Organic Salts and Merrifield Resin Supported [PM12O40]3− (M = Mo or W) as Catalysts for Adipic Acid Synthesis




Abstract

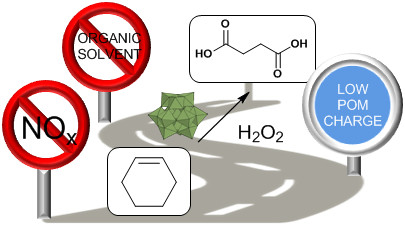
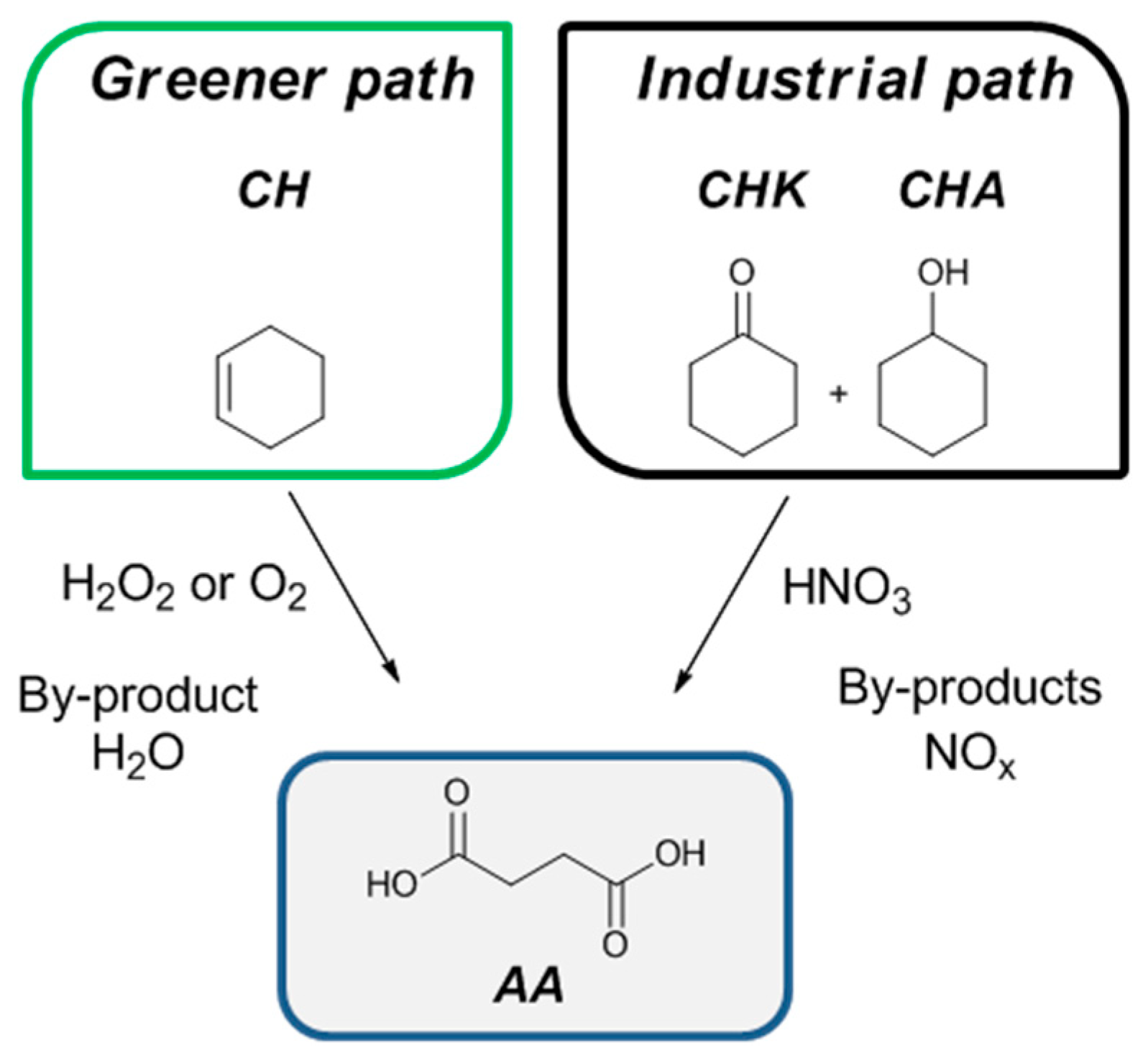
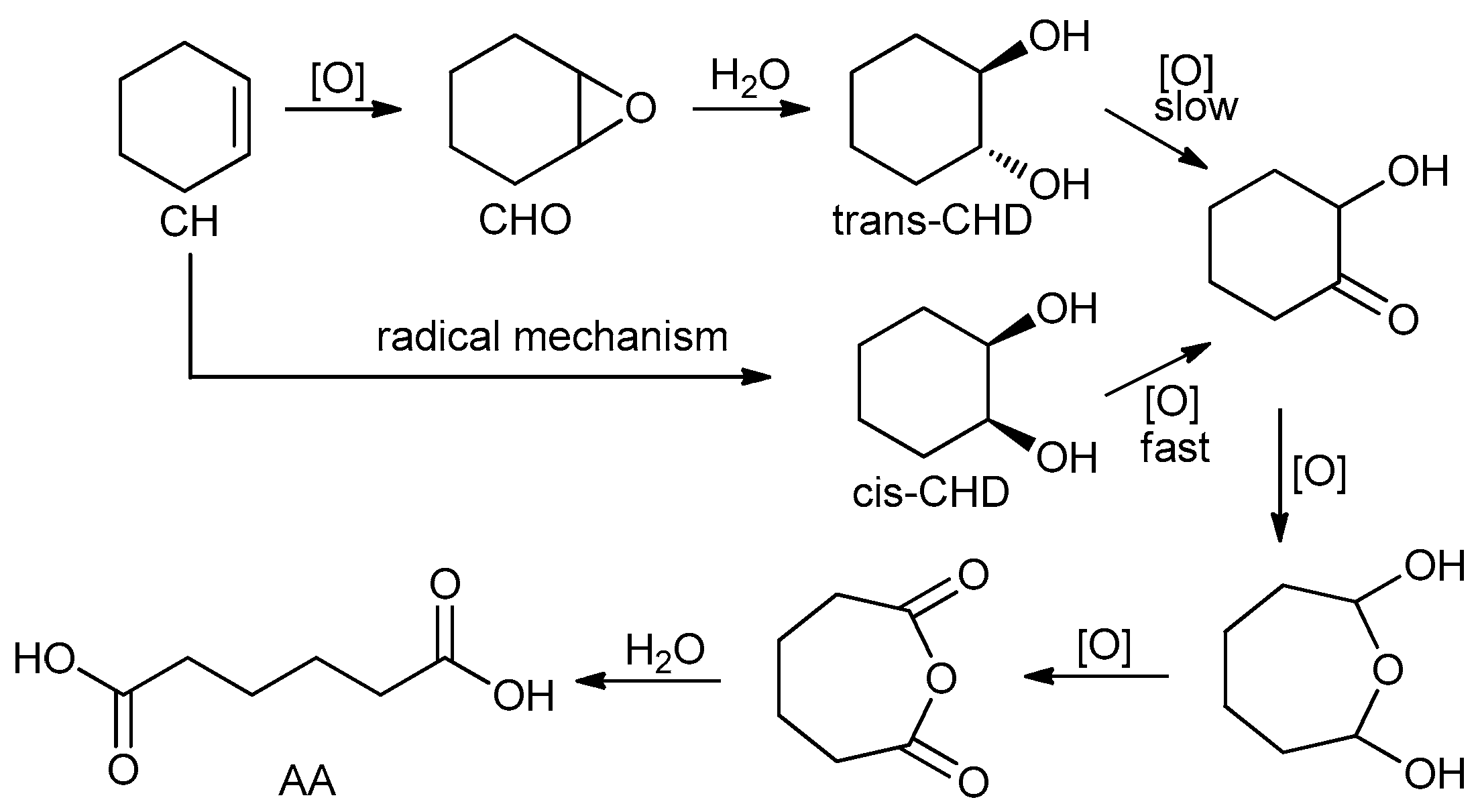
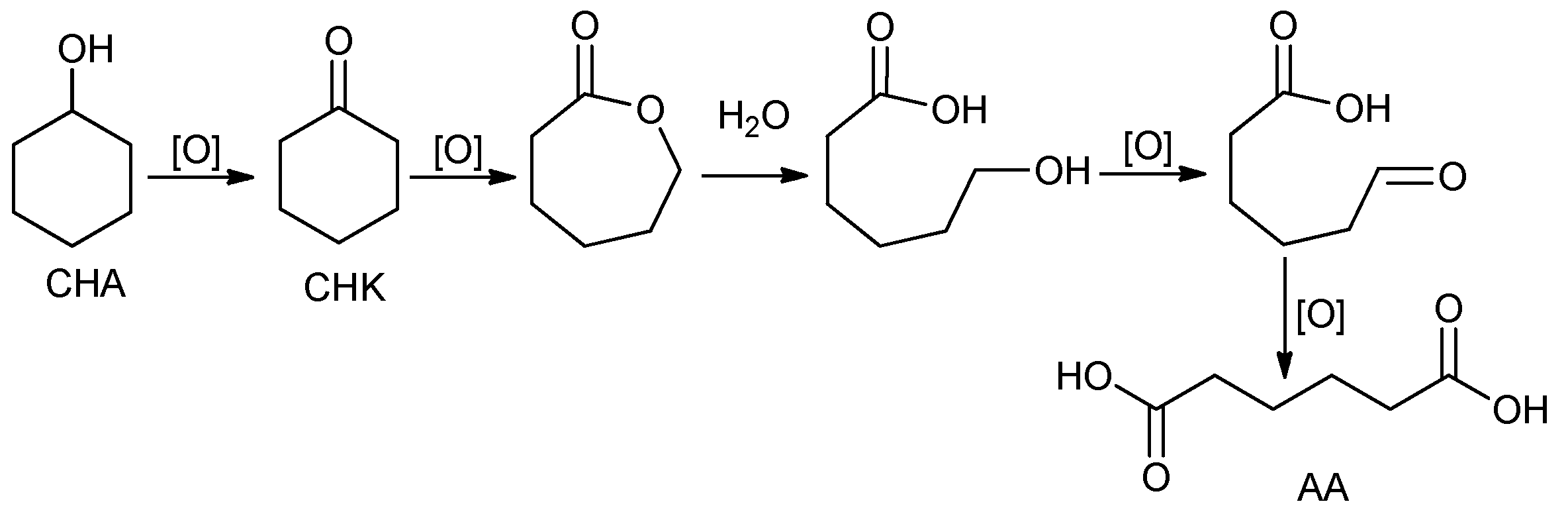
1. Introduction
2. Results and Discussion
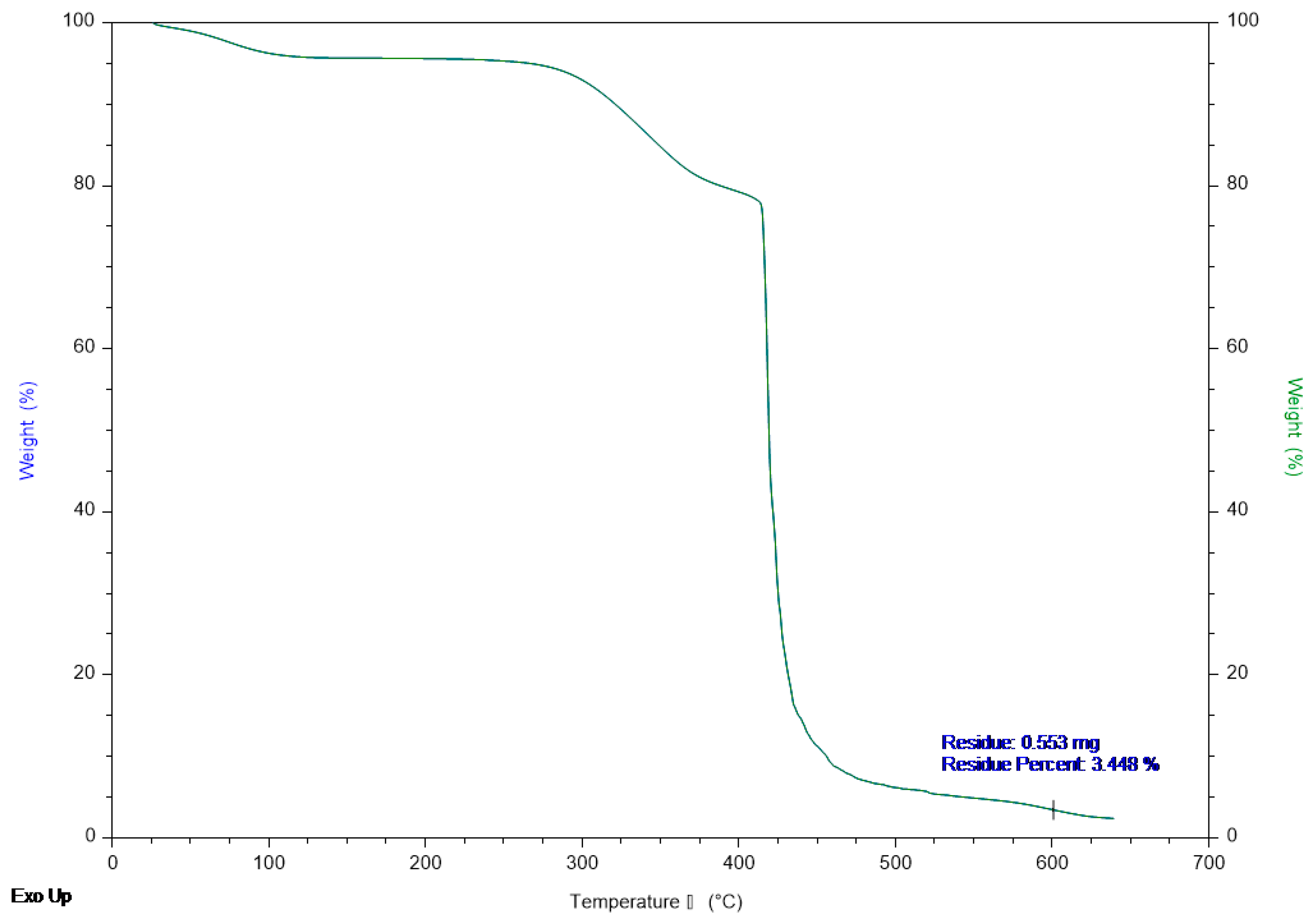
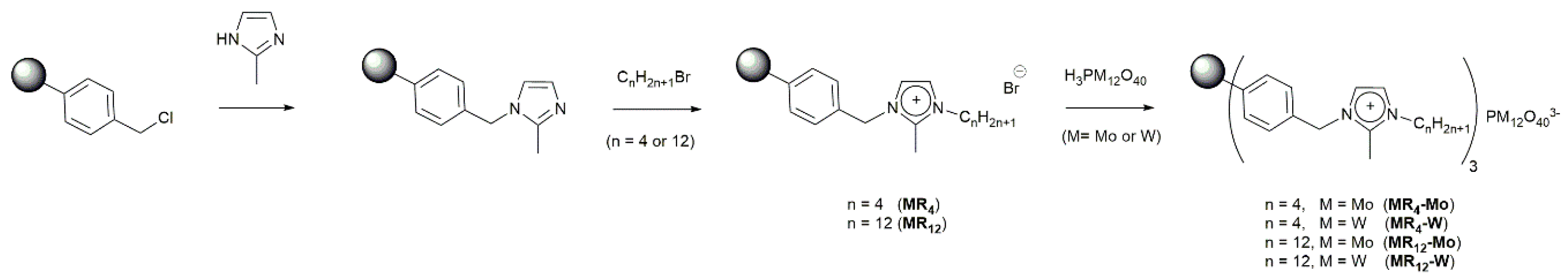
2.1. Synthesis and Characterization of the Prepared Catalysts
2.1.1. Synthesis
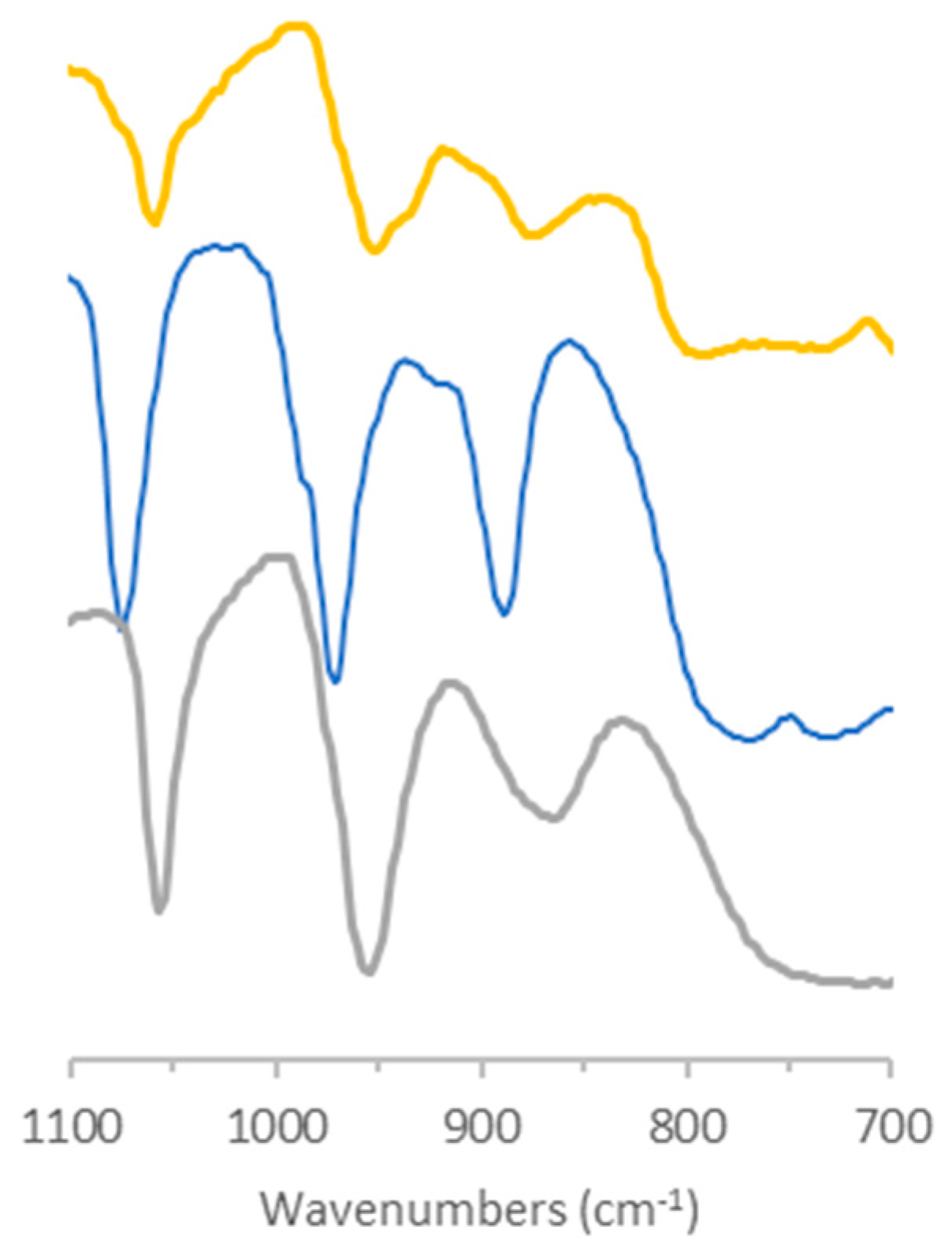
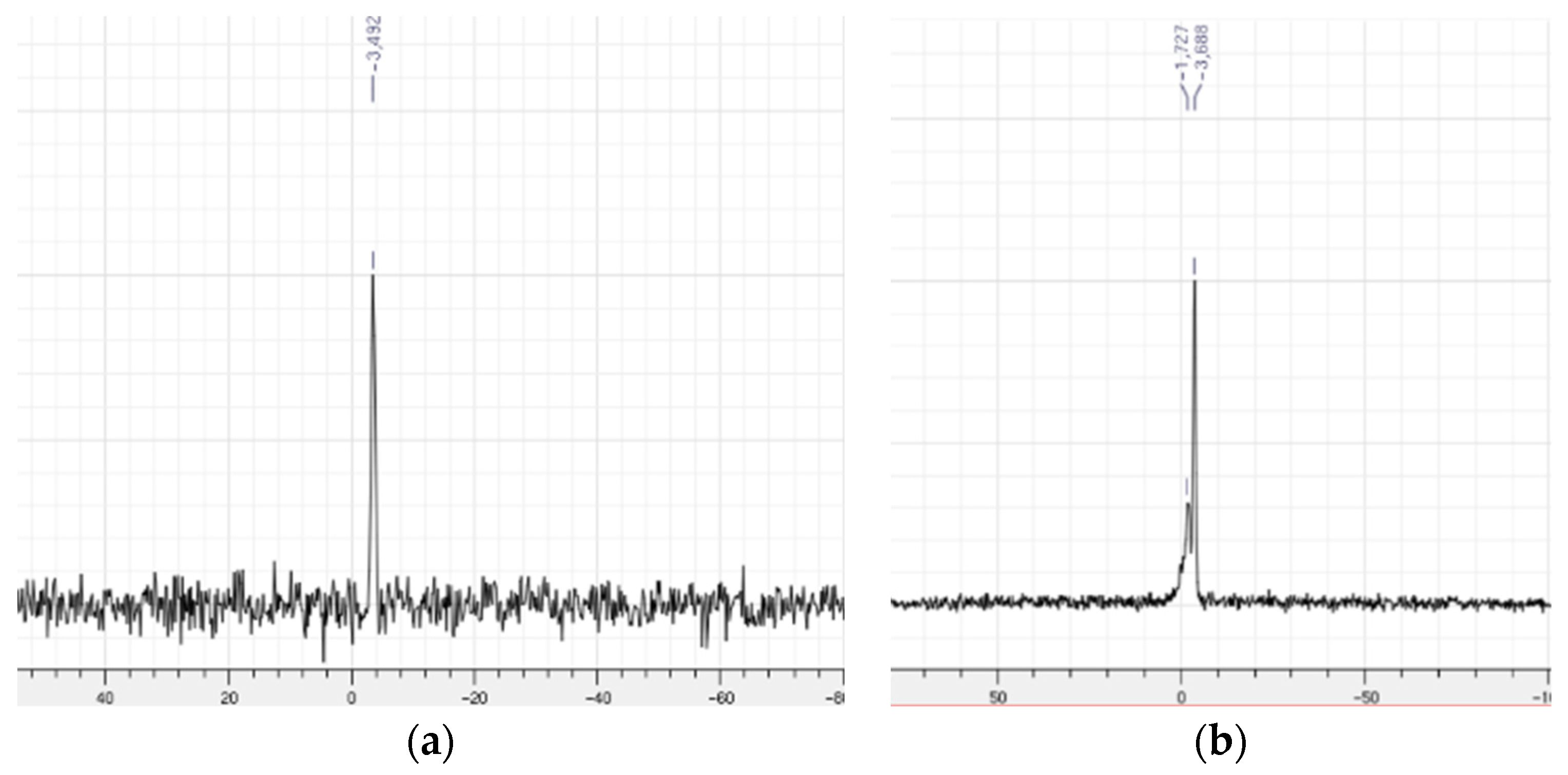
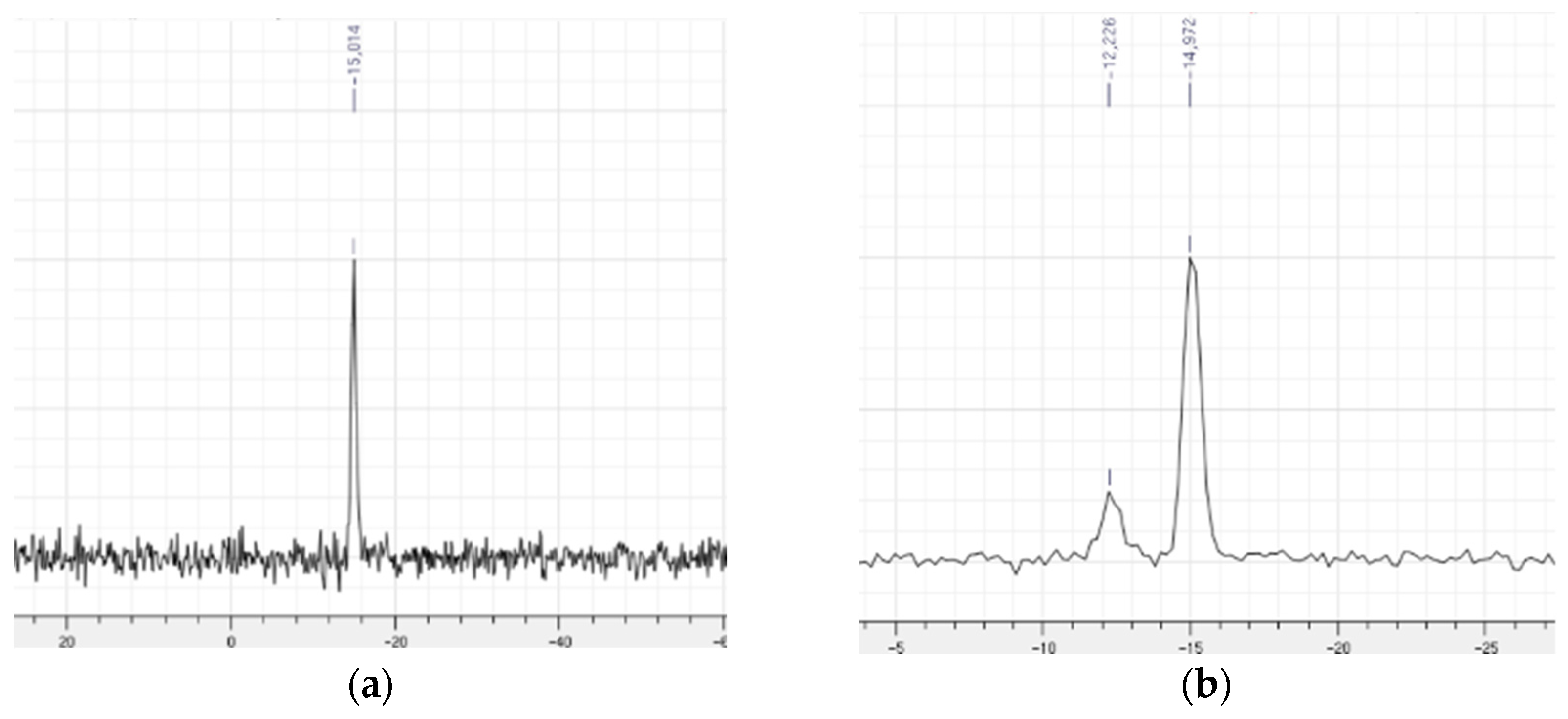
2.1.2. Characterization
2.2. Catalysis
3. Materials and Methods
3.1. Materials
3.2. Physical Methods
3.3. Preparative Part
3.4. Swelling Test for MR
3.5. General Procedure for Catalytic Oxidation to AA
3.5.1. CH Oxidation (Procedure A)
3.5.2. CHO Oxidation (Procedure B)
3.5.3. CHD Oxidation (Procedure C)
3.5.4. Recycling of the Resins
3.5.5. CHK or CHA Oxidation (Procedure D)
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Viers, V.; Brendt, D. Polymer Data Handbook; Oxford University Press: Oxford, UK, 1999; p. 189. [Google Scholar]
- Van de Vyver, S.; Roman-Leshkov, Y. Emerging catalytic processes for the production of adipic acid. Catal. Sci. Technol. 2013, 3, 1465–1479. [Google Scholar] [CrossRef]
- Cavani, F.; Alini, S. Synthesis of Adipic Acid: On the Way to More Sustainable Production in Sustainable Industrial Processes; Cavani, F., Centi, G., Perathoner, S., Trifiró, F., Eds.; WILEY-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2009; pp. 367–425. ISBN 978-3-527-31552-9. [Google Scholar]
- Buchner, W.; Schliebs, R.; Winter, G.; Buchel, K.H. Industrielle Anorganische Chemie, 2nd ed.; VCH Weinheim: Weinheim, Germany, 1986. [Google Scholar]
- Kroeze, C.; Mosier, A. New estimates for Emissions of Nitrous Oxide. In Non-CO2 Greenhouse Gases: Scientific Understanding, Control and Implementation; van Ham, J.M., Baede, A.P., Meyer, L.A., Ybema, R., Eds.; Kluwer Academic Publishers: Dordrecht, The Netherlands, 1999; pp. 45–64. ISBN 978-0-7923-6199-2. [Google Scholar]
- Hermans, I.; Spier, S.E.; Neuenschwander, U.; Turra, N.; Baiker, A. Selective oxidation catalysis: Opportunities and challenges. Top. Catal. 2009, 52, 1162–1174. [Google Scholar] [CrossRef]
- Shimizu, A.; Tanaka, K.; Fujimori, M. Abatement technologies for N2O emissions in the adipic acid industry. Chemosphere Glob. Chang. Sci. 2000, 2, 425–434. [Google Scholar] [CrossRef]
- Reimer, R.A.; Slaten, C.S.; Seapan, M.; Lower, M.W.; Tomlinson, P.E. Abatement of N2O emissions produced in the adipic acid industry. Environ. Prog. 1994, 13, 134–137. [Google Scholar] [CrossRef]
