Multi-Steps Fragmentation-Ion Trap Mass Spectrometry Coupled to Liquid Chromatography Diode Array System for Investigation of Olaparib Related Substances


Abstract
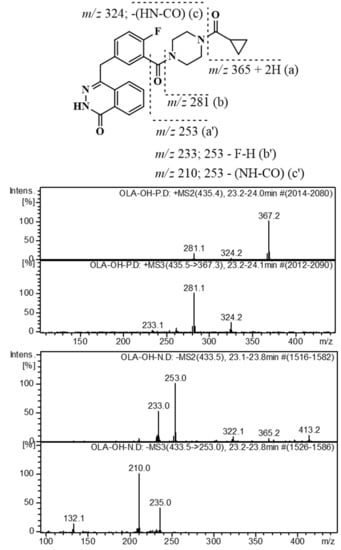
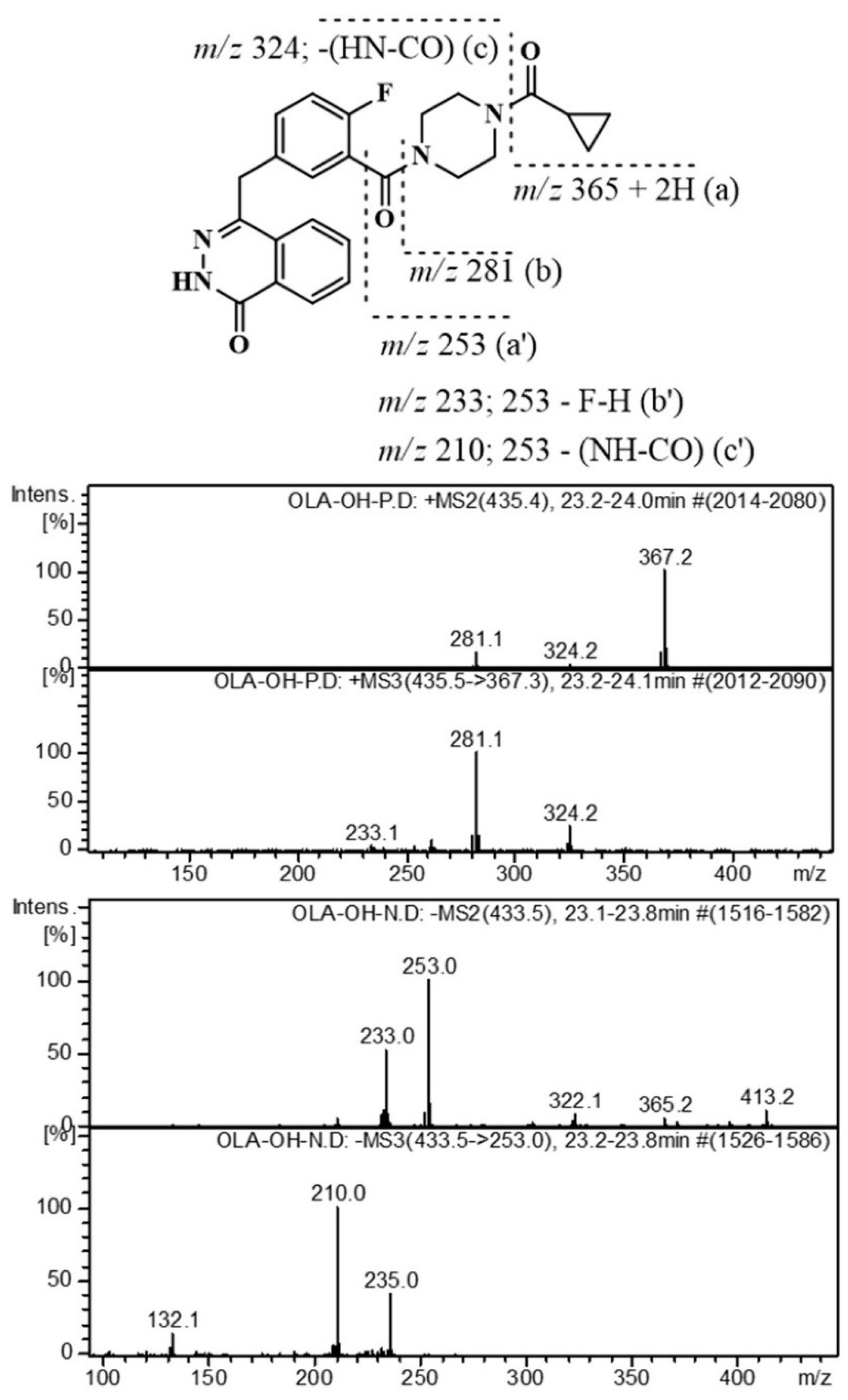
:
1. Introduction
2. Results and Discussion
2.1. Optimization of Chromatographic and QQQ-MS Conditions
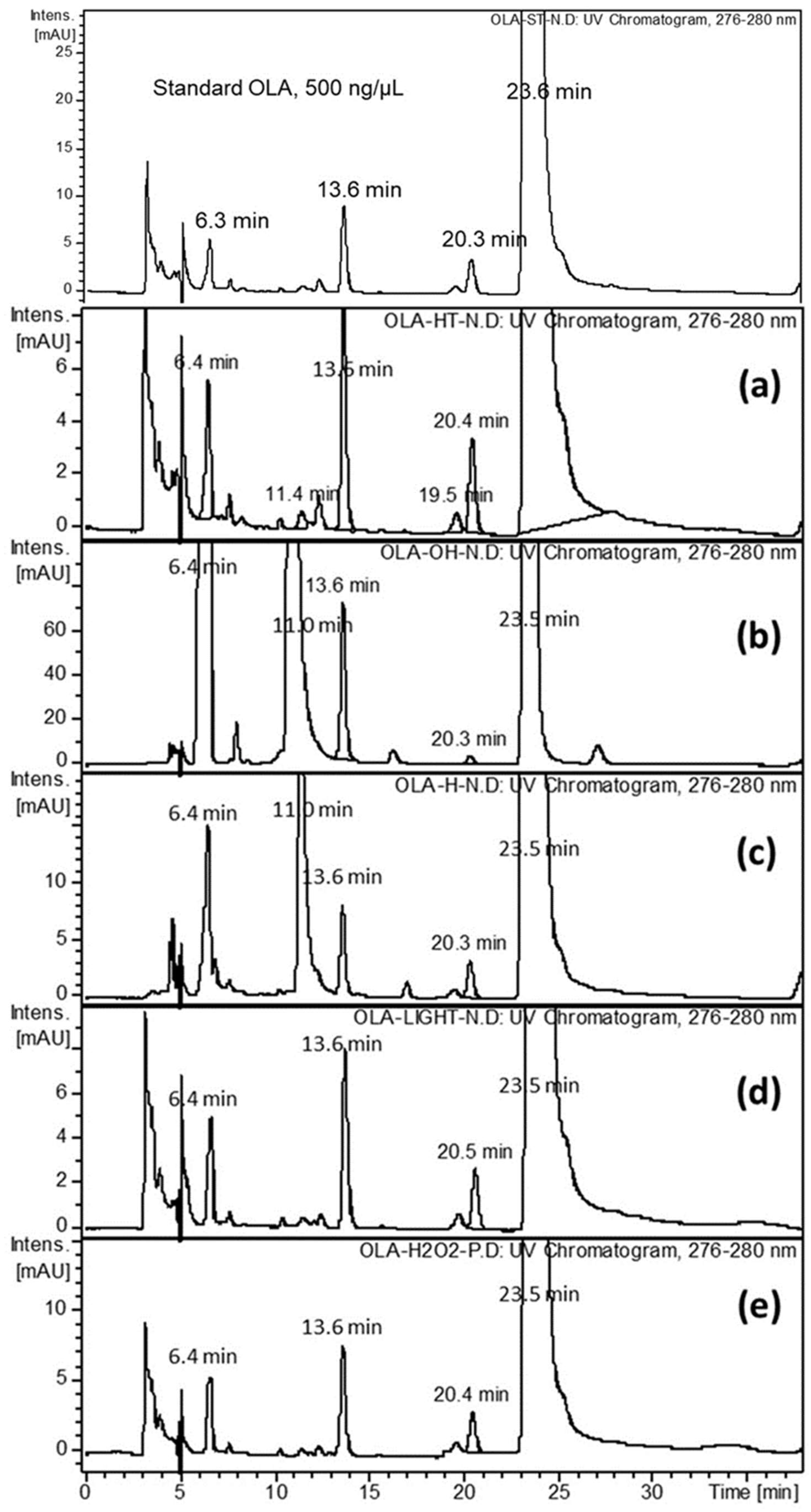
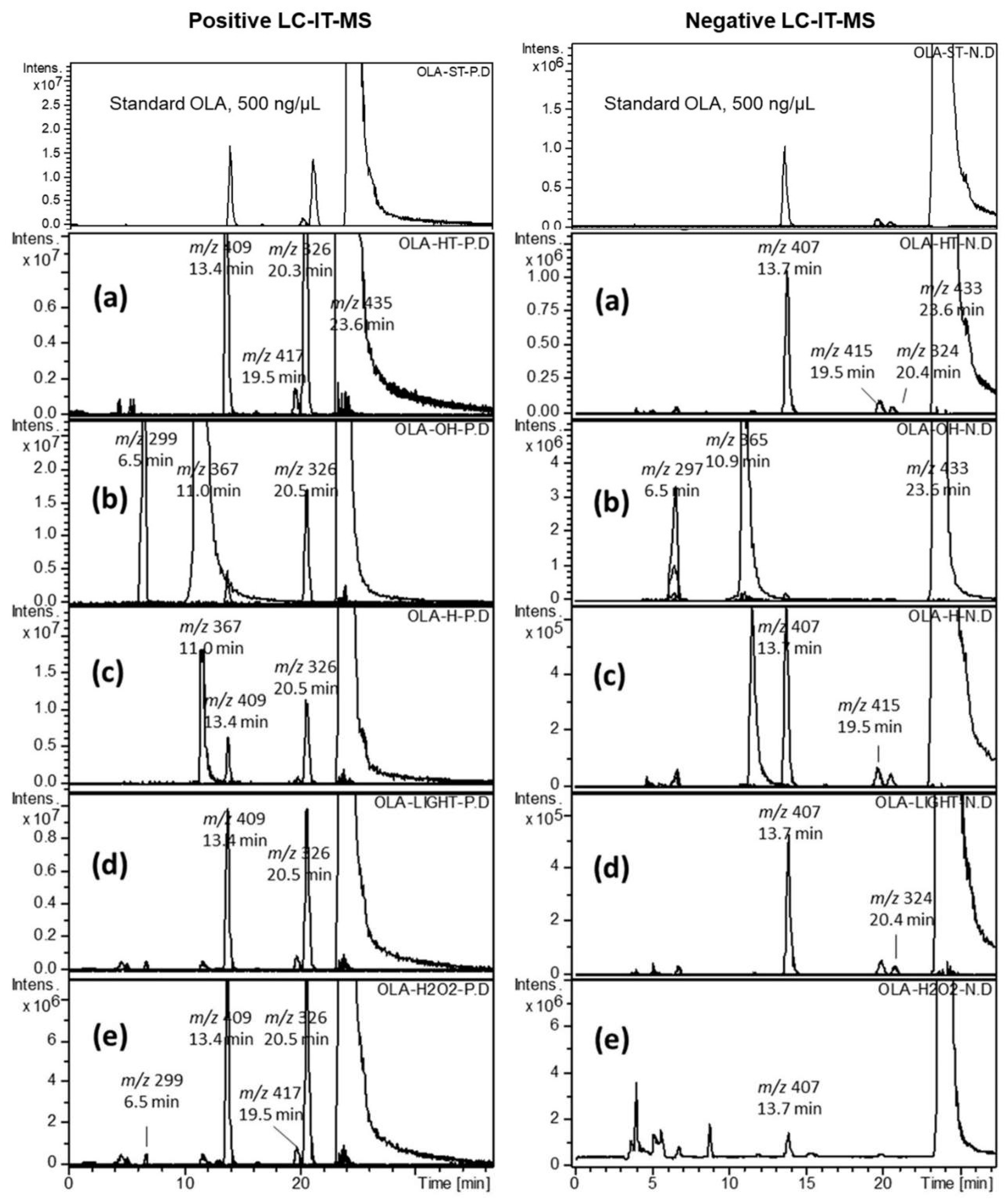
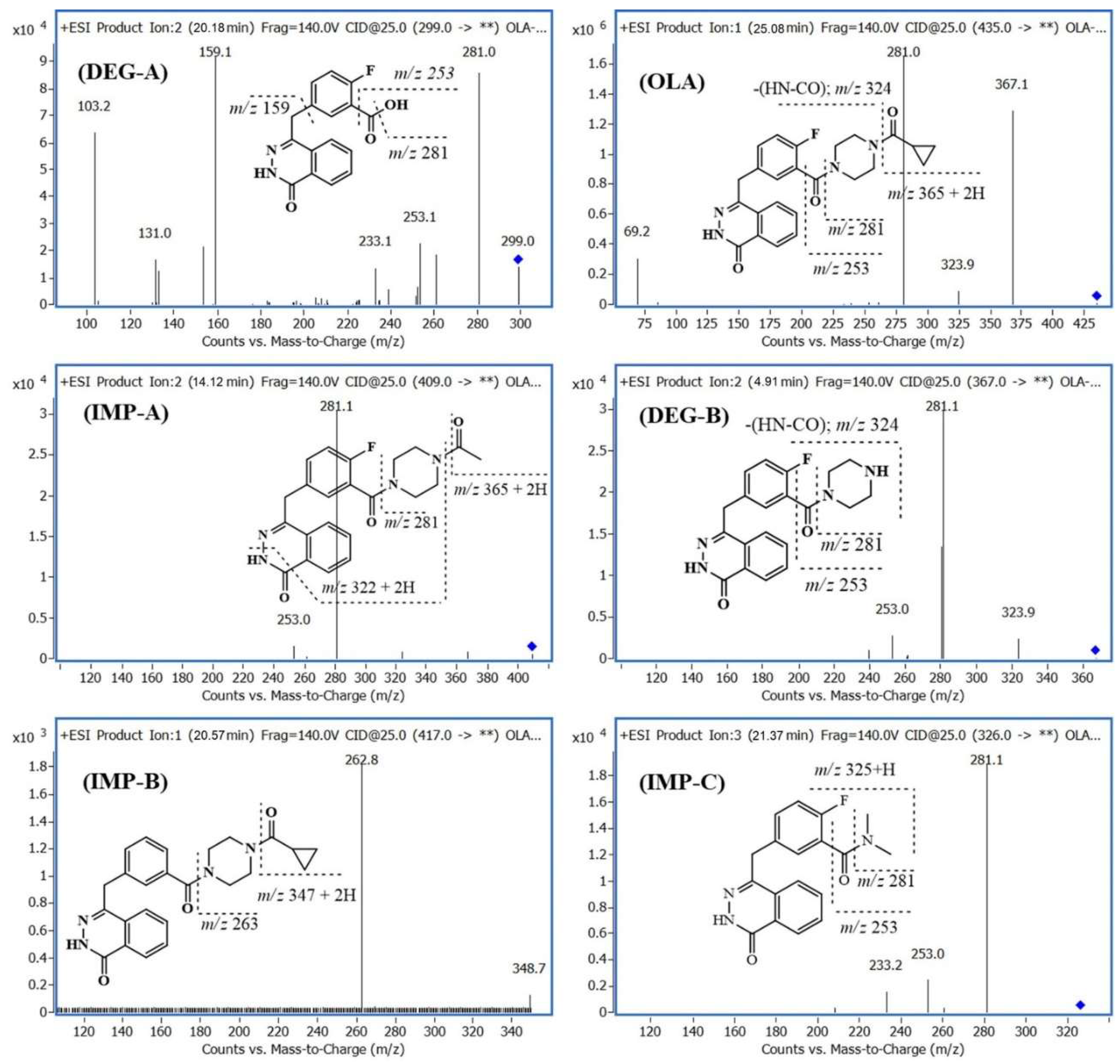
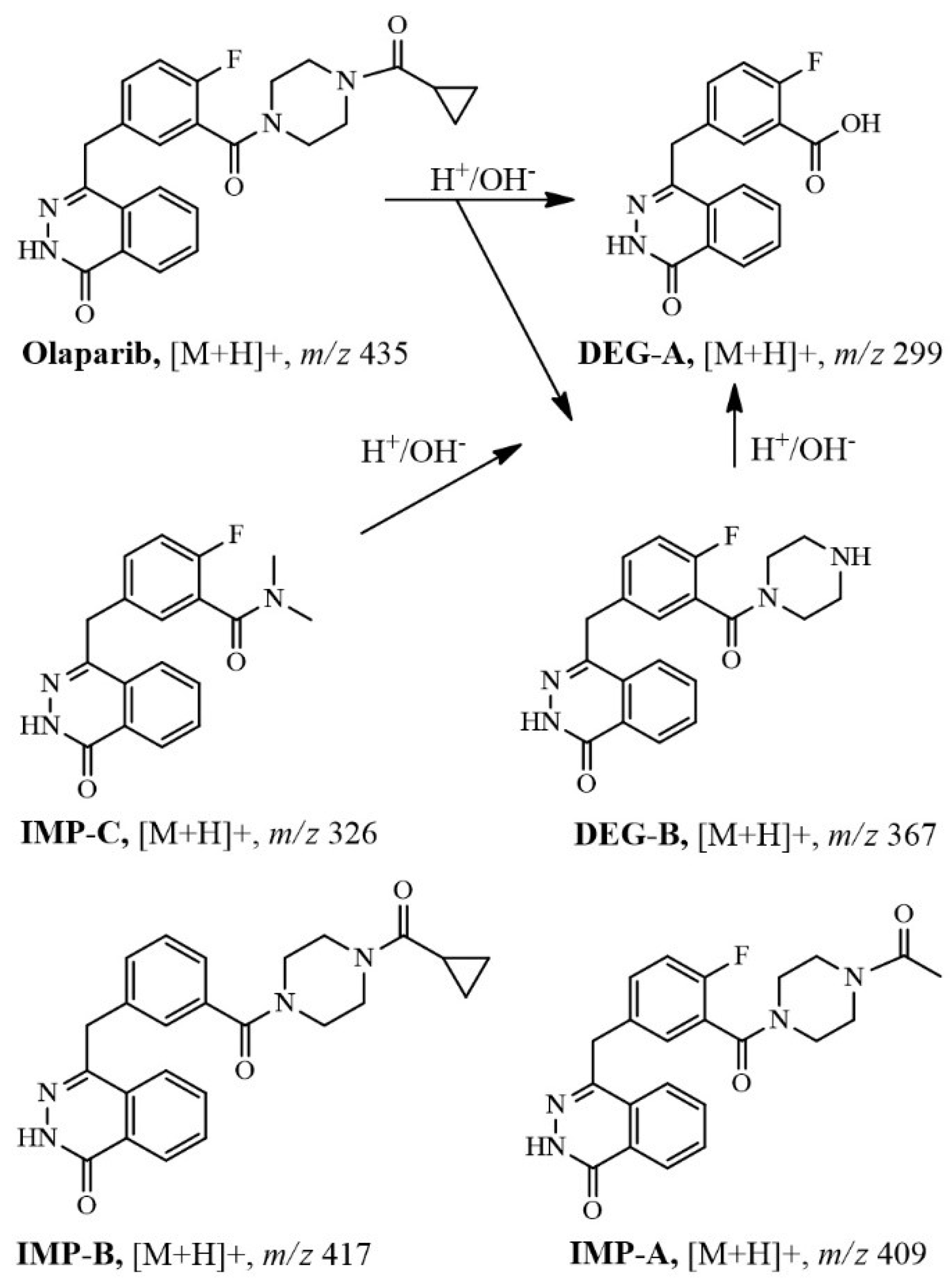
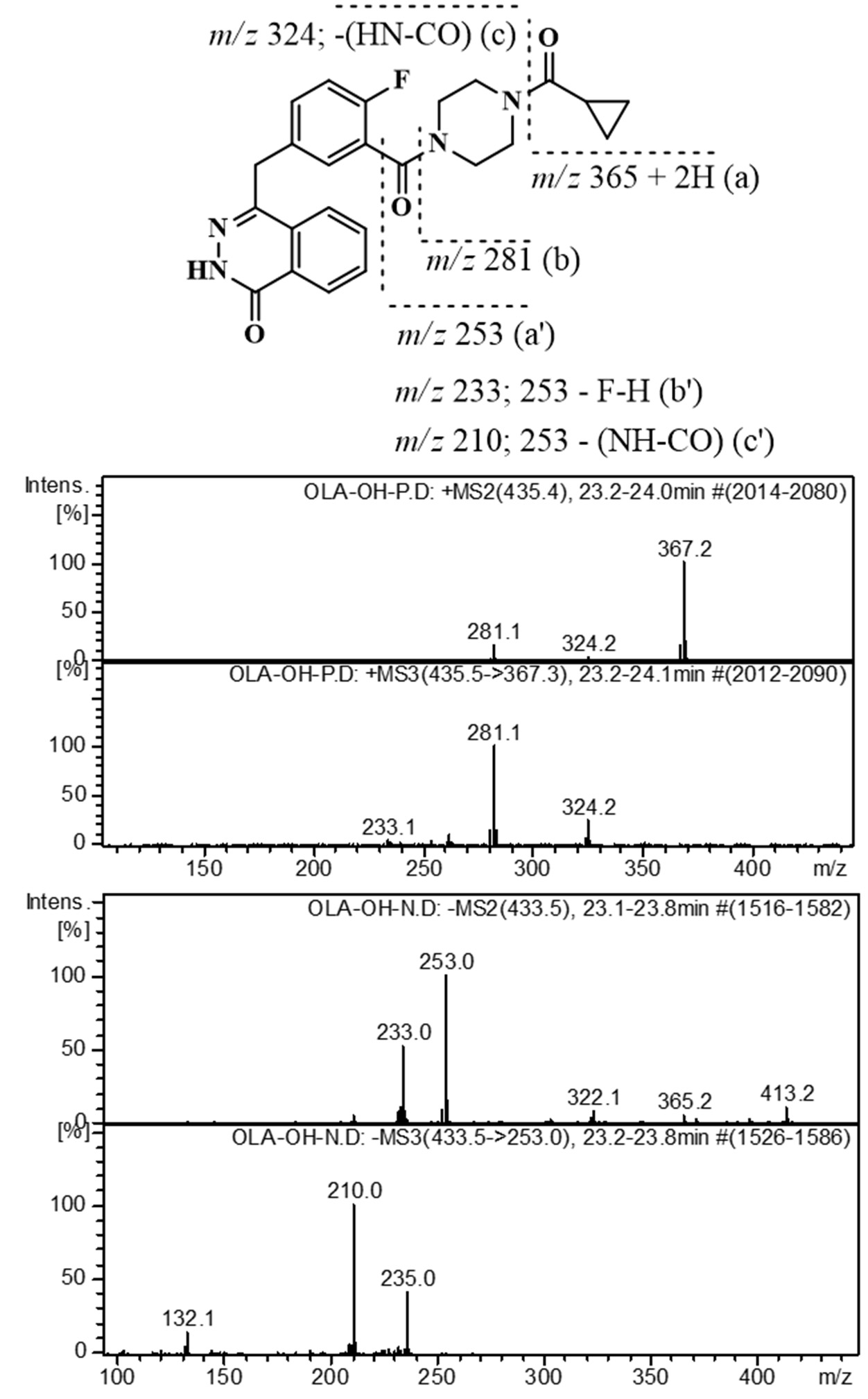
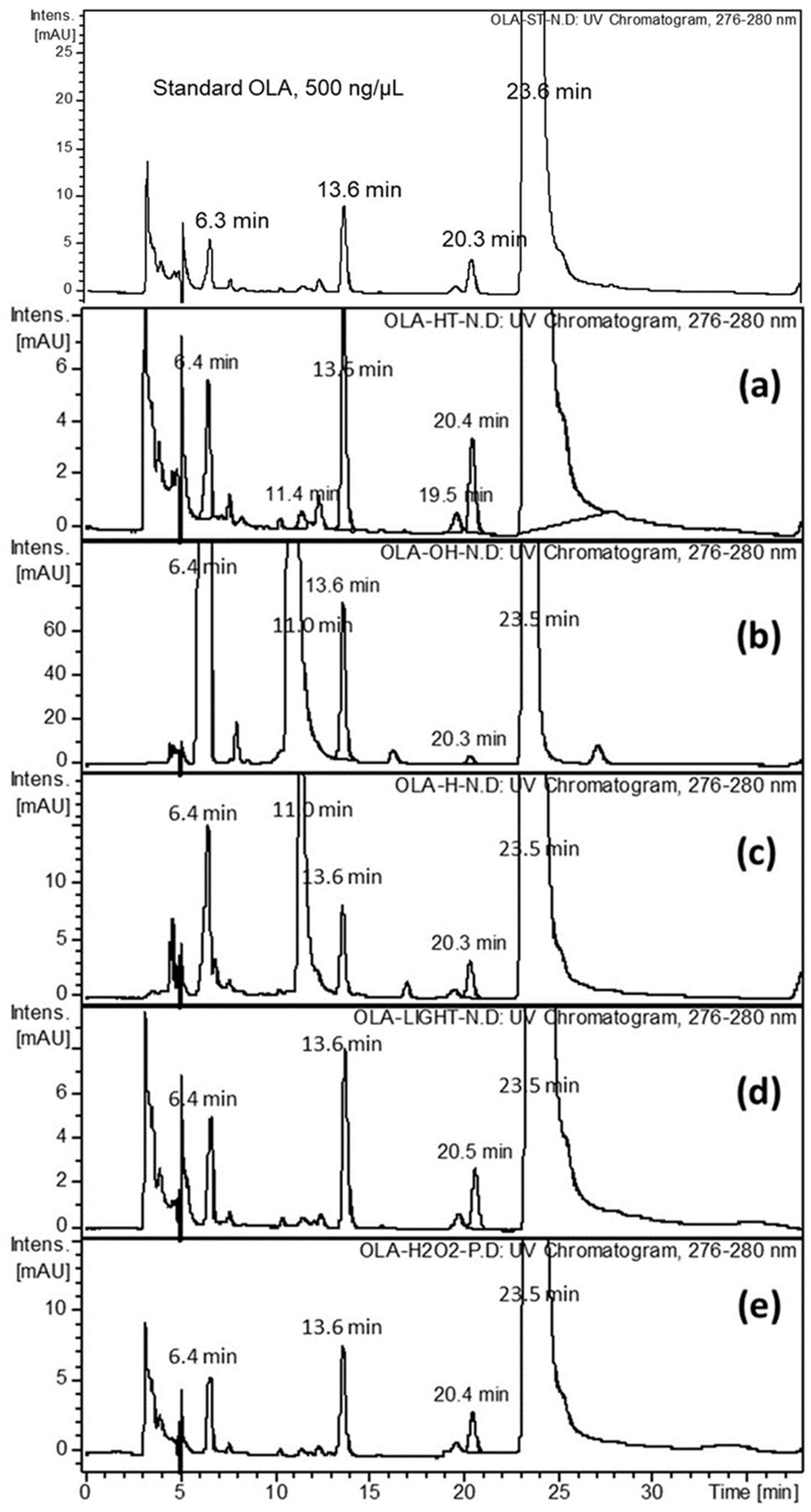
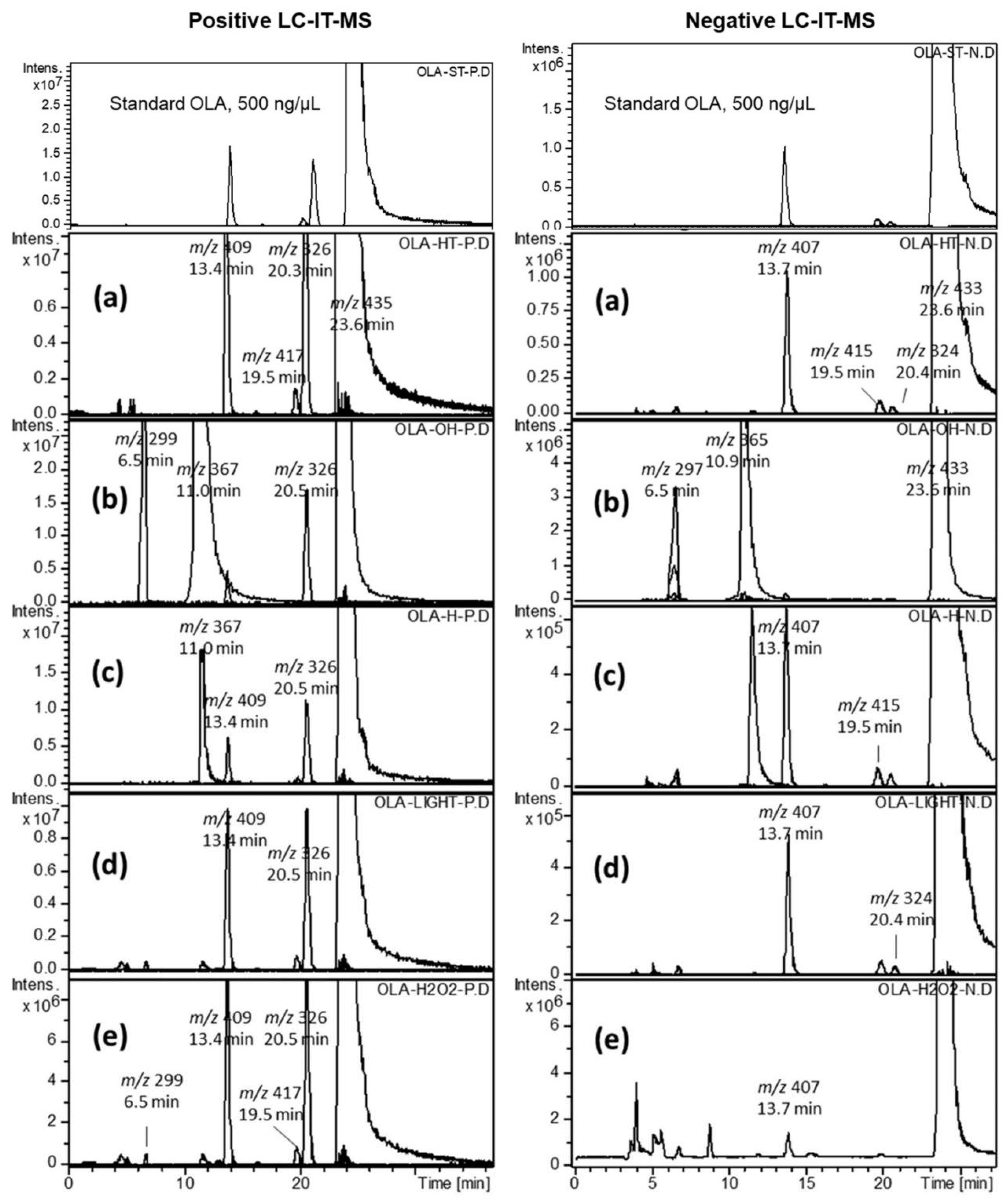
2.2. Forced Degradation of OLA
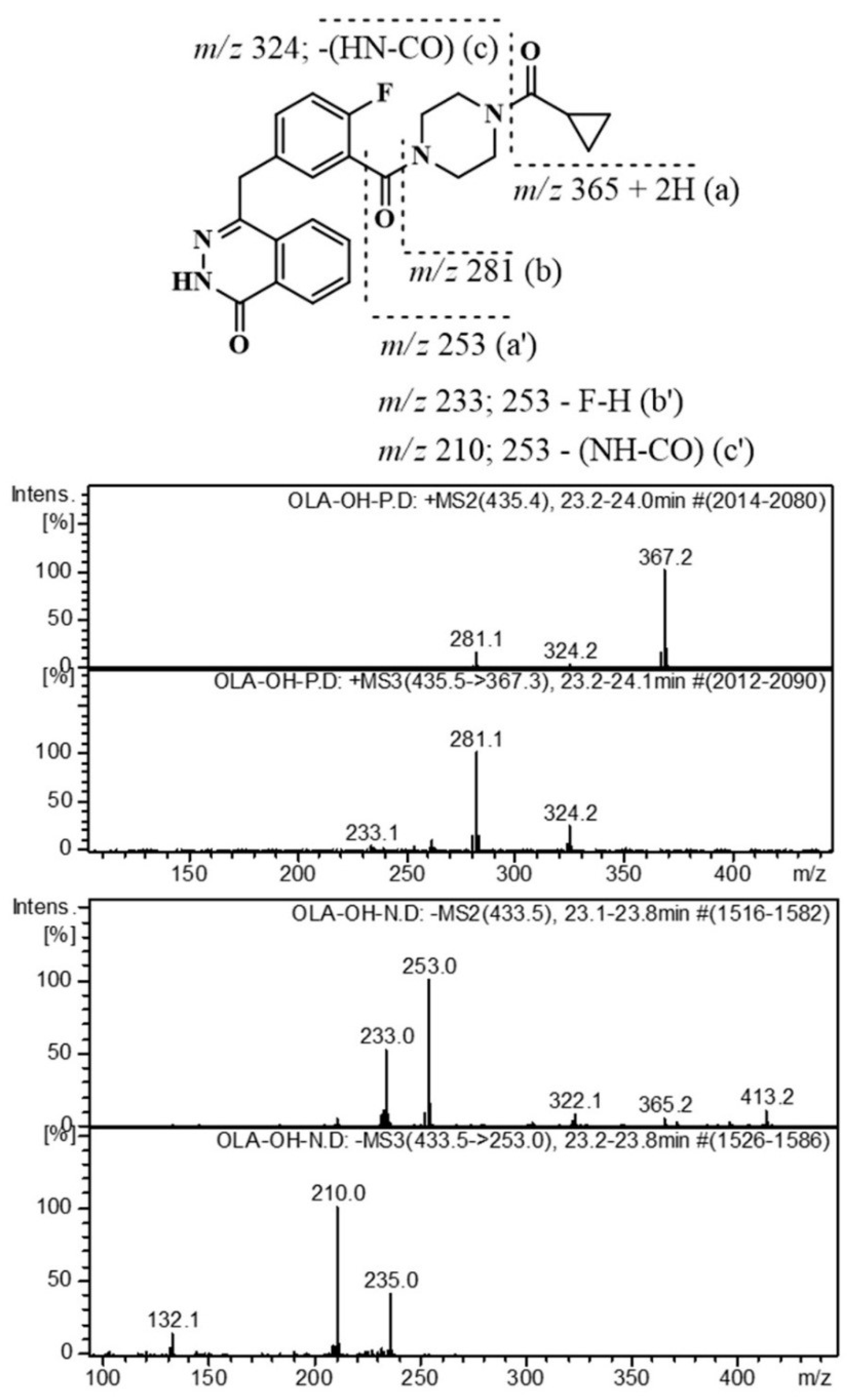
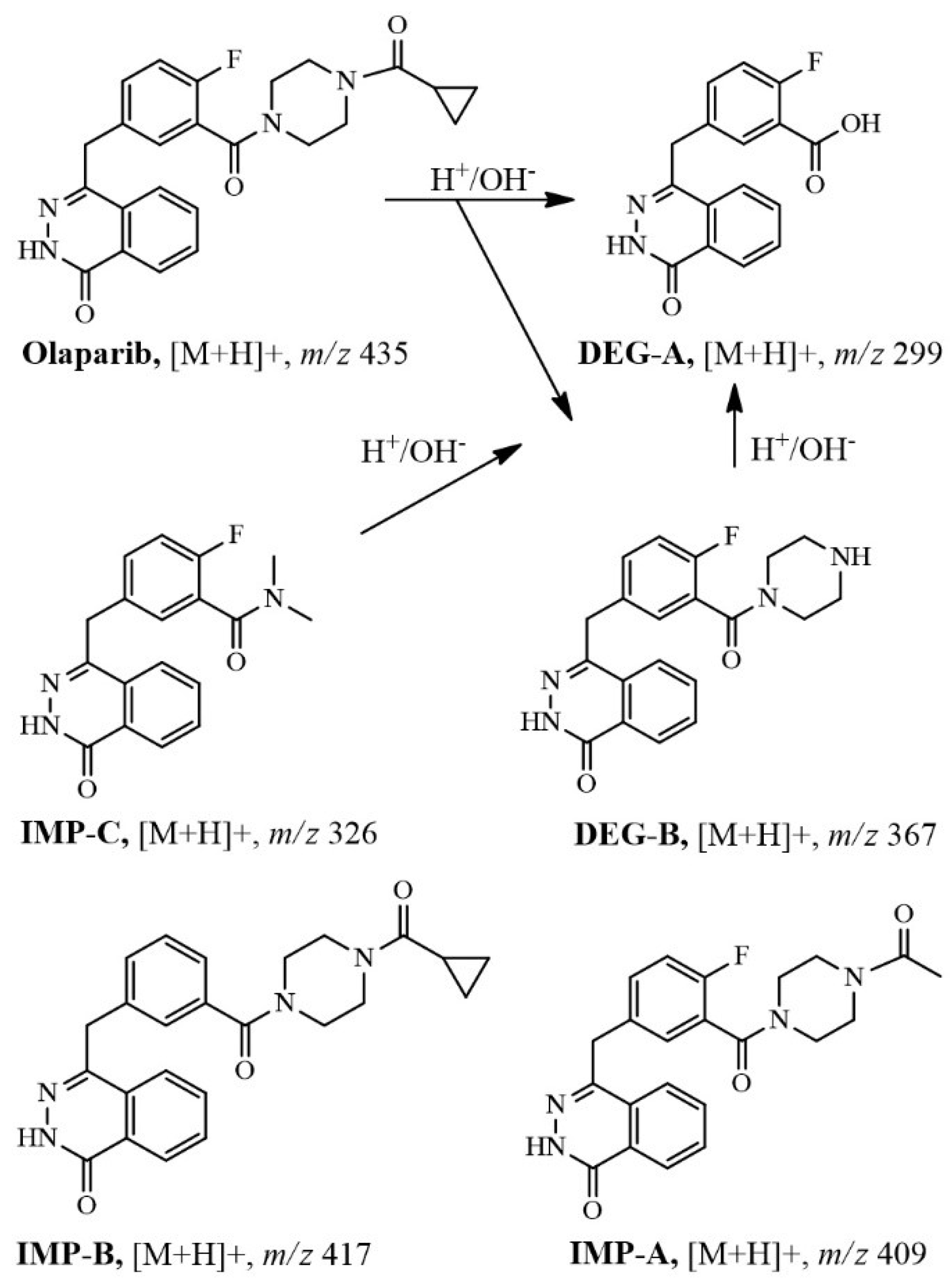
2.3. Identification of Related Substances
2.4. Method Validation
2.4.1. Linearity
