Li and Na Adsorption on Graphene and Graphene Oxide Examined by Density Functional Theory, Quantum Theory of Atoms in Molecules, and Electron Localization Function

 ,
,  ,
,
Abstract

1. Introduction
2. Computational Methods
2.1. Periodic Structure Modeling
2.2. DFT Parameters
2.3. QTAIM Parameters
2.4. ELF Parameters
3. Results and Discussion
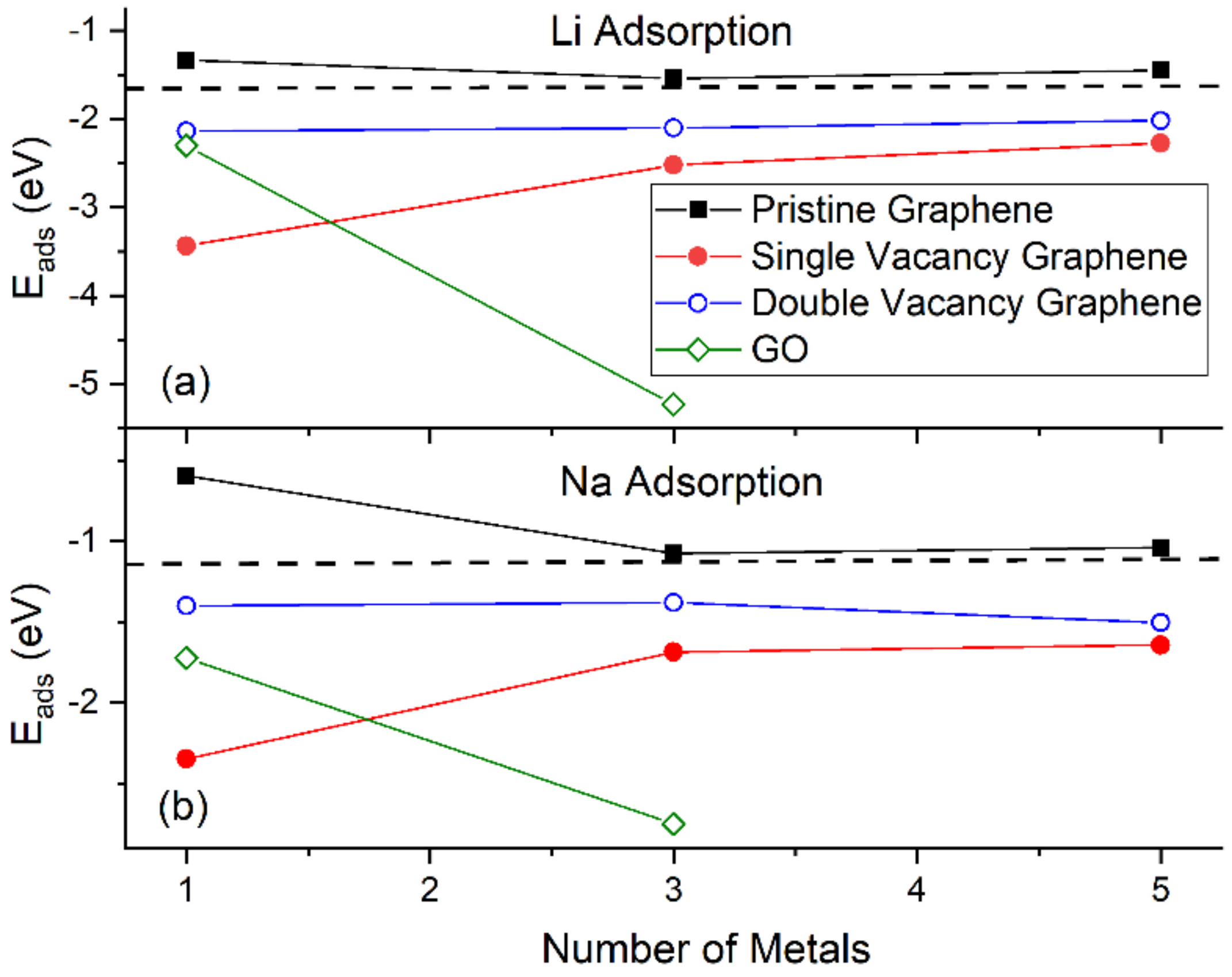
3.1. Structural Properties, Eads, and Ec for Li and Na Adsorbed on Graphene and Graphene Oxide
3.1.1. Adsorption on Pristine Graphene
3.1.2. Adsorption on Defective Graphene
3.1.3. Adsorption on Graphene Oxide
3.2. Charge transfers and COOP
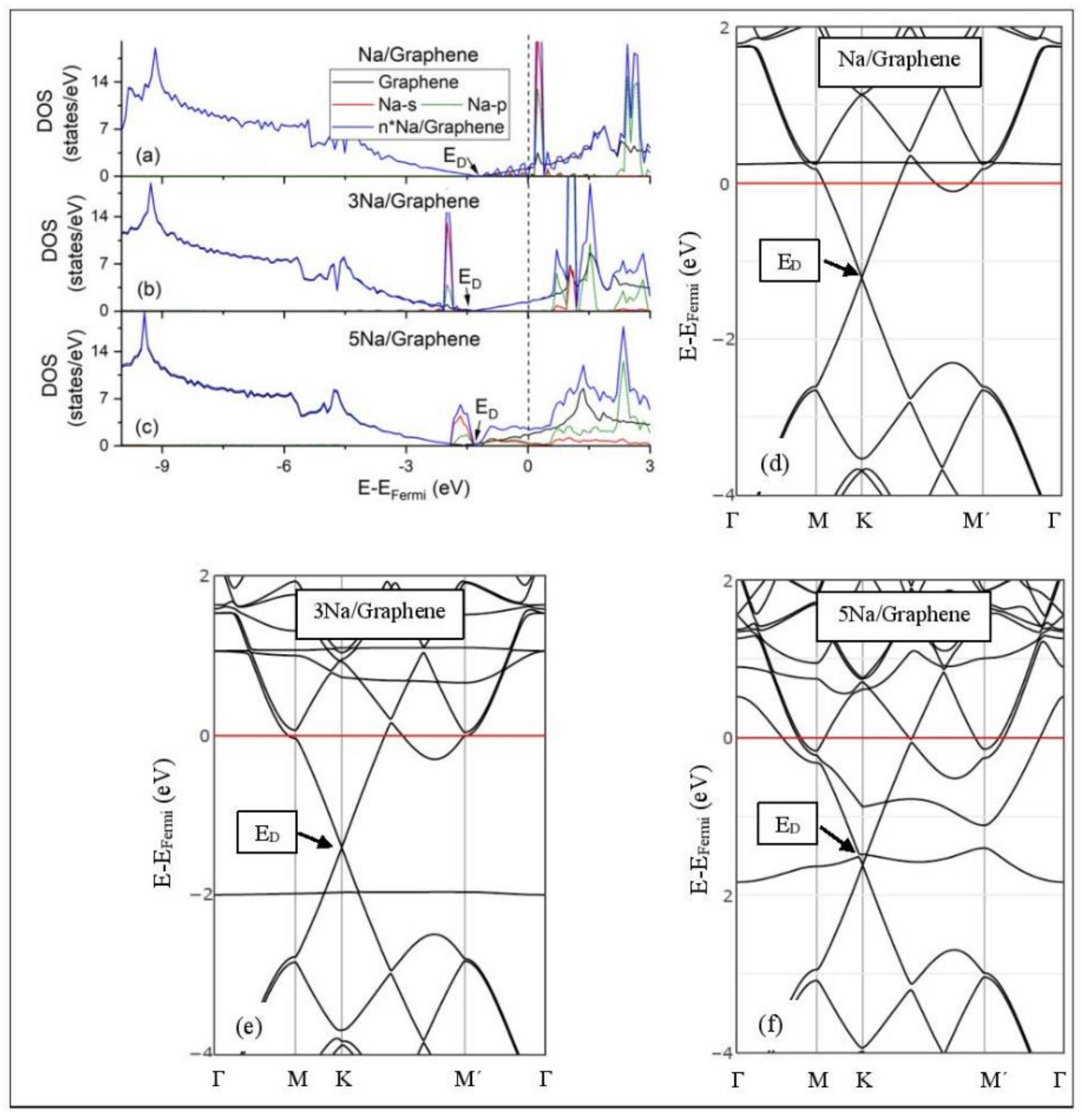
3.3. DOS and electronic structures
3.3.1. Adsorption on Pristine Graphene
3.3.2. Adsorption on Defective Graphene.
3.3.3. Adsorption on GO
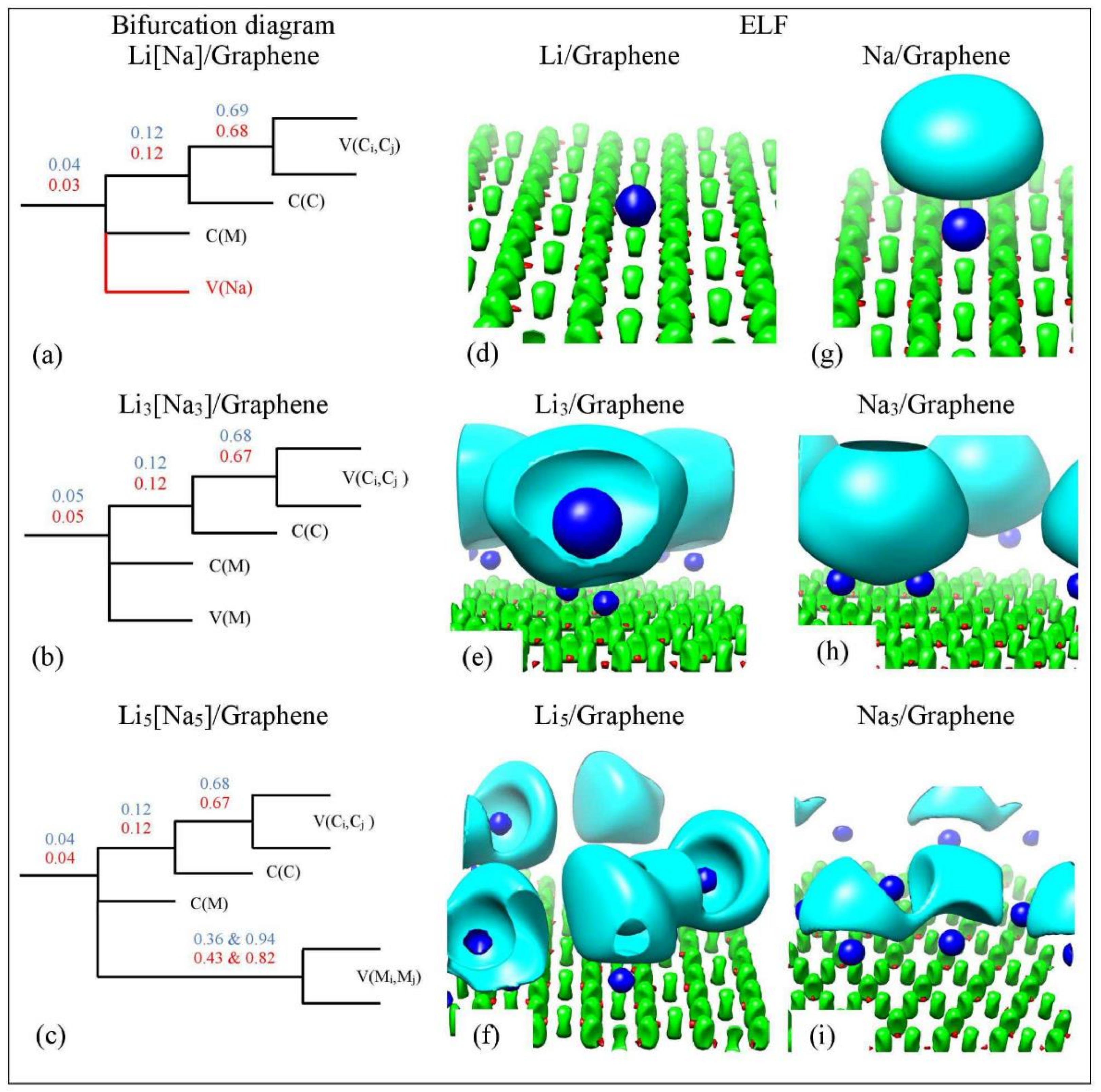
3.4. QTAIM analysis
3.4.1. The Metal–C Bond
3.4.2. The Metal–metal Bond
3.5. ELF Analysis
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Tarascon, J.-M.; Armand, M. Issues and challenges facing rechargeable lithium batteries. Nature 2001, 414, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Goodenough, J.B.; Park, K.-S. The Li-Ion Rechargeable Battery: A Perspective. J. Am. Chem. Soc. 2013, 135, 1167–1176. [Google Scholar] [CrossRef] [PubMed]
- Bruce, P.G.; Scrosati, B.; Tarascon, J.-M. Nanomaterials for Rechargeable Lithium Batteries. Angew. Chem. Int. Ed. 2008, 47, 2930–2946. [Google Scholar] [CrossRef] [PubMed]
- Dahn, J.R.; Zheng, T.; Liu, Y.H.; Xue, J.S. Mechanisms for Lithium Insertion in Carbonaceous Materials. Science 1995, 270, 590–593. [Google Scholar] [CrossRef]
- Kheirabadi, N.; Shafiekhani, A. Graphene/Li-ion battery. J. Appl. Phys. 2012, 112, 124323–124325. [Google Scholar] [CrossRef]
- Yang, S.; Li, S.; Tang, S.; Dong, W.; Sun, W.; Shen, D.; Wang, M. Sodium adsorption and intercalation in bilayer graphene from density functional theory calculations. Theor. Chem. Acc. 2016, 135, 164. [Google Scholar] [CrossRef]
- Winter, M.; Besenhard, J.O.; Spahr, M.E.; Novak, P. Insertion Electrode Materials for Rechargeable Lithium Batteries. Adv. Mater. 1998, 10, 725–763. [Google Scholar] [CrossRef]
- Geim, A.K.; Novoselov, K.S. The rise of graphene. Nat. Mater. 2007, 6, 183–191. [Google Scholar] [CrossRef]
- Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.; Katsnelson, M.I.; Grigorieva, I.V.; Firsov, A.A. Electric field effect in anatomically thin carbon films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef]
- Yoo, E.; Kim, J.; Hosono, E.; Zhou, H.-S.; Kudo, T.; Honma, I. Large Reversible Li Storage of Graphene Nanosheet Families for Use in Rechargeable Lithium Ion Batteries. Nano Lett. 2008, 8, 2277–2282. [Google Scholar] [CrossRef]
- Liu, Y.; Xie, B.; Zhang, Z.; Zheng, Q.; Xu, Z. Mechanical properties of graphene papers. J. Mech. Phys. Solids 2012, 60, 591–605. [Google Scholar] [CrossRef]
- Pop, E.; Varshney, V.; Roy, A. Thermal properties of graphene: Fundamentals and applications. MRS Bull. 2012, 37, 1273–1281. [Google Scholar] [CrossRef]
- Castro Neto, A.H.; Guinea, F.; Peres, N.M.R.; Novoselov, K.S.; Geim, A.K. The electronic properties of graphene. Rev. Mod. Phys. 2009, 81, 110–155. [Google Scholar] [CrossRef]
- Wu, M.; Cao, C.; Jiang, J.Z. Light non-metallic atom (B, N, O and F)-doped graphene: A first-principles study. Nanotechnology 2010, 21, 505202–505206. [Google Scholar] [CrossRef] [PubMed]