- Bart, J.C.J.; Cavallaro, S. Transiting from Adipic Acid to Bioadipic Acid. 1, Petroleum-Based Processes. Ind. Eng. Chem. Res. 2015, 54, 1–46. [Google Scholar] [CrossRef]
- Cavani, F. Catalytic selective oxidation faces the sustainability challenge: Turning points, objectives reached, old approaches revisited and solutions still requiring further investigation. J. Chem. Technol. Biotechnol. 2010, 85, 1175–1183. [Google Scholar] [CrossRef]
- Brégeault, J.-M. Transition-metal complexes for liquid-phase catalytic oxidation: Some aspects of industrial reactions and of emerging technologies. Dalton Trans. 2003, 3289–3302. [Google Scholar] [CrossRef]
- Jones, C.W. Applications of Hydrogen Peroxide and Derivatives; Royal Society of Chemistry: Cambridge, UK, 1999; ISBN 978-0-85404-536-5. [Google Scholar]
- Strukul, G. (Ed.) Catalytic Oxidations with Hydrogen Peroxide as Oxidant; Kluwer Academic: Dordrecht, The Netherlands, 1992; ISBN 978-0-7923-1771-5. [Google Scholar]
- Sheldon, R.A.; Kochi, J.K. Metal Catalyzed Oxidations of Organic Compounds; Academic Press: New York, NY, USA, 1981; ISBN 978-0-12-639380-4. [Google Scholar]
- Damm, M.; Gutmann, B.; Kappe, C.O. Continuous-Flow Synthesis of Adipic Acid from Cyclohexene Using Hydrogen Peroxide in High-Temperature Explosive Regimes. ChemSusChem 2013, 6, 978–982. [Google Scholar] [CrossRef]
- Shang, M.; Noel, T.; Su, Y.; Hessel, V. High Pressure Direct Synthesis of Adipic Acid from Cyclohexene and Hydrogen Peroxide via Capillary Microreactors. Ind. Eng. Chem. Res. 2016, 55, 2669–2676. [Google Scholar] [CrossRef]
- Vural-Gursel, I.; Wang, Q.; Noel, T.; Hessel, V.; Tinge, J.T. Improving Energy Efficiency of Process of Direct Adipic Acid Synthesis in Flow Using Pinch Analysis. Ind. Eng. Chem. Res. 2013, 52, 7827–7835. [Google Scholar] [CrossRef]
- Shang, M.; Noël, T.; Wang, Q.; Su, Y.; Miyabayashi, K.; Hessel, V.; Hasebe, S. 2- and 3-Stage temperature ramping for the direct synthesis of adipic acid in micro-flow packed-bed reactors. Chem. Eng. J. 2015, 260, 454–462. [Google Scholar] [CrossRef]
- Wen, Y.; Wang, X.; Wei, H.; Li, B.; Jin, P.; Li, L. A large-scale continuous-flow process for the production of adipic acid via catalytic oxidation of cyclohexene with H2O2. Green Chem. 2012, 14, 2868–2875. [Google Scholar] [CrossRef]
- Buonomenna, M.G.; Golemme, G.; De Santo, M.P.; Drioli, E. Direct Oxidation of Cyclohexene with Inert Polymeric Membrane Reactor. Org. Process Res. Dev. 2010, 14, 252–258. [Google Scholar] [CrossRef]
- Freitag, J.; Nüchter, M.; Ondruschka, B. Oxidation of styrene and cyclohexene under microwave conditions. Green Chem. 2003, 5, 291–295. [Google Scholar] [CrossRef]
- Misono, M. Unique acid catalysis of heteropoly compounds(heteropolyoxometalates) in the solid state. Chem. Commun. 2001, 1141–1152. [Google Scholar] [CrossRef]