2.4.2. Precision and Accuracy
2.4.3. Accuracy
2.4.4. System Suitability and Selectivity
2.4.5. Robustness
3. Materials and Methods
3.1. Chemicals
3.2. Preparation of Calibration Solutions
3.3. Liquid Chromatography-DAD and Mass Spectrometry
3.4. Forced Degradation
3.4.1. Effect of UV-Irradiation
3.4.2. Effect of Oxidation
3.4.3. Effects of Acid and Base Hydrolysis
3.4.4. Effect of Heat
4. Conclusion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| OLA | Olaparib |
| DPs | Degradation products |
| DEG-A | Degradation product A |
| DEG-B | Degradation product B |
| IMP-A | Impurity A |
| IMP-B | Impurity B |
| IMP-C | Impurity C |
| DAD | Diode array detector |
| MS | Mass spectrometry |
| IT | Ion trap |
| QQQ | Triple quad |
References
- AstraZeneca AB. Lynparza 50 mg hard capsules: EU summary of product characteristics. Available online: http://www.ema.europa.eu/ema (accessed on 12 January 2015).
- AstraZeneca Pharmaceuticals LP. Lynparza™ (olaparib) capsules, for oral use: US prescribing information. Available online: https://www.fda.gov/ (accessed on 5 January 2015).
- Fong, P.C.; Boss, D.S.; Yap, T.A.; Tutt, A.; Wu, P.; Mergui-Roelvink, M.; Mortimer, P.; Swaisland, H.; Lau, A.; O’Connor, M.J.; et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N. Engl. J. Med. 2009, 361, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Nijenhuis, C.M.; Lucas, L.; Rosing, H.; Schellens, J.H.M.; Beijnen, J.H. Development and validation of a high-performance liquid chromatography–tandem mass spectrometry assay quantifying olaparib in human plasma. J. Chromatogr. B 2013, 940, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Sparidans, R.W.; Martens, I.; Valkenburg-van