- Pollak, E.; Geng, B.; Jeon, K.-J.; Lucas, I.T.; Richardson, T.J.; Wang, F.; Kostecki, R. The Interaction of Li+ with Single-Layer and Few-Layer Graphene. Nano Lett. 2010, 10, 3386–3388. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.T.; Neaton, J.B.; Cohen, M.L. First-Principles Study of Metal Adatom Adsorption on Graphene. Phys. Rev. B 2008, 77, 235430. [Google Scholar] [CrossRef]
- Dimakis, N.; Valdez, D.; Flor, F.A.; Salgado, A.; Adjibi, K.; Vargas, S.; Saenz, J. Density functional theory calculations on alkali and the alkaline Ca atoms adsorbed on graphene monolayers. Appl. Surf. Sci. 2017, 413, 197–208. [Google Scholar] [CrossRef]
- Nakada, K.; Ishii, A. DFT Calculation for adatom adsorption on Graphene. In Graphene Simulation; Gong, J.R., Ed.; InTech Inc.: Zagreb, Croatia, 2011; pp. 3–20. [Google Scholar]
- Medeiros, P.V.C.; de Brito Mota, F.; Mascarenhas, A.J.S.; de Castilho, C.M.C. Adsorption of Monovalent Metal Atoms on Graphene: A Theoretical Approach. Nanotechnology 2010, 21, 115701–115706. [Google Scholar] [CrossRef]
- Lee, E.; Persson, K.A. Li Absorption and Intercalation in Single Layer Graphene and Few Layer Graphene by First Principles. Nano Lett. 2012, 12, 4624–4628. [Google Scholar] [CrossRef]
- Okamoto, Y. Density Functional Theory Calculations of Lithium Adsorption and Insertion to Defect-Free and Defective Graphene. J. Phys. Chem. C 2016, 120, 14009–14014. [Google Scholar] [CrossRef]
- Yildirim, H.; Kinaci, A.; Zhao, Z.-J.; Chan, M.K.Y.; Greeley, J.P. First-Principles Analysis of Defect-Mediated Li Adsorption on Graphene. ACS Appl. Mater. Interfaces 2014, 6, 21141–21150. [Google Scholar] [CrossRef] [PubMed]
- Shiota, K.; Kawai, T. Li atom adsorption on graphene with various defects for large-capacity Li ion batteries: First-principles calculations. Jpn. J. Appl. Phys. 2017, 56, 06GE11. [Google Scholar] [CrossRef]
- Datta, D.; Li, J.; Koratkar, N.; Shenoy, V.B. Enhanced lithiation in defective graphene. Carbon 2014, 80, 305–310. [Google Scholar] [CrossRef]
- Yu, Y.-X. Can all nitrogen-doped defects improve the performance of graphene anode materials for lithium-ion batteries? Phys. Chem. Chem. Phys. 2013, 15, 16819–16827. [Google Scholar] [CrossRef] [PubMed]
- Slater, M.D.; Kim, D.; Lee, E.; Johnson, C.S. Sodium-ion batteries. Adv. Funct. Mater. 2013, 23, 947–958. [Google Scholar] [CrossRef]
- Teng, F.Z.; McDonough, W.F.; Rudnick, R.L.; Dalpé, C.; Tomascak, P.B.; Chappell, B.W.; Gao, S. Lithium isotopic composition and concentration of the upper continental crust. Geochim. Cosmochim. Acta 2004, 68, 4167–4178. [Google Scholar] [CrossRef]
- Palomares, V.; Serras, P.; Villaluenga, I.; Hueso, K.B.; Carretero-González, J.; Rojo, T. Na-ion batteries, recent advances and present challenges to become low cost energy storage systems. Energy Environ. Sci. 2012, 5, 5884–5901. [Google Scholar] [CrossRef]
- Harris, S.J.; Timmons, A.; Baker, D.R.; Monroe, C. Direct in situ Measurements of Li Transport in Li-Ion Battery Negative Electrodes. Chem. Phys. Lett. 2010, 485, 265–274. [Google Scholar] [CrossRef]
- Zhou, J.; Sun, Q.; Wang, Q.; Jena, P. Tailoring Li adsorption on graphene. Phys. Rev. B 2014, 90, 205427. [Google Scholar] [CrossRef]
- Liu, M.F.; Kutana, A.; Liu, Y.; Yakobson, B.I. First-Principles Studies of Li Nucleation on Graphene. J. Phys. Chem. Lett. 2014, 5, 1225–1229. [Google Scholar] [CrossRef]
- Gómez-Navarro, C.; Meyer, J.C.; Sundaram, R.S.; Chuvilin, A.; Kurasch, S.; Burghard, M.; Kern, K.; Kaiser, U.A. Atomic Structure of Reduced Graphene Oxide. Nano Lett. 2010, 10, 1144–1148. [Google Scholar] [CrossRef]
- Acik, M.; Lee, G.; Mattevi, C.; Pirkle, A.; Wallace, R.M.; Chhowalla, M.; Cho, K.; Chabal, Y. The Role of Oxygen during Thermal Reduction of Graphene Oxide Studied by Infrared Absorption Spectroscopy. J. Phys. Chem. C 2011, 115, 19761–19781. [Google Scholar] [CrossRef]
- Nobuhara, K.; Nakayama, H.; Nose, M.; Nakanishi, S.; Iba, H. First-Principles Study of Alkali Metal-Graphite Intercalation Compounds. J. Power Sources 2013, 243, 585–587. [Google Scholar] [CrossRef]
- Asher, R.C. A lamellar compound of sodium and graphite. J. Inorg. Nucl. Chem. 1959, 10, 238–249. [Google Scholar] [CrossRef]
- Datta, D.; Li, J.; Shenoy, V.B. Defective Graphene as a High-Capacity Anode Material for Na- and Ca-Ion Batteries. ACS Appl. Mater. Interfaces 2014, 6, 1788–1795. [Google Scholar] [CrossRef]
- Liang, Z.; Fan, X.; Zheng, W.; Singh, D.J. Adsorption and Formation of Small Na Clusters on Pristine and Double-Vacancy Graphene for Anodes of Na-Ion Batteries. ACS Appl. Mater. Interfaces 2017, 9, 17076–17084. [Google Scholar] [CrossRef] [PubMed]
- Bader, R.F.W. Atoms in Molecules. In Encyclopedia of Computational Chemistry; John Wiley and Sons: London, UK, 1998; Volume 1, pp. 64–86. [Google Scholar]
- Koritsanszky, T.S.; Coppens, P. Chemical Applications of X-ray Charge-Density Analysis. Chem. Rev. 2001, 101, 1583–1628. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules; Wiley Online Library: Hoboken, NJ, USA, 1990. [Google Scholar]
- Becke, A.D.; Edgecombe, K.E. A simple measure of electron localization in atomic and molecular systems. J. Chem. Phys. 1990, 92, 5397–5403. [Google Scholar] [CrossRef]
- Jabłoński, M.; Palusiak, M. Basis set and method dependence in atoms in molecules calculations. J. Phys. Chem. A 2010, 114, 2240–2244. [Google Scholar] [CrossRef]
- Fuentealba, P.; Chamorro, E.; Santos, J.C. Understanding and using the electron localization function. In Theoretical Aspects of Chemical Reactivity; Toro-Labbé, A., Ed.; Theoretical and Computational Chemistry Series; Elsevier: Amsterdam, The Netherlands, 2007; Volume 19, pp. 57–85. [Google Scholar]
- Gupta, S.; Carrizosa, S.B.; Jasisnki, J.; Dimakis, N. Charge Transfer Dynamical Processes at Graphene-Transition Metal Oxides/Electrolyte Interface for Energy Storage: Insights from In-Situ Raman Spectroelectrochemistry. AIP Adv.J. Raman Spectrosc. 2018, 8, 065225. [Google Scholar] [CrossRef]
- Mallia, G.; Orlando, R.; Roetti, C.; Ugliengo, P.; Dovesi, R. F center in LiF: A quantum mechanical ab initio investigation of the hyperfine interaction between the unpaired electron at the vacancy and its first seven neighbors. Phys. Rev. B 2001, 63, 235102–235107. [Google Scholar] [CrossRef]
- Zhou, S.; Bongiorno, A. Density Functional Theory Modeling of Multilayer “Epitaxial” Graphene Oxide. Acc. Chem. Res. 2014, 47, 3331–3339. [Google Scholar] [CrossRef] [PubMed]
- Dovesi, R.; Saunders, V.R.; Roetti, C.; Zicovich-Wilson, C.M.; Pascale, F.; Civalleri, B.; Doll, K.; Harrison, N.M.; Bush, I.J.; D’Arco, P.; et al. CRYSTAL17 User’s Manual; University Torino: Torino, Italy, 2017. [Google Scholar]
- Shi, H.; Eglitis, R.; Borstel, G. Ab initio calculations of the BaF2 bulk and surface F centres. J. Phys. Condens. Matter 2006, 18, 8367. [Google Scholar] [CrossRef]
- Dimakis, N.; Flor, F.A.; Salgado, A.; Adjibi, K.; Vargas, S.; Saenz, J. Density functional theory calculations on transition metal atoms adsorbed on graphene monolayers. Appl. Surf. Sci. 2017, 421, 252–259. [Google Scholar] [CrossRef]
- Gupta, S.; Dimakis, N. Computational Predictions of Electronic Properties of Graphene with Defects, Adsorbed Transition Metal-Oxides and Water using Density Functional Theory. Appl. Surf. Sci. 2019, 467, 760–772. [Google Scholar] [CrossRef]
- Gupta, S.; Meek, R.; Evans, B.; Dimakis, N. Graphene-based “hybrid” aerogels with carbon nanotubes: Mesoporous network–functionality promoted defect density and electrochemical activity correlations. J. Appl. Phys. 2018, 124, 124304–124317. [Google Scholar] [CrossRef]
- Ernzerhof, M.; Scuseria, G.E. Assessment of the Perdew–Burke–Ernzerhof exchange-correlation functional. J. Chem. Phys. 1999, 110, 5029–5036. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
- Peintinger, M.F.; Oliveira, D.V.; Bredow, T. Consistent Gaussian basis sets of triple-zeta valence with polarization quality for solid-state calculations. J. Comput. Chem. 2013, 34, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, A.; Erba, A.; El-Kelany, K.E.; Rérat, M.; Orlando, R. Low-temperature phase of BaTiO3: Piezoelectric, dielectric, elastic, and photoelastic properties from ab initio simulations. Phys. Rev. B 2014, 89, 045103–045109. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188. [Google Scholar] [CrossRef]
- Gilat, G.; Raubenheimer, L. Accurate numerical method for calculating frequency-distribution functions in solids. Phys. Rev. 1966, 144, 390. [Google Scholar] [CrossRef]
- Gilat, G. Analysis of methods for calculating spectral properties in solids. J. Comput. Phys. 1972, 10, 432–465. [Google Scholar] [CrossRef]
- Anderson, D.G. Iterative procedures for nonlinear integral equations. J. ACM 1965, 12, 547–560. [Google Scholar] [CrossRef]
- Hoffmann, R. A chemical and theoretical way to look at bonding on surfaces. Rev. Mod. Phys. 1988, 60, 601–628. [Google Scholar] [CrossRef]
- Mulliken, R.S. Electronic population analysis on LCAO-MO [linear combination of atomic orbital-molecular orbital] molecular wave functions. I. J. Chem. Phys. 1955, 23, 1833–1840. [Google Scholar] [CrossRef]
- Bultinck, P.; Van Alsenoy, C.; Ayers, P.W.; Carbó-Dorca, R. Critical analysis and extension of the Hirshfeld atoms in molecules. J. Chem. Phys. 2007, 126, 144111–144119. [Google Scholar] [CrossRef]
- Marenich, A.V.; Jerome, S.V.; Cramer, C.J.; Truhlar, D.G. Charge Model 5: An Extension of Hirshfeld Population Analysis for the Accurate Description of Molecular Interactions in Gaseous and Condensed Phases. J. Chem. Theory Comput. 2012, 8, 527–541. [Google Scholar] [CrossRef]
- Davidson, E.R.; Chakravorty, S. A test of the Hirshfeld definition of atomic charges and moments. Theor. Chim. Acta 1992, 83, 319–330. [Google Scholar] [CrossRef]
- Boys, S.F.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Dimakis, N.; Flor, F.A.; Navarro, N.E.; Salgado, A.; Smotkin, E.S. Adsorption of Carbon Monoxide on Platinum–Ruthenium, Platinum–Osmium, Platinum–Ruthenium–Osmium, and Platinum–Ruthenium–Osmium–Iridium Alloys. J. Phys. Chem. C 2016, 120, 10427–10441. [Google Scholar] [CrossRef]
- Gatti, C. TOPOND-96: An electron density topological program for systems periodic in N (N=0-3) dimensions, User’s Manual. In User’s Manual; CNR-CSRSRC: Milano, Italy, 1996. [Google Scholar]
- Macchi, P.; Proserpio, D.M.; Sironi, A. Experimental electron density in a transition metal dimer: Metal–metal and metal–ligand bonds. J. Am. Chem. Soc. 1998, 120, 13429–13435. [Google Scholar] [CrossRef]
- Gatti, C. Chemical bonding in crystals: New directions. Z. Kristallogr. Cryst. Mater. 2005, 220, 399–457. [Google Scholar] [CrossRef]
- Espinosa, E.; Alkorta, I.; Elguero, J.; Molins, E. From weak to strong interactions: A comprehensive analysis of the topological and energetic properties of the electron density distribution involving X–H⋯F–Y systems. J. Chem. Phys. 2002, 117, 5529–5542. [Google Scholar] [CrossRef]
- Azizi, A.; Momen, R.; Xu, T.; Kirk, S.R.; Jenkins, S. Non-nuclear attractors in small charged lithium clusters, Limq (m = 2–5, q = ±1), with QTAIM and the Ehrenfest force partitioning. Phys. Chem. Chem. Phys. 2018, 20, 24695–24707. [Google Scholar] [CrossRef]
- Ayers, P.W.; Jenkins, S. Bond metallicity measures. Comput. Theor. Chem. 2015, 1053, 112–122. [Google Scholar] [CrossRef]
- Jenkins, S.; Ayers, P.W.; Kirk, S.R.; Mori-Sánchez, P.; Martín Pendás, A. Bond metallicity of materials from real space charge density distributions. Chem. Phys. Lett. 2009, 471, 174–177. [Google Scholar] [CrossRef]
- Centeno, S.A.; Vila, A.; Barro, L. Characterization of unprocessed historic platinum photographic papers by Raman, FTIR and XRF. Microchem. J. 2014, 114, 8–15. [Google Scholar] [CrossRef]
- Contreras-García, J.; Pendás, Á.M.; Silvi, B.; Recio, J.M. Useful applications of the electron localization function in high pressure crystal chemistry. J. Phys. Chem. Solids 2008, 69, 2204–2207. [Google Scholar] [CrossRef]
- Silvi, B.; Savin, A. Classification of chemical bonds based on topological analysis of electron localization functions. Nature 1994, 371, 683–686. [Google Scholar] [CrossRef]
- Simas, A.M.; Sagar, R.P.; Ku, A.C.T.; Smith, V.H.J. The radial charge distribution and the shell structure of atoms and ions. Can. J. Chem. 1988, 66, 1923–1930. [Google Scholar] [CrossRef]
- Kohout, M.; Savin, A. Atomic shell structure and electron numbers. Int. J. Quantum Chem. 1996, 60, 875–882. [Google Scholar] [CrossRef]
- Savin, A.; Nesper, R.; Wengert, S.; Fassler, T. ELF: The Electron Localization Function. Angew. Chem. Int. Ed. Engl. 1997, 36, 1808–1832. [Google Scholar] [CrossRef]
- Matito, E.; Poater, J.; Bickelhaupt, F.M.; Solà, M. Bonding in Methylalkalimetals (CH3M)n (M = Li, Na, K; n = 1, 4). Agreement and Divergences between AIM and ELF Analyses. J. Phys. Chem. B 2006, 110, 7189–7198. [Google Scholar] [CrossRef]
- Savin, A.; Silvi, B.; Colonna, F. Topological analysis of the electron localization function applied to delocalized bonds. Can. J. Chem. 1996, 74, 1088–1096. [Google Scholar] [CrossRef]
- Marx, D.; Savin, A. Topological Bifurcation Analysis: Electronic Structure of CH5+. Angew. Chem. Int. Ed. Engl. 1997, 36, 2077–2080. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Nadler, M.R.; Kempier, C.P. Crystallographic Data 186. Lithium. Anal. Chem. 1959, 31, 2109. [Google Scholar] [CrossRef]
- Aruja, E.; Perlitz, H. Neubestimmung der Gitterkonstante von Natrium. Z. Kristallogr. Cryst. Mater. 2015, 100, 195–200. [Google Scholar] [CrossRef]
- Kittel, C. Introduction to Solid State Physics, 8th ed.; Wiley: New York, NY, USA, 2005. [Google Scholar]
- Maezono, R.; Towler, M.D.; Lee, Y.; Needs, R.J. Quantum Monte Carlo Study of Sodium. Phys. Rev. B 2003, 68, 165103–165109. [Google Scholar] [CrossRef]
- Asher, R.C.; Wilson, S.A. Lamellar Compound of Sodium with Graphite. Nature 1958, 181, 409–410. [Google Scholar] [CrossRef]
- Pašti, I.A.; Jovanović, A.; Dobrota, A.S.; Mentus, S.V.; Johansson, B.J.; Skorodumova, N.V. Atomic adsorption on pristine graphene along the Periodic Table of Elements—From PBE to non-local functionals. Appl. Surf. Sci. 2018, 436, 433–440. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Nakada, K.; Ishii, A. Migration of adatom adsorption on graphene using DFT calculation. Solid State Commun. 2011, 151, 13–16. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104–154118. [Google Scholar] [CrossRef] [PubMed]
- Pašti, I.A.; Jovanović, A.; Dobrota, A.S.; Mentus, S.V.; Johansson, B.J.; Skorodumova, N.V. Atomic adsorption on graphene with a single vacancy: Systematic DFT study through the periodic table of elements. Phys. Chem. Chem. Phys. 2018, 20, 858–865. [Google Scholar] [CrossRef]
- Lee, K.; Murray, E.D.; Kong, L.; Lundqvist, B.I.; Langreth, D.C. Higher-accuracy van der Waals density functional. Phys. Rev. B 2010, 82, 081101–081104. [Google Scholar] [CrossRef]
- Dobrota, A.S.; Pašti, I.A.; Mentus, S.V.; Johansson, B.J.; Skorodumova, N.V. Functionalized graphene for sodium battery applications: The DFT insights. Electrochim. Acta 2017, 250, 185–195. [Google Scholar] [CrossRef]
- Doan, J.; Navarro, N.E.; Kumari, D.; Anderson, K.; Kingston, E.; Johnson, C.; Vong, A.; Dimakis, N.; Smotkin, E.S. Symmetry-based IR group modes as dynamic probes of Nafion ion exchange site structure. Polymer 2015, 73, 34–41. [Google Scholar] [CrossRef]
- Grechnev, A.; Ahuja, R.; Eriksson, O. Balanced crystal orbital overlap population—A tool for analysing chemical bonds in solids. J. Phys. Condens. Matter 2003, 15, 7751. [Google Scholar] [CrossRef]
- Rani, P.; Jindal, V.K. Designing band gap of graphene by B and N dopant atoms. RSC Adv. 2013, 3, 802–812. [Google Scholar] [CrossRef]
- Bader, R.F.W.; MacDougall, P.J.; Lau, C.D.H. Bonded and nonbonded charge concentrations and their relation to molecular geometry and reactivity. J. Am. Chem. Soc. 1984, 106, 1594–1605. [Google Scholar] [CrossRef]
- Aray, Y.; Rodriguez, J. Study of CO adsorption on the Fe (100) surface using the Laplacian of the electronic charge density. Surf. Sci. 1998, 405, L532–L541. [Google Scholar] [CrossRef]
Sample Availability: Input CRYSTAL17 files are available upon request. |

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | Vacancies | Current Work | Past work | |
|---|---|---|---|---|
| Eads (eV) | Method | |||
| Li /Graphene | 0 | −1.34 (−1.33) | −1.19 [90], −1.096 [16] | PBE [91] |
| −1.36 [92] | LDA | |||
| −1.56 [90] | PBE+D2 [54,91] | |||
| −1.23 [90] | PBE+D3 [91,93] | |||
| −1.05 [90] | PBE+vdW-DF2 [91] | |||
| 1 | (−3.44) | −2.65 [94] | PBE [91] | |
| −2.94 [94] | PBE+D2 [54,91] | |||
| −2.72 [94] | PBE+D3 [91,93] | |||
| −2.59 [94] | PBE+vdW-DF2 [91,95] | |||
| Na /Graphene | 0 | −0.59 (−0.59) | −0.55 [90], −0.462 [16] | PBE [91] |
| −0.72 [92] a | LDA | |||
| −0.93 [90] | PBE+D2 [54,91] | |||
| −0.64 [90] | PBE+D3 [91,93] | |||
| −0.49 [90] | PBE+vdW-DF2 [91,95] | |||
| −0.88 [37] | PBE+D2 [91] | |||
| 1 | (−2.35) | −1.88 [94] | PBE [91] | |
| −2.20 [94] | PBE+D2 [54,91] | |||
| −2.00 [94] | PBE+D3 [91,93] | |||
| −1.74 [94] | PBE+vdW-DF2 [91,95] | |||
| Metals | Support | Vacancies | Metal charge (e) | Metal–C COOP a | |
|---|---|---|---|---|---|
| Hirshfeld | Mulliken | ||||
| Li | Graphene | 0 | 2.27 (2.27) | 2.66 (2.64) | 0.041 (0.040) |
| 1 | (2.37) | (2.73) | (0.138) 3 | ||
| 2 | (2.31) | (2.67) | (0.039) 6 | ||
| GO | 0 | 2.13 (2.14) | 2.43 (2.45) | 0.085 (0.082) 1 | |
| Li3 | Graphene | 0 | 2.98 (2.98) | 2.97 (2.97) | 0.024 (0.024) |
| 1 | (2.64) | (2.84) | (0.036) 6 | ||
| 2 | (2.76) | (2.84) | (0.006) 4 | ||
| GO | 0 | 2.12 (2.13) | 2.42 (2.43) | 0.088 (0.090) 1 | |
| Li5 | Graphene | 0 | 3.01 (3.02) | 2.99 (2.99) | 0.015 (0.022) |
| 1 | (2.81) | (2.91) | (0.029) 6 | ||
| 2 | (2.84) | (2.43) | (0.036) 6 | ||
| Na | Graphene | 0 | 10.53 (10.53) | 10.81 (10.58) | 0.043 (0.042) |
| 1 | (10.32) | (10.76) | (0.116) 3 | ||
| 2 | (10.21) | (10.68) | (0.043) 6 | ||
| GO | 0 | 10.13 (10.13) | 10.53 (10.54) | 0.051(0.050) 1 | |
| Na3 | Graphene | 0 | 10.85 (10.85) | 10.97 (10.98) | 0.039 (0.039) |
| 1 | (10.72) | (10.97) | (0.039) 6 | ||
| 2 | (10.85) | (10.97) | (0.035) 6 | ||
| GO | 0 | 10.72 (10.52) | 10.84 (10.64) | 0.018 (0.032) | |
| Na5 | Graphene | 0 | 10.95 (10.95) | 11.03 (11.04) | 0.031 (0.037) 6 |
| 1 | (10.85) | (11.01) | (0.039) 6 | ||
| 2 | (10.86) | (11.00) | (0.035) 6 | ||
| Metals | Support | Vacancies | Distances (Å) | QTAIM properties (a.u.) | |||
|---|---|---|---|---|---|---|---|
| Li-C [Li-O] | |||||||
| Li | Graphene | 0 | 2.16 | 0.025 | 0.133 | 0.095 | 1.224 |
| (2.22) | (0.023) | (0.117) | (0.101) | (1.192) | |||
| 1 | (1.98) | (0.044) | (0.197) | (< 0.01) | (1.111) | ||
| 2 | (2.12) | (0.026) | (0.132) | (0.094) | (1.185) | ||
| GO | 0 | 2.33 | 0.017 | 0.079 | 0.127 | 1.020 | |
| (2.26) | (0.021) | (0.093) | (0.107) | (1.000) | |||
| [(1.84)] | [(0.039)] | [(0.276)] | [(0.136)] | [(1.569)] | |||
| Li3 | Graphene | 0 | 2.281 | 0.0221 | 0.1361 | 0.1881 | 1.3251 |
| (2.30) | (0.018) | (0.093) | (0.142) | (1.147) | |||
| 1 | (2.14) | (0.024) | (0.131) | (0.118) | (1.228) | ||
| 2 | (2.12) | (0.025) | (0.128) | (0.099) | (1.162) | ||
| GO | 0 | 2.362 | 0.016 | 0.078 | 0.157 | 1.057 | |
| (2.30) | (0.018) | (0.089) | (0.143) | (1.068) | |||
| [(1.82)] | [(0.042)] | [(0.294)] | [(0.186)] | [(1.565)] | |||
| Li5 | Graphene | 0 | 2.24 | 0.018 | 0.092 | 0.143 | 1.131 |
| (2.28) | (0.018) | (0.095) | (0.153) | (1.155) | |||
| 1 | (1.99) | (0.043) | (0.188) | (< 0.01) | (1.098) | ||
| 2 | (2.10) | (0.030) | (0.153) | (0.057) | (1.209) | ||
| Metals | Support | Vacancies | Distances (Å) | QTAIM properties (a.u.) | |||
|---|---|---|---|---|---|---|---|
| Na-C[Na-O] | |||||||
| Na | Graphene | 0 | 2.62 | 0.015 | 0.075 | 0.147 | 1.105 |
| (2.64) | (0.015) | (0.073) | (0.149) | (1.098) | |||
| 1 | (2.47) | (0.020) | (0.092) | (0.106) | (1.054) | ||
| 2 | (2.49) | (0.020) | (0.096) | (0.108) | (1.108) | ||
| GO | 0 | 2.54 | 0.019 | 0.087 | 0.116 | 1.048 | |
| (2.50) | (0.019) | (0.092) | (0.120) | (1.074) | |||
| [(2.37)] | [(0.019)] | [(0.162)] | [(0.237)] | [(1.321)] | |||
| Na3 | Graphene | 0 | 2.50 | 0.018 | 0.088 | 0.142 | 1.093 |
| (2.59) | (0.016) | (0.078) | (0.151) | (1.085) | |||
| 1 | (2.40) | (0.029) | (0.127) | (0.047) | (1.031) | ||
| 2 | (2.63) | (0.014) | (0.072) | (0.165) | (1.083) | ||
| GO | 0 | 2.87 | 0.009 | 0.043 | 0.277 | 1.005 | |
| (2.60) | (0.016) | (0.077) | (0.155) | (1.048) | |||
| [(2.27)] | [(0.023)] | [(0.152)] | [(0.237)] | [(1.426)] | |||
| Na5 | Graphene | 0 | 2.75 | 0.012 | 0.059 | 0.189 | 1.027 |
| (2.62) | (0.014) | (0.070) | (0.179) | (1.067) | |||
| 1 | (2.40) | (0.029) | (0.126) | (0.048) | (1.035) | ||
| 2 | (2.51) | (0.020) | (0.090) | (0.110) | (1.001) | ||
| System | Vacancies | Li-Li (Å) | QTAIM properties (a.u.) | ELF | |||
|---|---|---|---|---|---|---|---|
| Li3/Graphene | 0 | 2.92 | 0.0131 | −0.159 | 0.233 | 3.37 | 0.31 |
| 2.92 | 0.0131 | −0.158 | 0.234 | 3.28 | 0.31 | ||
| (2.85) | (0.014)1 | (−0.166) | (0.243) | (3.25) | (0.31) | ||
| (2.85) | (0.014)1 | (−0.165) | (0.243) | (3.21) | (0.31) | ||
| Li5/Graphene | 0 | 2.73 | 0.0112 | −0.174 | 0.093 | −3.09 | 0.69 |
| 2.97 | 0.0111 | −0.150 | 0.228 | 3.20 | 0.28 | ||
| 3.04 | 0.0131 | −0.153 | 0.249 | 2.55 | 0.30 | ||
| (2.77) | (<0.01)2 | (−0.148) | (0.044) | (−2.40) | (0.89) | ||
| (2.91) | (0.010)2 | (−0.170) | (0.164) | (−40.60) | (0.39) | ||
| (2.92) | (0.012)1 | (−0.163) | (0.211) | (5.27) | (0.34) | ||
| 1 | (2.70) | (<0.01)1 | (−0.115) | (0.227) | (2.24) | (0.22) | |
| (2.84) | (0.011)1 | (−0.159) | (0.215) | (4.24) | (0.32) | ||
| (3.18) | (<0.01)1 | (−0.128) | (0.246) | (2.13) | (0.21) | ||
| 2 | (2.87) | (0.010)1 | (−0.152) | (0.214) | (4.07) | (0.29) | |
| (3.37) | (<0.01)1 | (−0.115) | (0.251) | (1.84) | (0.18) | ||
| (3.49) | (<0.01)1 | (−0.087) | (0.233) | (1.71) | (0.11) | ||
| System | Vacancies | Na-Na (Å) | QTAIM properties (a.u) | ELF | |||
|---|---|---|---|---|---|---|---|
| Na3/Graphene | 0 | 3.35 | 0.091 | −0.103 | 0.186 | 3.02 | 0.31 |
| 3.37 | 0.091 | −0.096 | 0.197 | 2.50 | 0.28 | ||
| (3.23) | (0.10)1 | (−0.109) | (0.205) | (2.60) | (0.30) | ||
| (3.24) | (0.10)1 | (−0.108) | (0.207) | (2.51) | (0.29) | ||
| 1 | (3.82)3 | (0.04)2 | (−0.124) | (0.054) | (−3.59) | (0.65) | |
| 2 | (3.35) | (0.08)1 | (−0.099) | (0.190) | (2.75) | (0.28) | |
| (3.38) | (0.08)1 | (−0.093) | (0.198) | (2.38) | (0.26) | ||
| Na3/GO | 0 | 3.34 | 0.091 | −0.097 | 0.184 | 2.84 | 0.30 |
| 3.43 | 0.081 | −0.088 | 0.195 | 2.33 | 0.26 | ||
| (3.19) | 0.052 | (−0.151) | (0.137) | (−17.51) | (0.28) | ||
| Na5/Graphene | 0 | 3.66 | 0.062 | −0.114 | 0.061 | -4.71 | 0.71 |
| 4.10 | 0.061 | −0.104 | 0.114 | 26.06 | 0.43 | ||
| (3.54) | 0.072 | (−0.121) | (0.029) | (−2.71) | (0.93) | ||
| (4.05) | (0.07)1 | (−0.110) | (0.141) | (8.04) | (0.34) | ||
| 1 | (3.55) | (0.06)1 | (−0.113) | (0.115) | (201.62) | (0.41) | |
| (3.63) | (0.06)1 | (−0.089) | (0.151) | (4.017) | (0.28) | ||
| 2 | (3.34) | (0.07)1 | (−0.084) | (0.220) | (1.84) | (0.20) | |
| (3.41) | (0.07)1 | (−0.090) | (0.182) | (2.70) | (0.26) | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dimakis, N.; Salas, I.; Gonzalez, L.; Vadodaria, O.; Ruiz, K.; Bhatti, M.I. Li and Na Adsorption on Graphene and Graphene Oxide Examined by Density Functional Theory, Quantum Theory of Atoms in Molecules, and Electron Localization Function. Molecules 2019, 24, 754. https://doi.org/10.3390/molecules24040754
Dimakis N, Salas I, Gonzalez L, Vadodaria O, Ruiz K, Bhatti MI. Li and Na Adsorption on Graphene and Graphene Oxide Examined by Density Functional Theory, Quantum Theory of Atoms in Molecules, and Electron Localization Function. Molecules. 2019; 24(4):754. https://doi.org/10.3390/molecules24040754
Chicago/Turabian StyleDimakis, Nicholas, Isaiah Salas, Luis Gonzalez, Om Vadodaria, Korinna Ruiz, and Muhammad I. Bhatti. 2019. "Li and Na Adsorption on Graphene and Graphene Oxide Examined by Density Functional Theory, Quantum Theory of Atoms in Molecules, and Electron Localization Function" Molecules 24, no. 4: 754. https://doi.org/10.3390/molecules24040754
APA StyleDimakis, N., Salas, I., Gonzalez, L., Vadodaria, O., Ruiz, K., & Bhatti, M. I. (2019). Li and Na Adsorption on Graphene and Graphene Oxide Examined by Density Functional Theory, Quantum Theory of Atoms in Molecules, and Electron Localization Function. Molecules, 24(4), 754. https://doi.org/10.3390/molecules24040754