- Kholdeeva, O.A.; Maksimchuk, N.V.; Maksimo, G.M. Polyoxometalate-based heterogeneous catalysts for liquid phase selective oxidations: Comparison of different strategies. Catal. Today 2010, 157, 107–113. [Google Scholar] [CrossRef]
- Mizuno, N.; Misono, M. Heterogeneous Catalysis. Chem. Rev. 1998, 98, 199–218. [Google Scholar] [CrossRef]
- Gupta, K.C.; Sutar, A.K.; Lin, C.-C. Polymer-supported Schiff base complexes in oxidation reactions. Coord. Chem. Rev. 2009, 253, 1926–1946. [Google Scholar] [CrossRef]
- McNamara, C.A.; Dixon, M.J.; Bradley, M. Recoverable Catalysts and Reagents Using Recyclable Polystyrene-Based Supports. Chem. Rev. 2002, 102, 3275–3300. [Google Scholar] [CrossRef]
- Mizuno, N.; Yamaguchi, K.; Kamata, K. Molecular design of polyoxometalate-based compounds for environmentally-friendly functional group transformations: From molecular catalysts to heterogeneous catalysts. Catal. Surv. Asia 2011, 15, 68–79. [Google Scholar] [CrossRef]
- Bentaleb, F.; Makrygenni, O.; Brouri, D.; Coelho Diogo, C.; Mehdi, A.; Proust, A.; Launay, F.; Villanneau, R. Efficiency of Polyoxometalate-Based Mesoporous Hybrids as Covalently Anchored Catalysts. Inorg. Chem. 2015, 54, 7607–7616. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.-Y.; Lin, K.-J.; Prasad, M.R.; Fu, S.-J.; Chang, S.-Y.; Shyu, S.-G.; Sheu, H.-S.; Chen, C.-H.; Chuang, C.-H.; Lin, M.-T. Synthesis of a reusable oxotungsten-containing SBA-15 mesoporous catalyst for the organic solvent-free conversion of cyclohexene to adipic acid. Catal. Commun. 2007, 8, 1060–1064. [Google Scholar] [CrossRef]
- Xiao, Y.; Chen, D.; Ma, N.; Hou, Z.Y.; Hu, M.B.; Wang, C.H.; Wang, W. Covalent immobilization of a polyoxometalate in a porous polymer matrix: A heterogeneous catalyst towards sustainability. RSC Adv. 2013, 3, 21544–21551. [Google Scholar] [CrossRef]
- Merrifield, R.B. Solid Phase Peptide Synthesis. I. The Synthesis of a Tetrapeptide. J. Am. Chem. Soc. 1963, 85, 2149–2154. [Google Scholar] [CrossRef]
- Maurya, M.R. Catalytic Applications of Polymer-Supported Molybdenum Complexes in Organic Transformations. Curr. Org. Chem. 2012, 16, 73–88. [Google Scholar] [CrossRef]
- Da Silva, J.A.L.; Fraústo da Silva, J.J.R.; Pombeiro, A.J.L. Oxovanadium complexes in catalytic oxidations. Coord. Chem. Rev. 2011, 255, 2232–2248. [Google Scholar] [CrossRef]
- Alini, S.; Babini, P. The Industrial Oxidation of KA Oil to Adipic Acid. In Handbook of Advanced Methods and Processes in Oxidation Catalysis; Imperial College Press: London, UK, 2014; pp. 320–333. [Google Scholar] [CrossRef]
- Usui, Y.; Sato, K. A green method of adipic acid synthesis: Organic solvent- and halide-free oxidation of cycloalkanones with 30% hydrogen peroxide. Green Chem. 2003, 5, 373–375. [Google Scholar] [CrossRef]
- Mazari, T.; Benadji, S.; Tahar, A.; Dermeche, L.; Rabia, C. Liquid-Phase Synthesis of Adipic Acid Using Keggin-Type Phosphomolybdates Catalysts. J. Mater. Sci. Eng. B 2013, 3, 146–152. [Google Scholar] [CrossRef]
- Benadji, S.; Mazari, T.; Dermeche, L.; Salhi, N.; Cadot, E.; Rabia, C. Clean Alternative for Adipic Acid Synthesis Via Liquid-Phase Oxidation of Cyclohexanone and Cyclohexanol Over H3−2xCoxPMo12O40 Catalysts with Hydrogen Peroxide. Catal. Lett. 2013, 143, 749–755. [Google Scholar] [CrossRef]
- Tahar, A.; Benadji, S.; Mazari, T.; Dermeche, L.; Roch-Marchal, C.; Rabia, C. Preparation, Characterization and Reactivity of Keggin Type Phosphomolybdates, H3−2xNixPMo12O40 and (NH4)3−2xNixPMo12O40, for Adipic Acid Synthesis. Catal. Lett. 2015, 145, 569–575. [Google Scholar] [CrossRef]
- Nomiya, K.; Miwa, M.; Sugaya, Y. Catalysis by heteropolyacid—VII. catalytic oxidation of cyclohexanol by dodecamolybdate. Polyhedron 1984, 3, 607–610. [Google Scholar] [CrossRef]
- Sato, K.; Aoki, M.; Noyori, R. A “Green” Route to Adipic Acid: Direct Oxidation of Cyclohexenes with 30 Percent Hydrogen Peroxide. Science 1998, 281, 1646–1647. [Google Scholar] [CrossRef] [PubMed]
- Cavani, F.; Teles, J.H. Sustainability in catalytic oxidation: An alternative approach or a structural evolution? ChemSusChem 2009, 2, 508–534. [Google Scholar] [CrossRef] [PubMed]
- Schindler, G.P.; Bartl, P.; Hoelderich, W.F. Oxidative cleavage of cyclohexane derivatives over titanium-containing Y zeolites. Appl. Catal. A 1998, 166, 267–279. [Google Scholar] [CrossRef]
- Wang, W.; Liu, H.; Ding, G.; Zhang, P.; Wu, T.; Jiang, T.; Han, B. Ru–Cd/Bentonite for the Partial Hydrogenation of Benzene: A Catalyst without Additives. ChemCatChem 2012, 4, 1836–1843. [Google Scholar] [CrossRef]
- Schwab, F.; Lucas, M.; Claus, P. Ruthenium-catalyzed selective hydrogenation of benzene to cyclohexene in the presence of an ionic liquid. Angew. Chem. Int. Ed. 2011, 50, 10453–10456. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Jiang, T.; Han, B.; Liang, S.; Wang, W.; Wu, T.; Yang, G. Highly selective benzene hydrogenation to cyclohexene over supported Ru catalyst without additives. Green Chem. 2011, 13, 1106–1109. [Google Scholar] [CrossRef]
- Jin, M.; Cheng, Z.-M. Oxidative Dehydrogenation of Cyclohexane to Cyclohexene over Mg-V-O Catalysts. Catal. Lett. 2009, 131, 266–278. [Google Scholar] [CrossRef]
- Patcas, F.; Patcas, F.C. Reaction pathways and kinetics of the gas-phase oxidation of cyclohexane on NiO/γ-Al2O3 catalyst. Catal. Today 2006, 117, 253–258. [Google Scholar] [CrossRef]
