
Design, Synthesis, Docking Studies and Monoamine Oxidase Inhibition of a Small Library of 1-acetyl- and 1-thiocarbamoyl-3,5-diphenyl-4,5-dihydro-(1H)-pyrazoles


 ,
, 


Abstract
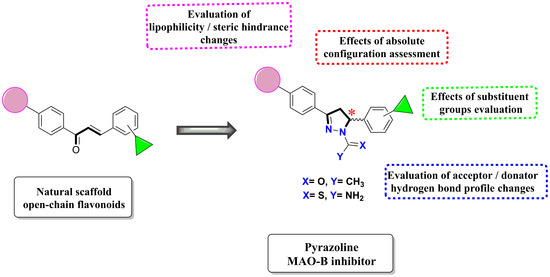
1. Introduction
2. Results and Discussion
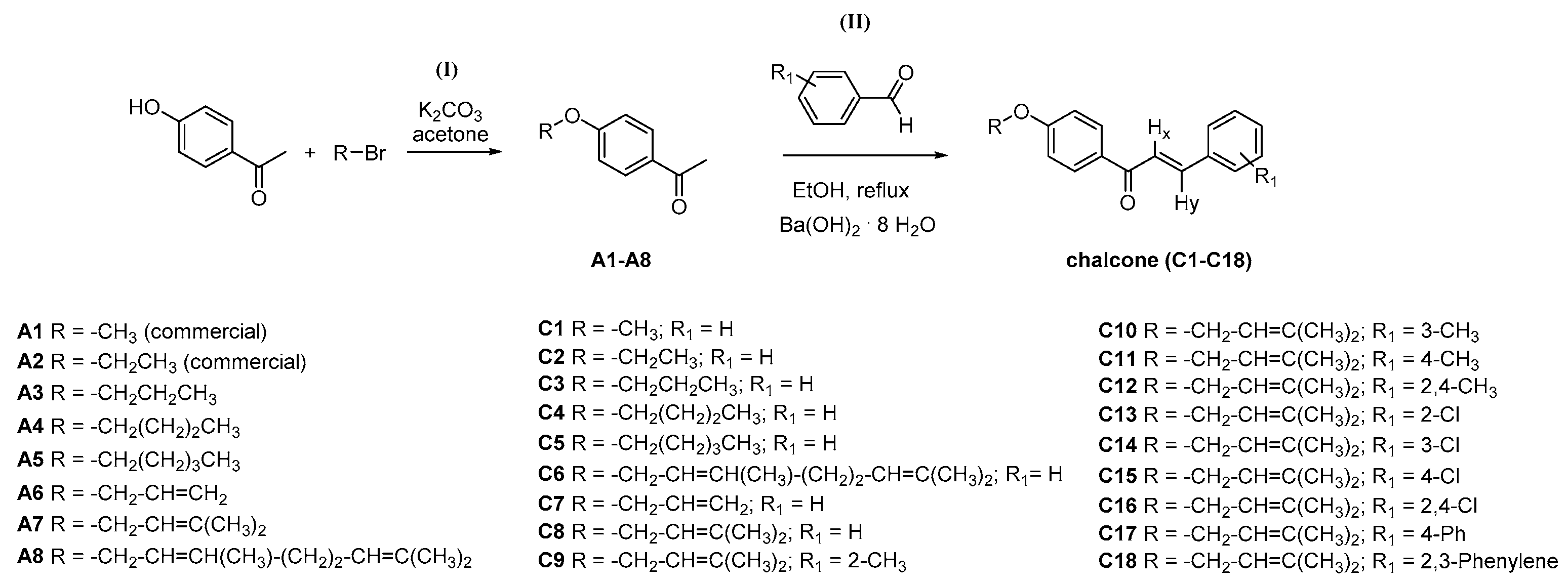
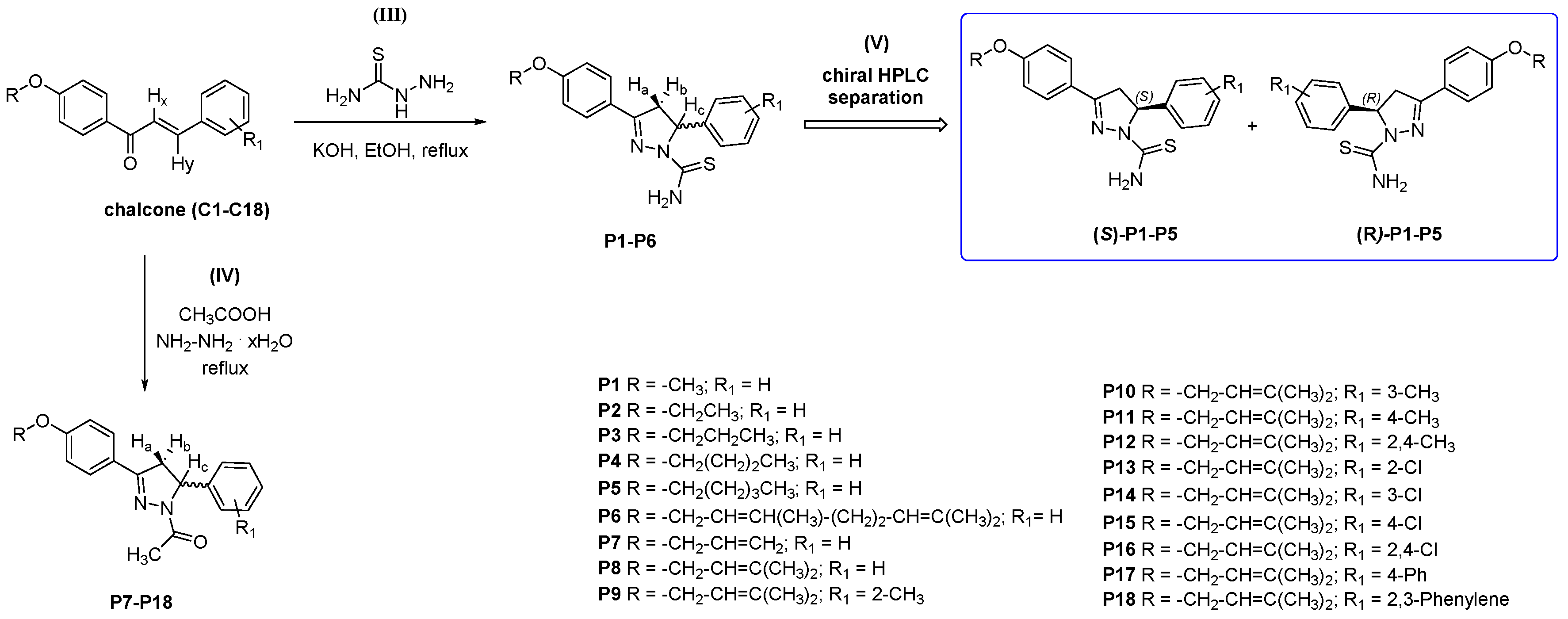
2.1. Chemistry and HPLC Enantioseparation
2.2. hMAO-A and hMAO-B Inhibition Studies
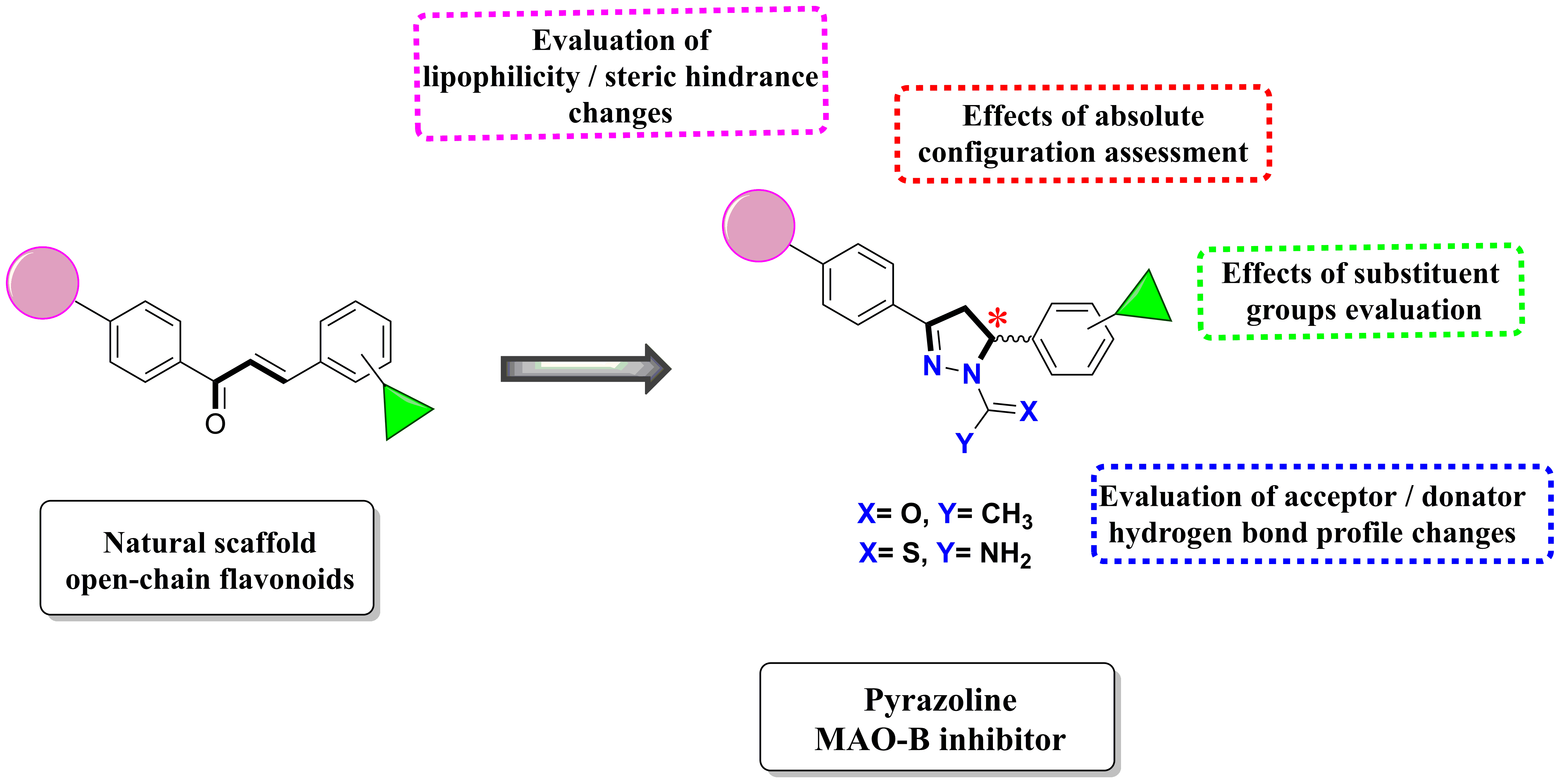
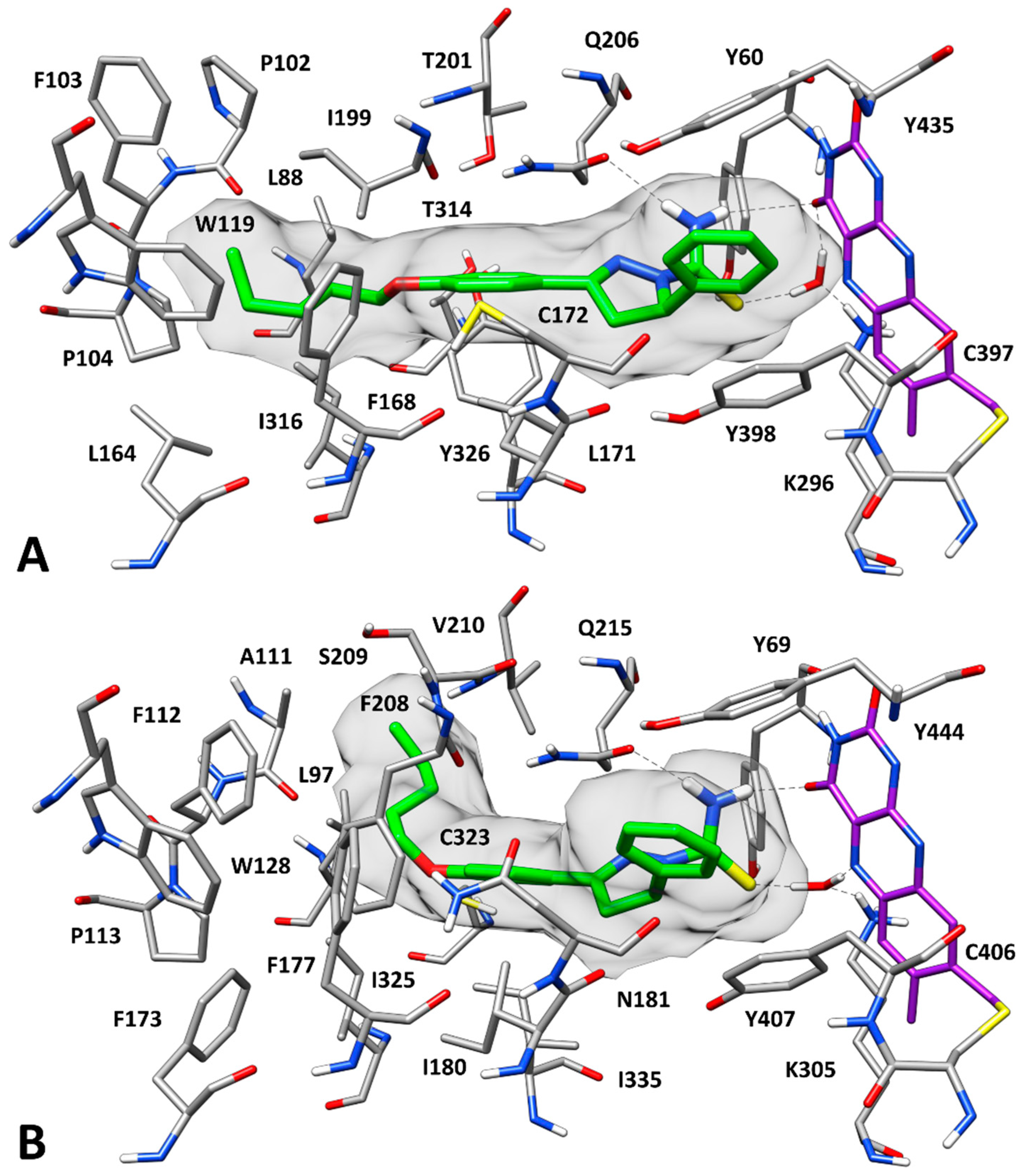
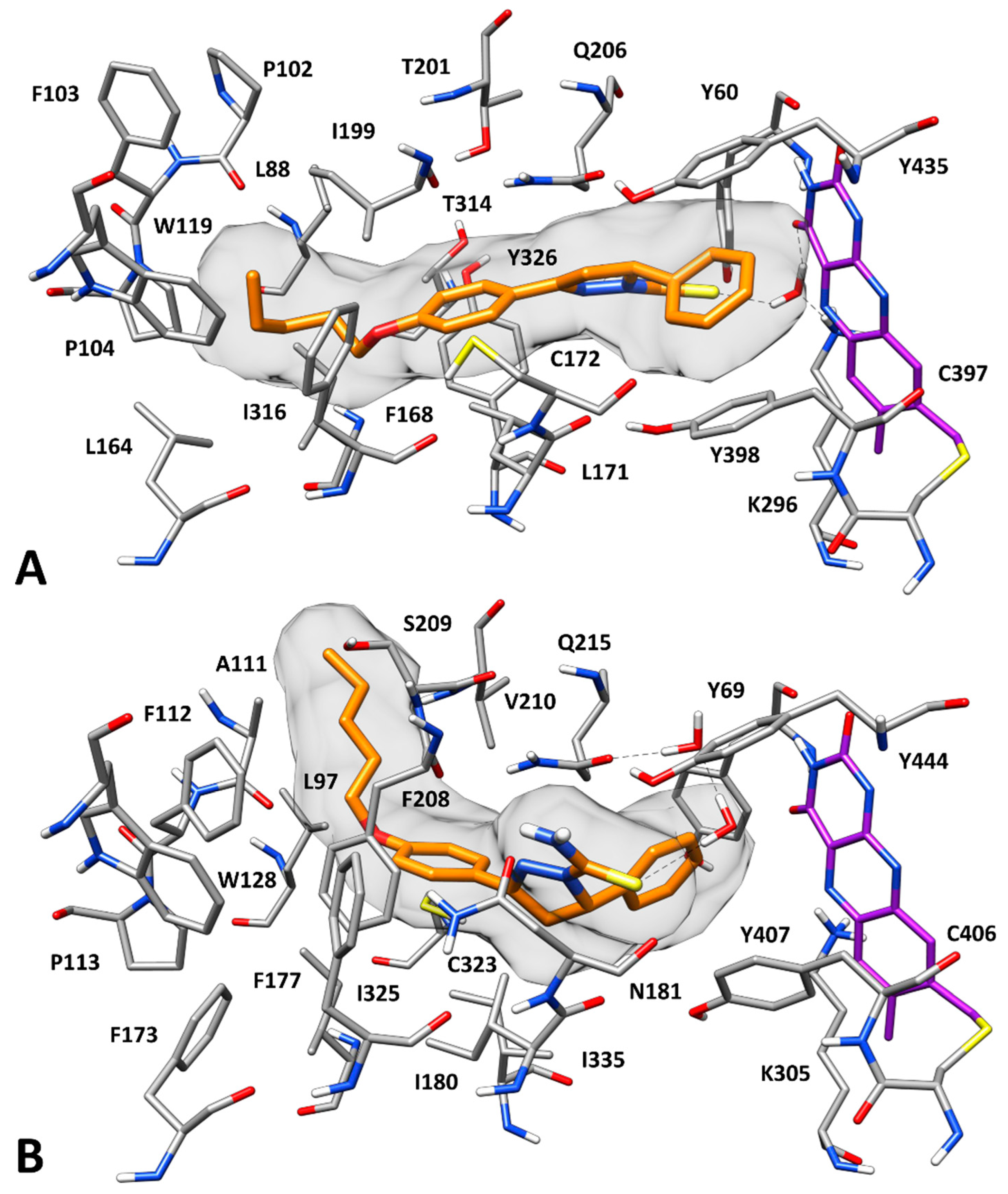
2.3. Docking Studies
3. Materials and Methods
3.1. General
3.2. General Synthesis for Acetophenone Derivatives A3–A8
3.2.1. General Synthesis and Characterization Data for Chalcones C1–C18
3.2.2. Synthesis and Characterization Data for 3,5-disubstituted-N-thiocarbamoylated Pyrazolines P1–P6
3.2.3. Synthesis and Characterization Data for 3,5-disubstituted-N-acetylated Pyrazolines P7–P18
3.3. hMAO-A and hMAO-B Inhibition Studies
3.4. Enantioselective HPLC
3.5. Molecular Modeling Studies
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Binde, C.D.; Tvete, I.F.; Gåsemyr, J.; Natvig, B.; Klemp, M. A multiple treatment comparison meta-analysis of monoamine oxidase type B inhibitors for Parkinson’s disease. Br. J. Clin. Pharmacol. 2018, 84, 1917–1927. [Google Scholar] [CrossRef]
- Shih, J.C. Monoamine oxidase isoenzymes: Genes, functions and targets for behavior and cancer therapy. J. Neural Transm. 2018, 125, 1553–1566. [Google Scholar] [CrossRef] [PubMed]
- Iacovino, L.G.; Magnani, F.; Binda, C. The structure of monoamine oxidases: Past, present, and future. J. Neural Transm. 2018, 125, 1567–1579. [Google Scholar] [CrossRef] [PubMed]
- Carradori, S.; Secci, D.; Bolasco, A.; Chimenti, P.; D’Ascenzio, M. Patent-related survey on new monoamine oxidase inhibitors and their therapeutic potential. Expert Opin. Ther. Pat. 2012, 22, 759–801. [Google Scholar] [CrossRef] [PubMed]
- Carradori, S.; Petzer, J.P. Novel monoamine oxidase inhibitors: A patent review (2012–2014). Expert Opin. Ther. Pat. 2014, 25, 91–110. [Google Scholar] [CrossRef] [PubMed]
- Carradori, S.; Secci, D.; Petzer, J.P. MAO inhibitors and their wider applications: A patent review. Expert Opin. Ther. Pat. 2018, 28, 211–226. [Google Scholar] [CrossRef]
- Bolasco, A.; Carradori, S.; Fioravanti, R. Focusing on new monoamine oxidase inhibitors. Expert Opin. Ther. Pat. 2010, 20, 909–939. [Google Scholar] [CrossRef]
- Bolasco, A.; Fioravanti, R.; Carradori, S. Recent development of monoamine oxidase inhibitors. Expert Opin. Ther. Pat. 2005, 15, 1763–1782. [Google Scholar] [CrossRef]
- Mathew, B.; Suresh, J.; Anbazhagan, S.; Mathew, G. Pyrazoline: A promising scaffold for the inhibition of monoamine oxidase. Cent. Nerv. Syst. Agents Med. Chem. 2014, 13, 195–206. [Google Scholar] [CrossRef]
- Chimenti, F.; Bolasco, A.; Manna, F.; Secci, D.; Chimenti, P.; Befani, O.; Turini, P.; Giovannini, V.; Mondovì, B.; Cirilli, R.; La Torre, F. Synthesis and selective inhibitory activity of 1-acetyl-3,5-diphenyl-4,5-dihydro-(1H)-pyrazole derivatives against monoamine oxidase. J. Med. Chem. 2004, 47, 2071–2074. [Google Scholar] [CrossRef]
- Chimenti, F.; Secci, D.; Bolasco, A.; Chimenti, P.; Granese, A.; Carradori, S.; Befani, O.; Turini, P.; Alcaro, S.; Ortuso, F. Synthesis, molecular modeling studies, and selective inhibitory activity against monoamine oxidase of N,N′-bis[2-oxo-2H-benzopyran]-3-carboxamides. Bioorganic Med. Chem. Lett. 2006, 16, 4135–4140. [Google Scholar] [CrossRef] [PubMed]
- Chimenti, F.; Bolasco, A.; Manna, F.; Secci, D.; Chimenti, P.; Granese, A.; Befani, O.; Turini, P.; Alcaro, S.; Ortuso, F. Synthesis and molecular modelling of novel substituted-4,5-dihydro-(1H)-pyrazole derivatives as potent and highly selective monoamine oxidase-A inhibitors. Chem. Biol. Drug Des. 2006, 67, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Chimenti, F.; Fioravanti, R.; Bolasco, A.; Manna, F.; Chimenti, P.; Secci, D.; Befani, O.; Turini, P.; Ortuso, F.; Alcaro, S. Monoamine oxidase isoform-dependent tautomeric influence in the recognition of 3,5-diaryl pyrazole inhibitors. J. Med. Chem. 2007, 50, 425–428. [Google Scholar] [CrossRef] [PubMed]
- Secci, D.; Carradori, S.; Bolasco, A.; Bizzarri, B.; D’Ascenzio, M.; Maccioni, E. Discovery and optimization of pyrazoline derivatives as promising monoamine oxidase inhibitors. Curr. Top. Med. Chem. 2012, 12, 2240–2257. [Google Scholar] [CrossRef] [PubMed]
- Maccioni, E.; Alcaro, S.; Orallo, F.; Cardia, M.C.; Distinto, S.; Costa, G.; Yáñez, M.; Sanna, M.L.; Vigo, S.; Meleddu, R. Synthesis of new 3-aryl-4,5-dihydropyrazole-1-carbothioamide derivatives. An investigation on their ability to inhibit monoamine oxidase. Eur. J. Med. Chem. 2010, 45, 4490–4498. [Google Scholar] [CrossRef]
- Brezani, V.; Smejkal, K.; Hosek, J.; Tomasova, V. Anti-inflammatory natural prenylated phenolic compounds—Potential lead substances. Curr. Med. Chem. 2018, 25, 1094–1159. [Google Scholar] [CrossRef]
- Chi, Y.S.; Jong, H.G.; Son, K.H.; Chang, H.W.; Kang, S.S.; Kim, H.P. Effects of naturally occurring prenylated flavonoids on enzymes metabolizing arachidonic acid: Cyclooxygenases and lipoxygenases. Biochem. Pharmacol. 2001, 62, 1185–1191. [Google Scholar] [CrossRef]
- Goksen, U.S.; Sarigul, S.; Bultinck, P.; Herrebout, W.; Dogan, I.; Yelekci, K.; Ucar, G.; Gokhan Kelekci, N. Absolute configuration and biological profile of pyrazoline enantiomers as MAO inhibitory activity. Chirality 2019, 31, 21–33. [Google Scholar] [CrossRef]
- Kitawat, B.S.; Singh, M. Synthesis, characterization, antibacterial, antioxidant, DNA binding and SAR study of a novel pyrazine moiety bearing 2-pyrazoline derivatives. New J. Chem. 2014, 38, 4290–4299. [Google Scholar] [CrossRef]
- Carradori, S.; Secci, D.; Bolasco, A.; De Monte, C.; Yáñez, M. Synthesis and selective inhibitory activity against human COX-1 of novel 1-(4-substituted-thiazol-2-yl)-3,5-di(hetero)aryl-pyrazoline derivatives. Arch. Pharm. 2012, 345, 973–979. [Google Scholar] [CrossRef]
- Jagrat, M.; Behera, J.; Yabanoglu, S.; Ercan, A.; Ucar, G.; Sinha, B.N.; Sankaran, V.; Basu, A.; Jayaprakash, V. Pyrazoline based MAO inhibitors: Synthesis, biological evaluation and SAR studies. Bioorg. Med. Chem. Lett. 2011, 21, 4296–4300. [Google Scholar] [CrossRef]
- Jayaprakash, V.; Sinha, B.N.; Ucar, G.; Ercan, A. Pyrazoline-based mycobactin analogues as MAO-inhibitors. Bioorg. Med. Chem. Lett. 2008, 18, 6362–6368. [Google Scholar] [CrossRef]
- Narender, T.; Venkateswarlu, K.; Madhur, G.; Reddy, K.P. Highly efficient and selective deprotection method for prenyl, geranyl, and phytyl ethers and esters using borontrifluoride-etherate. Synth. Commun. 2013, 43, 26–33. [Google Scholar] [CrossRef]
- Aguilera, A.; Alcantara, A.R.; Marinas, J.M.; Sinisterra, J.V. Ba(OH)2 as the catalyst in organic reactions. Part XIV. Mechanism of Claisen–Schmidt condensation in solid–liquid conditions. Can. J. Chem. 1987, 65, 1165–1171. [Google Scholar] [CrossRef]
- Gökhan, N.; Yeşilada, A.; Uçar, G.; Erol, K.; