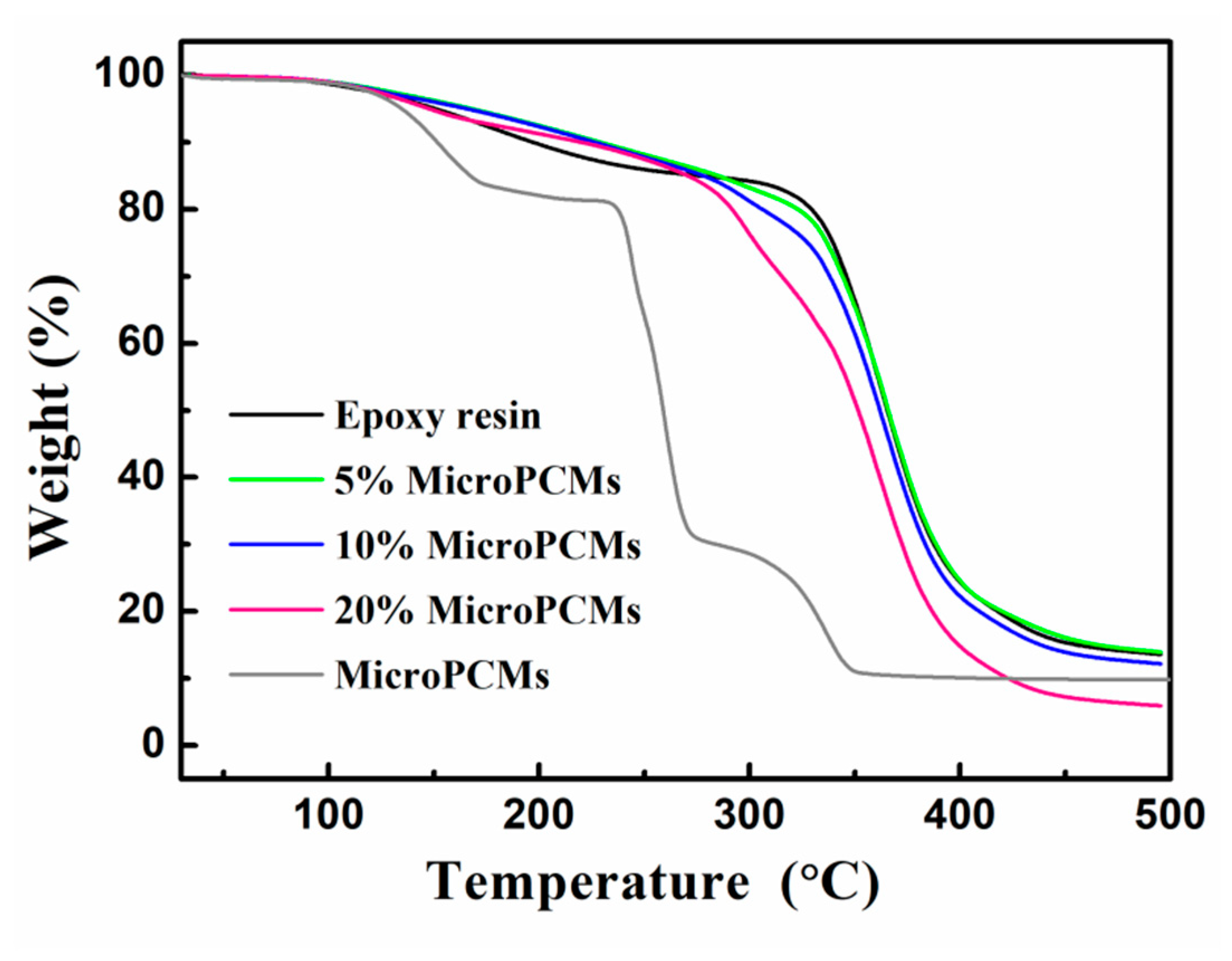
2.4. Thermal Stability
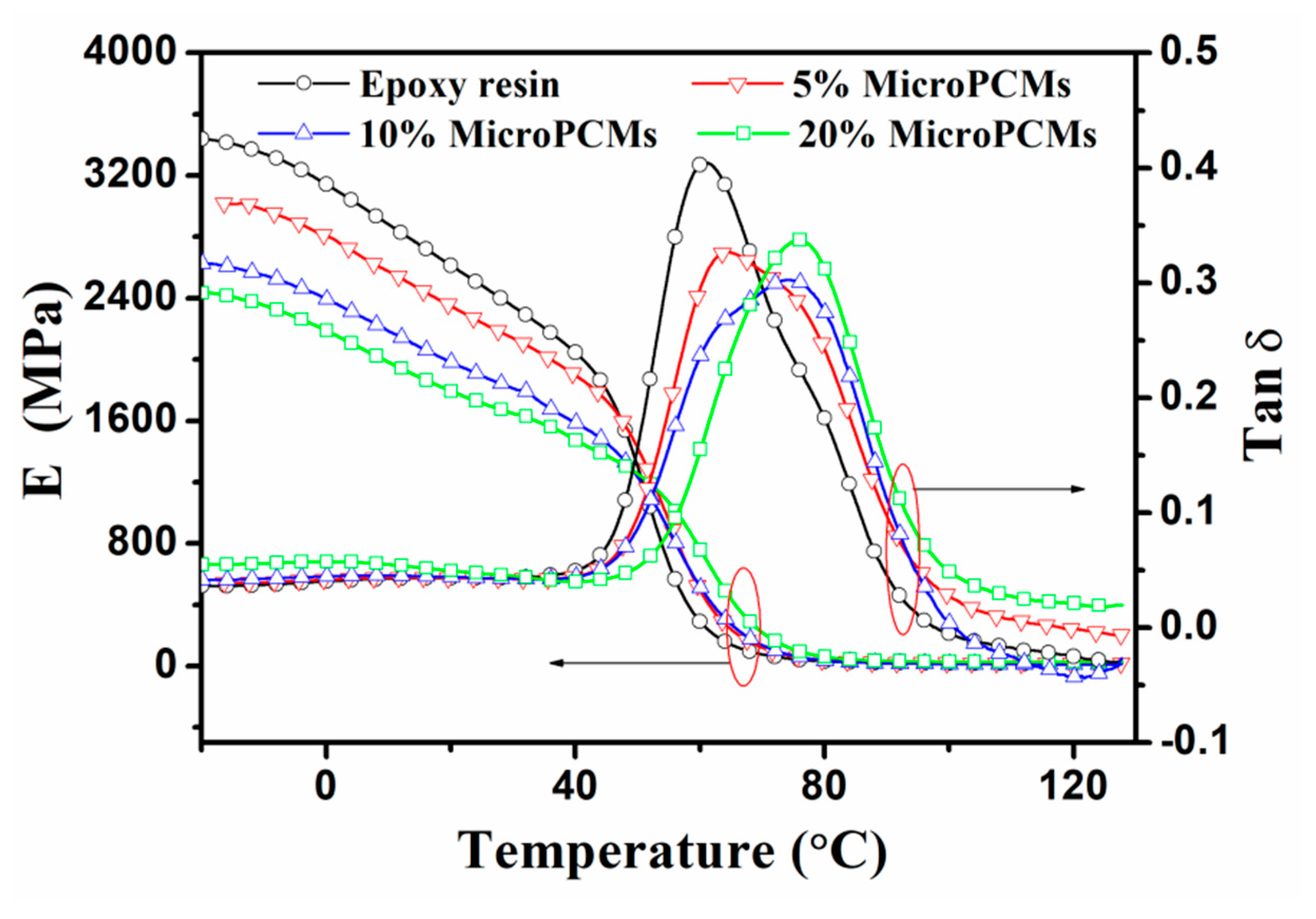
2.5. Dynamic Mechanical Properties
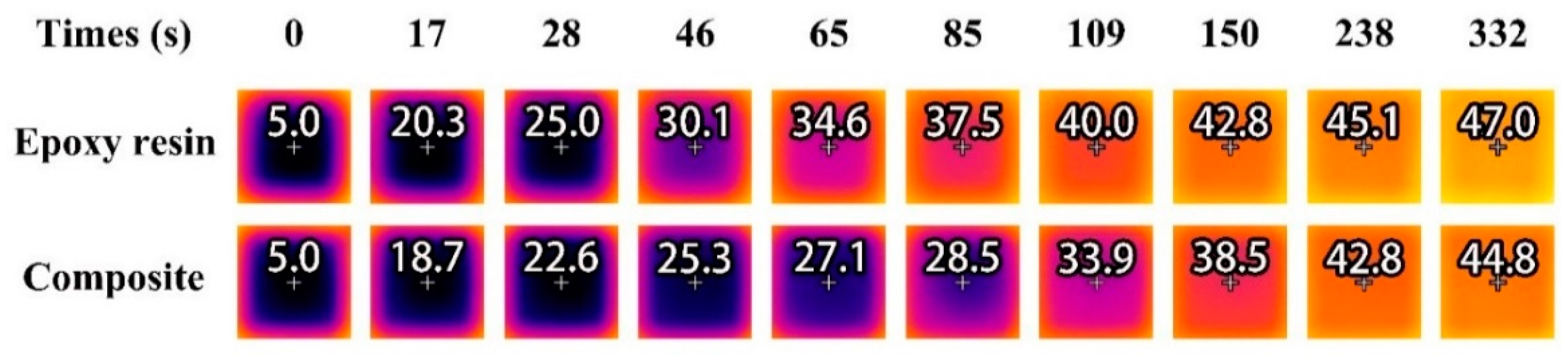
2.6. Evaluation of Temperature Regulation Capacity
3. Experimental
3.1. Materials
3.2. Preparation of PU-Shelled n-Octadecane MicroPCMs
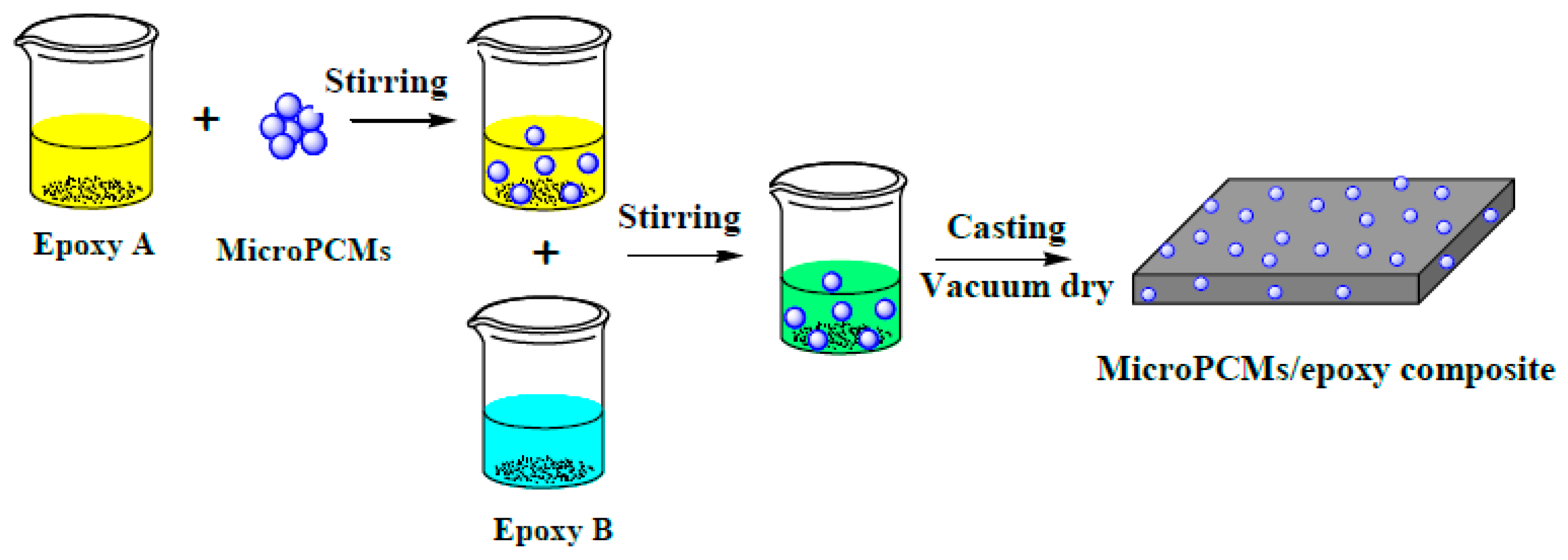
3.3. Fabrication of PU-Shelled n-Octadecane MicroPCMs Filled Epoxy Composites
3.4. Morphological Observation
3.5. Size Measurement of MicroPCMs
3.6. FTIR Measurement
3.7. Measurement of Phase Change Properties
3.8. Thermal Stability Test
3.9. Measurement of Dynamic Mechanical Properties
3.10. Thermal Conductivity Test
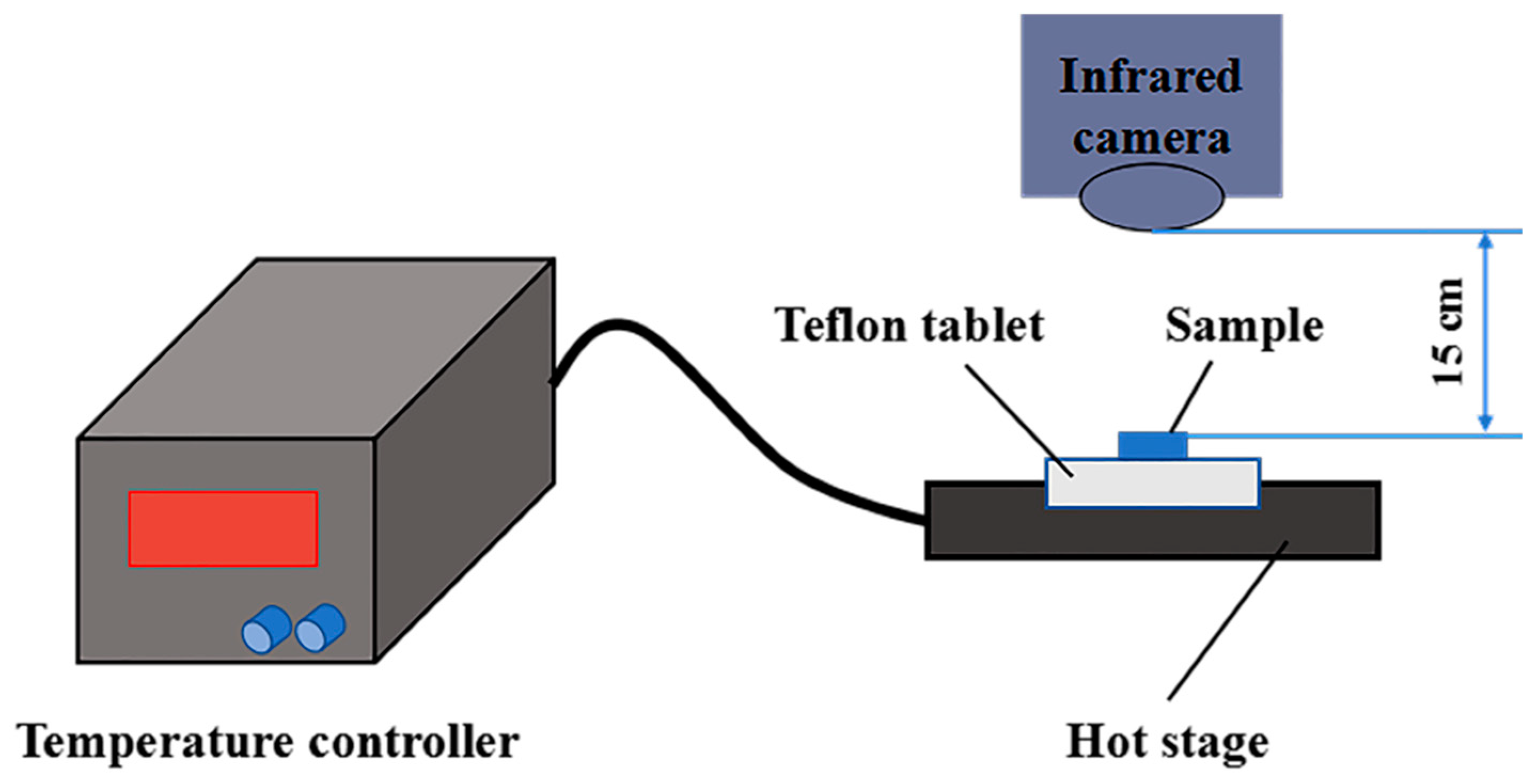
3.11. Infrared Thermography
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chandel, S.S.; Agarwal, T. Review of current state of research on energy storage, toxicity, health hazards and commercialization of phase changing materials. Renew. Sustain. Energy Rev. 2017, 67, 581–596. [Google Scholar] [CrossRef]
- Pielichowska, K.; Pielichowski, K. Phase change materials for thermal energy storage. Prog. Mater. Sci. 2014, 65, 67–123. [Google Scholar] [CrossRef]
- Zhang, H.Z.; Xu, Q.Y.; Zhao, Z.M.; Zhang, J.; Sun, Y.J.; Sun, L.X.; Xu, F.; Sawada, Y. Preparation and thermal performance of gypsum boards incorporated with microencapsulated phase change materials for thermal regulation. Sol. Energy Mater. Sol. Cells 2012, 102, 93–102. [Google Scholar] [CrossRef]
- Zhang, H.; Xing, F.; Cui, H.Z.; Chen, D.Z.; Ouyang, X.; Xu, S.Z.; Wang, J.X.; Huang, Y.T.; Zuo, J.D.; Tang, J.N. A novel phase-change cement composite for thermal energy storage: Fabrication, thermal and mechanical properties. Appl. Energy 2016, 170, 130–139. [Google Scholar] [CrossRef]
- Cui, H.Z.; Liao, W.Y.; Mi, X.; Lo, T.Y.; Chen, D.Z. Study on functional and mechanical properties of cement mortar with graphite-modified microencapsulated phase-change materials. Energy Build. 2015, 105, 273–284. [Google Scholar] [CrossRef]
- Giro-Paloma, J.; Martínez, M.; Cabeza, L.F.; Fernández, A.I. Types, methods, techniques, and applications for microencapsulated phase change materials (MPCM): A review. Renew. Sustain. Energy Rev. 2016, 53, 1059–1075. [Google Scholar] [CrossRef]
- Cao, V.D.; Pilehvar, S.; Salas-Bringas, C.; Szczotok, A.M.; Rodriguez, J.F.; Carmona, M.; Al-Manasir, N.; Kjøniksen, A.L. Microencapsulated phase change materials for enhancing the thermal performance of Portland cement concrete and geopolymer concrete for passive building applications. Energy Convers. Manag. 2017, 133, 56–66. [Google Scholar] [CrossRef]
- Akeiber, H.; Nejat, P.; Majid, M.Z.A.; Wahid, M.A.; Jomehzadeh, F.; Famileh, I.Z.; Zaki, S.A. A review on phase change material (PCM) for sustainable passive cooling in building envelopes. Renew. Sustain. Energy Rev. 2016, 60, 1470–1497. [Google Scholar] [CrossRef]
- Sharma, A.; Sharma, S.D.; Buddhi, D. Accelerated thermal cycle test of acet-amide, stearic acid and paraffin wax for solar thermal latent heat storage applications. Energy Convers. Manag. 2002, 43, 1923–1930. [Google Scholar] [CrossRef]
- Ventola, L.; Diarte, M.A.C.; Calvet, T.; Angulo, I.; Vivanco, M.; Bernar, M. Molecular alloys as phase change materials (MAPCM) for energy storage and thermal protection at temperatures from 70 °C to 80 °C. J. Phys. Chem. Solids 2005, 66, 1668–1674. [Google Scholar] [CrossRef]
- Su, J.F.; Wang, L.X.; Ren, L. Synthesis of polyurethane microPCMs containing n-octadecane by interfacial polycondensation: Influence of styrene-maleic anhydride as a surfactant. Colloid. Surf. A 2007, 299, 268–275. [Google Scholar] [CrossRef]
- Sarı, A.; Alkan, C.; Döğüşcü, D.K.; Kızıl, Ç. Micro/nano encapsulated n-tetracosane and n-octadecane eutectic mixture with polystyrene shell for low-temperature latent heat thermal energy storage applications. Sol. Energy 2015, 115, 195–203. [Google Scholar] [CrossRef]
- Yang, Y.; Ye, X.; Luo, J.; Song, G.; Liu, Y.; Tang, G. Polymethylmethacrylate based phase change microencapsulation for solar energy storage with silicon nitride. Sol. Energy 2015, 115, 289–296. [Google Scholar] [CrossRef]
- Su, J.F.; Wang, X.Y.; Dong, H. Influence of temperature on the deformation behaviors of melamine–formaldehyde microcapsules containing phase change material. Mater. Lett. 2012, 84, 158–161. [Google Scholar] [CrossRef]
- Zhang, X.X.; Fan, Y.F.; Tao, X.M.; Yick, K.L. Crystallization and prevention of supercooling of microencapsulated n-alkanes. J. Colloid Interface Sci. 2005, 281, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.; Li, Q.; Zhu, Y.; Chen, K.; Tian, C.; Wang, J.; Bai, R. Nanoencapsulation of n-octadecane phase change material with silica shell through interfacial hydrolysis and polycondensation in miniemulsion. Energy 2015, 93, 1684–1692. [Google Scholar] [CrossRef]
- Yu, S.; Wang, X.; Wu, D. Microencapsulation of n-octadecane phase change material with calcium carbonate shell for enhancement of thermal conductivity and serving durability: Synthesis, microstructure, and performance evaluation. Appl. Energy 2014, 114, 632–643. [Google Scholar] [CrossRef]
- Salaun, F.; Devaux, E.; Bourbigot, S.; Rumeau, P. Thermoregulating response of cotton fabric containing microencapsulated phase change materials. Thermochim. Acta 2010, 506, 82–93. [Google Scholar] [CrossRef]
- Su, J.F.; Wang, S.B.; Huang, Z.; Liang, J.S. Polyurethane microPCMs containing n-octadecane applied in building materials synthesized by interfacial polycondensation: Thermal stability and heat absorption simulation. Adv. Mater. Res. 2010, 96, 121–127. [Google Scholar] [CrossRef]
- Tomizawa, Y.; Sasaki, K.; Kuroda, A.; Takeda, R.; Kaito, Y. Experimental and numerical study on phase change material (PCM) for thermal management of mobile devices. Appl. Therm. Eng. 2016, 98, 320–329. [Google Scholar] [CrossRef]
- Wang, Z.Y.; Situ, W.F.; Li, X.X.; Zhang, G.Q.; Huang, Z.; Yuang, W.Z.; Yang, C.Z.; Yang, C.X. Novel shape stabilized phase change material based on epoxy matrix with ultrahigh cycle life for thermal energy storage. Appl. Therm. Eng. 2017, 123, 1006–1012. [Google Scholar] [CrossRef]
- Su, J.F.; Wang, X.Y.; Huang, Z.; Zhao, Y.H.; Yuan, X.Y. Thermal conductivity of microPCMs-filled epoxy matrix composites. Colloid Polym. Sci. 2011, 289, 1535–1542. [Google Scholar] [CrossRef]
- Su, J.F.; Zhao, Y.H.; Wang, X.Y.; Dong, H.; Wang, S.B. Effect of interface debonding on the thermal conductivity of microencapsulated paraffin filled epoxy matrix composites. Compos. Part A-Appl. Sci. 2012, 43, 325–332. [Google Scholar] [CrossRef]
- Su, J.F.; Wang, S.B.; Zhou, J.W.; Huang, Z.; Zhao, Y.H.; Yuan, X.Y.; Zhang, Y.Y.; Kou, J.B. Fabrication and interfacial morphologies of methanol–melamine–formaldehyde (MMF) shell microPCMs/epoxy composites. Colloid Polym. Sci. 2011, 289, 169–177. [Google Scholar] [CrossRef]
- Su, J.F.; Wang, X.Y.; Wang, S.B.; Zhao, Y.H.; Zhu, K.Y.; Yuan, X.Y. Interface stability behaviors of methanol-melamine-formaldehyde shell microPCMs/epoxy matrix composites. Polym. Compos. 2011, 32, 810–820. [Google Scholar] [CrossRef]
- Wang, X.Y.; Su, J.F.; Wang, S.B.; Zhao, Y.H. The effect of interface debonding behaviors on the mechanical properties of microPCMs/epoxy composites. Polym. Compos. 2011, 32, 1439–1450. [Google Scholar] [CrossRef]
- Zhang, H.Z.; Wang, X.D. Synthesis and properties of microencapsulated n-octadecane with polyuria shells containing different soft segments for heat energy storage and thermal regulation. Sol. Energy Mater. Sol. Cells 2009, 93, 1366–1376. [Google Scholar] [CrossRef]
- Zhang, X.X.; Fan, Y.F.; Tao, X.M.; Yick, K.L. Fabrication and properties of microcapsules and nanocapsules containing n-octadecane. Mater. Chem. Phys. 2004, 88, 300–307. [Google Scholar] [CrossRef]
- Xie, B.; Liu, G.; Jiang, S.; Zhao, Y.; Wang, D. Crystallization behaviors of n-octadecane in confined space: Crossover of rotator phase from transient to metastable induced by surface freezing. J. Phys. Chem. B 2008, 112, 13310–13315. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.T.; Zhang, H.; Wan, X.J.; Chen, D.Z.; Chen, X.F.; Ye, X.; Ouyang, X.; Qin, S.Y.; Wen, H.X.; Tang, J.N. Carbon nanotube-enhanced double-walled phase-change microcapsules for thermal energy storage. J. Mater. Chem. A 2017, 5, 7482–7493. [Google Scholar] [CrossRef]
- Chen, D.Z.; Chen, Y.; Ouyang, X.; Zuo, J.; Ye, X. Influence of MicroPCMs on thermal and dynamic mechanical properties of a biodegradable P3HB4HB composite. Compos. Part B-Eng. 2014, 56, 245–248. [Google Scholar] [CrossRef]
- Wertzberger, B.E.; Steere, J.T.; Pfeifer, R.M.; Nensel, M.A.; Latta, M.A.; Gross, S.M. Physical characterization of a self-healing dental restorative material. J. Appl. Polym. Sci. 2010, 118, 428–434. [Google Scholar] [CrossRef]
- Chen, D.Z.; Tang, C.Y.; Chan, K.C.; Tsui, C.P.; Yu, P.H.F.; Leung, M.C.P.; Uskokovic, P.S. Dynamic mechanical properties and in vitro bioactivity of PHBHV/HA nanocomposite. Compos. Sci. Technol. 2007, 67, 1617–1626. [Google Scholar] [CrossRef]
- Chen, L.; Tang, C.Y.; Tsui, C.P.; Chen, D.Z. Mechanical properties and in vitro evaluation of bioactivity and degradation of dexamethasone-releasing poly-d-l-lactide/nano-hydroxyapatite composite scaffolds. J. Mech. Behav. Biomed. Mater. 2012, 22, 41–55. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Tang, C.Y.; Chen, D.Z.; Wong, C.T.; Tsui, C.P. Fabrication and characterization of poly-d-l-lactide/nano-hydroxyapatite composite scaffolds with poly (ethylene glycol) coating and dexamethasone releasing. Compos. Sci. Technol. 2011, 71, 1842–1849. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Melting Process | Freezing Process | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Tm,s (°C) | Tm (°C) | Tm,e (°C) | ΔHm (J g−1) | Tf,s (°C) | Tα (°C) | Tβ (°C) | Tγ (°C) | ΔHf (J g−1) | |
| Epoxy resin MicroPCMs | 24.10 | 28.14 | 34.70 | 182.40 | 24.51 | 22.58 | 14.67 | 9.90 | 183.20 |
| 5%MicroPCMs | 14.71 | 27.41 | 31.36 | 7.55 | 24.17 | 24.08 | 23.46 | 17.57 | 7.58 |
| 10%MicroPCMs | 14.85 | 27.31 | 31.13 | 15.77 | 24.54 | 24.25 | 23.46 | 15.30 | 14.92 |
| 20%MicroPCMs | 16.50 | 27.95 | 31.60 | 33.24 | 24.64 | 24.02 | 23.79 | 16.01 | 32.27 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, Q.; Chen, Y.; Hong, J.; Jin, S.; Zou, G.; Chen, L.; Chen, D.-Z. A Smart Epoxy Composite Based on Phase Change Microcapsules: Preparation, Microstructure, Thermal and Dynamic Mechanical Performances. Molecules 2019, 24, 916. https://doi.org/10.3390/molecules24050916
Hu Q, Chen Y, Hong J, Jin S, Zou G, Chen L, Chen D-Z. A Smart Epoxy Composite Based on Phase Change Microcapsules: Preparation, Microstructure, Thermal and Dynamic Mechanical Performances. Molecules. 2019; 24(5):916. https://doi.org/10.3390/molecules24050916
Chicago/Turabian StyleHu, Qinghong, Yan Chen, Jiaoling Hong, Shan Jin, Guangjin Zou, Ling Chen, and Da-Zhu Chen. 2019. "A Smart Epoxy Composite Based on Phase Change Microcapsules: Preparation, Microstructure, Thermal and Dynamic Mechanical Performances" Molecules 24, no. 5: 916. https://doi.org/10.3390/molecules24050916
APA StyleHu, Q., Chen, Y., Hong, J., Jin, S., Zou, G., Chen, L., & Chen, D.-Z. (2019). A Smart Epoxy Composite Based on Phase Change Microcapsules: Preparation, Microstructure, Thermal and Dynamic Mechanical Performances. Molecules, 24(5), 916. https://doi.org/10.3390/molecules24050916