- Zhu, H.; Ge, Q.; Li, W.; Liu, X.; Xu, H. Study of Mn-based Catalysts for Oxidative Dehydrogenation of Cyclohexane to Cyclohexene. Catal. Lett. 2005, 105, 29–33. [Google Scholar] [CrossRef]
- Soares, J.C.S.; Gonçalves, A.H.A.; Zotin, F.M.Z.; Raddi de Araújo, L.R.; Gaspar, A.B. Cyclohexene to adipic acid synthesis using heterogeneous polyoxometalate catalysts. Mol. Catal. 2018, 458, 223–229. [Google Scholar] [CrossRef]
- Bohström, Z.; Rico-Lattes, I.; Holmberg, K. Oxidation of cyclohexene into adipic acid in aqueous dispersions of mesoporous oxides with built-in catalytical sites. Green Chem. 2010, 12, 1861–1869. [Google Scholar] [CrossRef]
- Timofeeva, M.N.; Kholdeeva, O.A.; Jhung, S.H.; Chang, J.-S. Titanium and cerium-containing mesoporous silicate materials as catalysts for oxidative cleavage of cyclohexene with H2O2: A comparative study of catalytic activity and stability. Appl. Catal. A 2008, 345, 195–200. [Google Scholar] [CrossRef]
- Lee, S.O.; Raja, R.; Harris, K.D.M.; Tomas, J.M.; Johnson, B.F.J.; Sankar, G. Mechanistic Insights into the Conversion of Cyclohexene to Adipic Acid by H2O2 in the Presence of a TAPO-5 Catalyst. Angew. Chem. Int. Ed. 2003, 42, 1520–1523. [Google Scholar] [CrossRef] [PubMed]
- Lapisardi, G.; Chiker, F.; Launay, F.; Nogier, J.P.; Bonardet, J.L. Preparation, characterisation and catalytic activity of new bifunctional Ti–AlSBA15 materials. Application to a “one-pot” green synthesis of adipic acid from cyclohexene and organic hydroperoxides. Microporous Mesoporous Mater. 2005, 78, 289–295. [Google Scholar] [CrossRef]
- Lapisardi, G.; Chiker, F.; Launay, F.; Nogier, J.P.; Bonardet, J.L. A “one-pot” synthesis of adipic acid from cyclohexene under mild conditions with new bifunctional Ti-AlSBA mesostructured catalysts. Catal. Commun. 2004, 5, 277–281. [Google Scholar] [CrossRef]
- Gui, J.; Liu, D.; Cong, X.; Zhang, X.; Jiang, H.; Hu, Z.; Sun, Z. Clean synthesis of adipic acid by direct oxidation of cyclohexene with H2O2 catalysed by Na2WO4·2H2O and acidic ionic liquids. J. Chem. Res. 2005, 8, 520–522. [Google Scholar] [CrossRef]
- Wang, B.; Zhang, Z.; Zhang, X.; Sun, S.; Wu, L.; Xing, R. Efficient and convenient oxidation of cyclohexene to adipic acid with H2O2 catalyzed by H2WO4 in acidic ionic liquids. Chem. Pap. 2018, 72, 643–649. [Google Scholar] [CrossRef]
- Lin, Z.-P.; Wan, L. Synthesis of Adipic Acid Oxidized by H2O2-Silicotungstic Acid Under Ultrasonication. Asian J. Chem. 2013, 25, 6008–6010. [Google Scholar] [CrossRef]
- Chiker, F.; Launay, F.; Nogier, J.P.; Bonardet, J.L. Green epoxidation on Ti-mesoporous catalysts. Environ. Chem. Lett. 2003, 1, 117–120. [Google Scholar] [CrossRef]
- Santini, R.; Griffith, M.C.; Qi, M. A measure of solvent effects on swelling of resins for solid phase organic synthesis. Tetrahedron Lett. 1998, 39, 8951–8954. [Google Scholar] [CrossRef]
- Rocchiccioli-Deltcheff, C.; Fournier, M.; Franck, R.; Thouvenot, R. Vibrational investigations of polyoxometalates. 2. Evidence for anion-anion interactions in molybdenum(VI) and tungsten(VI) compounds related to the Keggin structure. Inorg. Chem. 1983, 22, 207–216. [Google Scholar] [CrossRef]
- Rocchiccioli-Deltcheff, C.; Fournier, M. Catalysis by polyoxometalates. Part 3.—Influence of vanadium(V) on the thermal stability of 12-metallophosphoric acids from in situ infrared studies. J. Chem. Soc. Faraday Trans. 1991, 87, 3913–3920. [Google Scholar] [CrossRef]
- Aouissi, A.; Abdullah Al-Othman, Z.; Al-Anezi, H. Reactivity of Heteropolymolybdates and Heteropolytungstates in the Cationic Polymerization of Styrene. Molecules 2010, 15, 3319–3328. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, T.; Kimizuka, N. Interfacial Synthesis of Hollow TiO2 Microspheres in Ionic Liquids. J. Am. Chem. Soc. 2003, 125, 6386–6387. [Google Scholar] [CrossRef] [PubMed]
- Ganapathy, S.; Fournier, M.; Paul, J.F.; Delevoye, L.; Guelton, M.; Amoureux, J.P. Location of Protons in Anhydrous Keggin Heteropolyacids H3PMo12O40 and H3PW12O40 by 1H{31P}/31P{1H} REDOR NMR and DFT Quantum Chemical Calculations. J. Am. Chem. Soc. 2002, 26, 7821–7828. [Google Scholar] [CrossRef]
- Chen, Y.; Tan, R.; Zheng, W.; Zhang, Y.; Zhao, G.; Yin, D. Dendritic phosphotungstate hybrids efficiently catalyze the selective oxidation of alcohols with H2O2. Catal. Sci. Technol. 2014, 4, 4084–4092. [Google Scholar] [CrossRef]
- Chang, T. NMR characterization of the supported 12-heteropoly acids. J. Chem. Soc. Faraday Trans. 1995, 91, 375–379. [Google Scholar] [CrossRef]
- Edwards, J.C.; Thiel, C.Y.; Benac, B.; Knifton, J.F. Solid-state NMR and FT-IR investigation of 12-tungstophosphoric acid on TiO2. Catal. Lett. 1998, 51, 77–83. [Google Scholar] [CrossRef]
- Ren, S.; Xie, Z.; Cao, L.; Xie, X.; Qin, G.; Wang, J. Clean synthesis of adipic acid catalyzed by complexes derived from heteropoly acid and glycine. Catal. Commun. 2009, 10, 464–467. [Google Scholar] [CrossRef]
- Guérin, B.; Mesquita Fernandez, D.; Daran, J.-C.; Agustin, D.; Poli, R. Investigation of induction times, activity, selectivity, interface and mass transport in solvent-free epoxidation by H2O2 and TBHP: A study with organic salts of the [PMo12O40]3− anion. New J. Chem. 2013, 37, 3466–3475. [Google Scholar] [CrossRef]
- Leclercq, L.; Mouret, A.; Proust, A.; Schmitt, V.; Bauduin, P.; Aubry, J.M.; Nardello-Rataj, V. Pickering Emulsion Stabilized by Catalytic Polyoxometalate Nanoparticles: A New Effective Medium for Oxidation Reactions. Chem.-Eur. J. 2012, 18, 14352–14358. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Leclercq, L.; Schmitt, V.; Pera-Titus, M.; Nardello-Rataj, V. Colloidal tectonics for tandem synergistic Pickering interfacial catalysis: Oxidative cleavage of cyclohexene oxide into adipic acid. Chem. Sci. 2019, 10, 501–507. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.-R.; Liu, J.; Yang, Y.; Deng, L. A general approach towards efficient catalysis in Pickering emulsions stabilized by amphiphilic RGO–Silica hybrid materials. RSC Adv. 2014, 4, 35744–35749. [Google Scholar] [CrossRef]
- Moudjahed, M.; Dermeche, L.; Benadji, S.; Mazari, T.; Rabia, C. Dawson-type polyoxometalates as green catalysts for adipic acid synthesis. J. Mol. Catal. A Chem. 2016, 414, 72–77. [Google Scholar] [CrossRef]
- Mouheb, L.; Dermeche, L.; Mazari, T.; Benadji, S.; Essayem, N.; Rabia, C. Clean Adipic Acid Synthesis from Liquid-Phase Oxidation of Cyclohexanone and Cyclohexanol Using (NH4)xAyPMo12O40 (A: Sb, Sn, Bi) Mixed Heteropolysalts and Hydrogen Peroxide in Free Solvent. Catal. Lett. 2018, 148, 612–620. [Google Scholar] [CrossRef]
- Huddleston, J.G.; Visser, A.E.; Reichert, W.M.; Willauer, H.D.; Broker, G.A.; Rogers, R.D. Characterization and comparison of hydrophilic and hydrophobic room temperature ionic liquids incorporating the imidazolium cation. Green Chem. 2001, 3, 156–164. [Google Scholar] [CrossRef]
- Cha, S.; Ao, M.; Sung, W.; Moon, B.; Ahlström, B.; Johansson, P.; Ouchi, Y.; Kim, D. Structures of ionic liquid–water mixtures investigated by IR and NMR spectroscopy. Phys. Chem. Chem. Phys. 2014, 16, 9591–9601. [Google Scholar] [CrossRef]
- Poli, E.; Clacens, J.-M.; Pouilloux, Y. Synthesis of peroxophosphotungstate immobilized onto polymeric support as heterogeneous catalyst for the epoxidation of unsaturated fatty esters. Catal. Today 2011, 164, 429–435. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sub. | Cat. | Cat/Sub Loading | Ox. Agent (eq) | T (°C) | Time (h) | AA Yield (%) | Sub Conv (%) | Ref. |
|---|---|---|---|---|---|---|---|---|
| CH | Na2WO4·2H2O [CH3(n-C8H17)3N]HSO4 | 1% | A | 75–90 | 8 | 90 | >99 | [40] |
| CH | Na2WO4 + ILs | 2.5% | 4.4 A | reflux | 10 | 100 | 89–97 | [55] |
| CHK CHA | H2WO4 + ILs | 1% | 3.3 A 4.4 A | 90 | 20 | 90 | >99 | [35] |
| CH | H2WO4 + IL | 2% | 4.4 A | 73–87 | 12 | 85–96 | n.d. | [56] |
| CH | H4SiW12O40 | 2% | 5 A | US (25 kHz) | 4 | 92 | [57] | |
| CHA | (C4H9N)3[PMo12O40] | 0.02% | 3 A | reflux | 17 | 18 | >99 | [39] |
| CHA CHK | H3PMo12O40 NiPMo12O40 CoPMo12O40 | 0.5% | 1 A | 90 | 14 | 17–30 17–32 | >99 | [36] |
| CHK CHA | H3−2xNixPMo12O40 (NH4)3−2xNixPMo12O40 (x = 0.0–1.5) | 0.1–0.4% | 1 A | 90 | 20 | 34–45 | >99 | [38] |