Iersel, L.B.J.; den Hartigh, J.; Schellens, J.H.M.; Beijnen, J.H. Liquid chromatography–tandem mass spectrometric assay for the PARP-1 inhibitor olaparib in combination with the nitrogen mustard melphalan in human plasma. J.Chromatogr. B 2011, 879, 1851–1856. [Google Scholar] [CrossRef] [PubMed]
- Thummar, M.; Kuswah, B.S.; Samanthula, G.; Bulbake, U.; Gour, J.; Khan, W. Validated stability indicating assay method of olaparib: LC-ESI-Q-TOF-MS/MS and NMR studies for characterization of its new hydrolytic and oxidative forced degradation products. J. Pharm. Biomed. Anal. 2018, 160, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Menear, K.; Ottridge, A.; Londesbrough, D.; Hallett, M.; Mullholland, K.; Pittam, J.; Laffan, D.; Ashworth, I.; Jones, M.; Cheryman, J. Method for the Preparation of 4-[3-(4-cyclopropanecarbonylpiperazine-1-carbonyl)-4-fluoro-benzyl]-2H-phthalazin-1-one. Patent EP 2 824 098 B1, 2016. [Google Scholar]
- Yinghua, Y.; Ji, Y.; Fangfang, C.; Yi, D. Preparation Method of High-Purity Olaparib. Patent CN105503739A, 24 February 2016. [Google Scholar]
- European Medicines Agency. International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use. In ICH Q3A (R2), Impurities in New Drug Substances; European Medicines Agency: Geneva, Switzerland, 2006; pp. 1–15. [Google Scholar]
- Iliou, K.; Malenovic, A.; Loukas, Y.L.; Dotsikas, Y. Analysis of potential genotoxic impurities in rabeprazole active pharmaceutical ingredient via Liquid Chromatography-tandem Mass Spectrometry, following quality-by-design principles for method development. J. Pharm. Biomed. Anal. 2018, 149, 410–418. [Google Scholar] [CrossRef] [PubMed]
- Kumar, T.; Ramya, M.; Srinivasan, V.; Xavier, N. A Simple and Direct LC-MS Method for Determination of Genotoxic Impurity Hydroxylamine in Pharmaceutical compounds. J. Chromatogr. Sci. 2017, 55, 683–689. [Google Scholar] [CrossRef] [PubMed]
- Paraskevas, D.T. Recent Advances in the Analysis of Organic Impurities of Active Pharmaceutical Ingredients and Formulations: A Review. Curr. Org. Chem. 2010, 14, 2348–2364. [Google Scholar]
- Wang, J.; Xu, Y.; Wen, C.; Wang, Z. Application of a trap-free two-dimensional liquid chromatography combined with ion trap/time-of-flight mass spectrometry for separation and characterization of impurities and isomers in cefpiramide. Anal. Chim. Acta 2017, 992, 42–54. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Liu, A.P.; Yan, Y.; Daly, T.J.; Li, N. Characterization of product-related low molecular weight impurities in therapeutic monoclonal antibodies using hydrophilic interaction chromatography coupled with mass spectrometry. J. Pharm. Biomed. Anal. 2018, 154, 468–475. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, T.; Zhang, W.; Cao, L.; He, K.; Wang, Y.; Li, J.; Chen, L.; Liu, B.; Zhang, G. Isolation, identification and characterization of two novel process-related impurities in olanzapine. J. Pharm. Biomed. Anal. 2018, 152, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Federal Register, Part II, Department of Health and Human Services, Food and Drug Administration, International Conference on Harmonisation; Guidelines for the Photostability Testing of New Drug Substances and Products, May 16/Notices. 1997. Available online: https://www.scribd.com/document/186240493/FDA-Photostability (accessed on 26 February 2019).
- International Conference on Harmonization (ICH) Guidelines Q2 (R1), Validation of Analytical Procedures: Test and Methodology, Geneva. 2005. Available online: http://www.ich.org/products/guidelines/quality/quality-single/article/validation-of-analytical-procedures-text-and-methodology.html (accessed on 26 February 2019).
- Q4B Regulatory Acceptance of Analytical Procedures and/or Acceptance Criteria, (RAAPAC), FDA/CDER/OPS. 8 June 2006. Available online: https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM218825.pdf (accessed on 26 February 2019).
- Dong, H.; Xiao, K. Modified QuEChERS combined with ultra high performance liquid chromatography tandem mass spectrometry to determine seven biogenic amines in Chinese traditional condiment soy sauce. Food Chem. 2017, 229, 502–508. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Xiao, K.; Xian, Y.; Wu, Y.; Zhu, L. A novel approach for simultaneous analysis of perchlorate (ClO4(-)) and bromate (BrO3(-)) in fruits and vegetables using modified QuEChERS combined with ultrahigh performance liquid chromatography-tandem mass spectrometry. Food Chem. 2019, 270, 196–203. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the investigated compounds are not available from the authors. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | [M + H]+ m/z | Rt, min | Bulk, % (w/w) | Heat, % (w/w) | H2O2, % (w/w) | Acid, % (w/w) | Base, % (w/w) | UV, % (w/w) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DAD | MS | DAD | MS | DAD | MS | DAD | MS | DAD | MS | DAD | MS | |||
| DEG-A | 299 | 6.5 | 0.03 | 0.04 | 0.33 | 0.32 | 0.81 | 0.83 | 0.30 | 0.29 | 20.85 | 21.22 | 9.40 | 9.31 |
| DEG-B | 367 | 11.0 | 0.00 | 0.00 | 0.11 | 0.09 | 0.0 | 0.0 | 1.20 | 1.21 | 13.51 | 13.62 | 7.30 | 7.61 |
| IMP-A | 409 | 13.6 | 0.23 | 0.16 | 0.24 | 0.23 | 0.19 | 0.17 | 0.20 | 0.20 | 0.00 | 0.12 | 0.00 | 0.06 |
| IMP-B | 417 | 19.5 | 0.02 | 0.13 | 0.14 | 0.16 | 0.0 | 0.0 | 0.02 | 0.02 | 0.00 | 0.00 | 0.00 | 0.00 |
| IMP-C | 326 | 20.3 | 0.09 | 0.21 | 0.11 | 0.13 | 0.10 | 0.10 | 0.10 | 0.10 | 0.00 | 0.00 | 0.00 | 0.00 |
| OLA | 435 | 23.5 | 99.15 | 99.21 | 99.00 | 98.87 | 98.20 | 98.15 | 97.78 | 97.38 | 65.02 | 64.30 | 81.24 | 81.02 |
| % of total related substances | 0.37 | 0.54 | 0.93 | 0.93 | 1.10 | 1.10 | 1.82 | 1.82 | 34.36 | 34.96 | 16.70 | 16.98 | ||
| Label | Retention Time, (min) | W(0.05), min | USP Tailing | k | α | USP Resolution | USP Plate Count |
|---|---|---|---|---|---|---|---|
| DEG-A | 6.5 | 0.42 | 1.00 | 1.3 | 2.2 | 15.5 | 3832 |
| DEG-B | 11.0 | 0.58 | 0.98 | 2.9 | 1.3 | 14.1 | 5755 |
| IMP-A | 13.6 | 0.57 | 1.02 | 3.9 | 1.5 | 22.7 | 9109 |
| IMP-B | 19.5 | 0.55 | 1.03 | 6.0 | 1.0 | 19.7 | 20112 |
| IMP-C | 20.3 | 0.52 | 0.97 | 6.3 | 1.2 | 17.3 | 24384 |
| OLA | 23.5 | 1.02 | 1.03 | 7.39 | 8493 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khedr, A.; El-Hay, S.S.A.; Kammoun, A.K. Multi-Steps Fragmentation-Ion Trap Mass Spectrometry Coupled to Liquid Chromatography Diode Array System for Investigation of Olaparib Related Substances. Molecules 2019, 24, 843. https://doi.org/10.3390/molecules24050843
Khedr A, El-Hay SSA, Kammoun AK. Multi-Steps Fragmentation-Ion Trap Mass Spectrometry Coupled to Liquid Chromatography Diode Array System for Investigation of Olaparib Related Substances. Molecules. 2019; 24(5):843. https://doi.org/10.3390/molecules24050843
Chicago/Turabian StyleKhedr, Alaa, Soad S. Abd El-Hay, and Ahmed K. Kammoun. 2019. "Multi-Steps Fragmentation-Ion Trap Mass Spectrometry Coupled to Liquid Chromatography Diode Array System for Investigation of Olaparib Related Substances" Molecules 24, no. 5: 843. https://doi.org/10.3390/molecules24050843
APA StyleKhedr, A., El-Hay, S. S. A., & Kammoun, A. K. (2019). Multi-Steps Fragmentation-Ion Trap Mass Spectrometry Coupled to Liquid Chromatography Diode Array System for Investigation of Olaparib Related Substances. Molecules, 24(5), 843. https://doi.org/10.3390/molecules24050843