| CHK CHA | H3−2xCoxPMo12O40 | 0.15–0.66% | 1 A | 90 | 20 | 30–50 | >99 | [37] |
| CHD CHO CHK CHA | Ti-Y zeolites | sub/cat mass ratio = 9.7 | 4 A | 80 | 24 | 13–39 44 78 70 | 24–60 59 36 | [42] |
| CH | Ti-MMM-2 Ce-SBA-15 | 5 mmol CH 50 mg cat | 3.6 A | 80 | 72 | 10–30 | >99 | [51] |
| CH CHO CHD | TAPO-5 | 62 mmol 0.5 g cat | 3.5 A | 80 | 72 24 24 | 30 40 42 | >99 >99 >72 | [52] |
| CHD CHO | Ti-AlSBA-15 + 10 mL CH3CN | 7 mmol sub 0.1 g | 3 B | 80 | 48 | 50–88 94 | 56–98 84 | [53] |
| CH | 7.8% | 3 A | 70 | 24 | 80 | 7 | [58] | |
| 1.5 B | 80 | 2 | 10 | 15 | ||||
| CHO | Ti(16)Al(DS)SBA + 10 mL CH3CN | 9.7% | 3 B | 80 | 48 | 94 | 84 | [54] |
| CHD | 24–48 | 76–96 | 33–88 | |||||
| CH | 9.7–19.4% | 4B | 40–84 |
| EA Data | C (%) | H (%) | N (%) | Cl (%) | M (Mo or W) Loading (µmol POM/g of Polymer) * |
|---|---|---|---|---|---|
| MR | 79.1 | 6.4 | - | 14.5 | - |
| MRim | 74.6 | 8.4 | 6.9 | 10.1 | - |
| MR4-Mo | 59.5 | 6.8 | 5.0 | n.d. | 66.7 |
| MR4-W | 55.9 | 6.0 | 4.5 | n.d. | 12.3 |
| MR12-Mo | 40.9 | 4.1 | 4.3 | n.d. | 55.6 |
| MR12-W | 30.0 | 3.0 | 3.5 | n.d. | 18.9 |
| Molecular Catalysts | Grafted Catalysts | |||||||
|---|---|---|---|---|---|---|---|---|
| C4 | C6 | MR4 | MR12 | |||||
| Mo | W | Mo | W | Mo | W | Mo | W | |
| %Cat a | 0.025 | 0.004 | 0.007 | 0.003 | 0.001 | |||
| CH as substrate | ||||||||
| CH conv.b | 65 | 75 | 61 | 68 | 58 | 61 | 56 | 73 |
| AA yield c | 46 | 61 | 42 | 50 | 33 | 43 | 30 | 51 |
| CHD form d | 15 | 12 | 11 | 17 | 24 | 17 | 19 | 20 |
| CHO as substrate | ||||||||
| CHO conv.e | >99 | >99 | ||||||
| AA yield c | 36 | 47 | 32 | 43 | 28 | 33 | 21 | 36 |
| CHD form d | 25 | 26 | 29 | 24 | 28 | 23 | 32 | 35 |
| CHD as substrate | ||||||||
| CHD conv.f | >99 | 71 | 94 | 79 | 95 | |||
| AA yield c | 74 | 72 | 58 | 63 | 46 | 60 | 41 | 56 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pisk, J.; Agustin, D.; Poli, R. Organic Salts and Merrifield Resin Supported [PM12O40]3− (M = Mo or W) as Catalysts for Adipic Acid Synthesis. Molecules 2019, 24, 783. https://doi.org/10.3390/molecules24040783
Pisk J, Agustin D, Poli R. Organic Salts and Merrifield Resin Supported [PM12O40]3− (M = Mo or W) as Catalysts for Adipic Acid Synthesis. Molecules. 2019; 24(4):783. https://doi.org/10.3390/molecules24040783
Chicago/Turabian StylePisk, Jana, Dominique Agustin, and Rinaldo Poli. 2019. "Organic Salts and Merrifield Resin Supported [PM12O40]3− (M = Mo or W) as Catalysts for Adipic Acid Synthesis" Molecules 24, no. 4: 783. https://doi.org/10.3390/molecules24040783
APA StylePisk, J., Agustin, D., & Poli, R. (2019). Organic Salts and Merrifield Resin Supported [PM12O40]3− (M = Mo or W) as Catalysts for Adipic Acid Synthesis. Molecules, 24(4), 783. https://doi.org/10.3390/molecules24040783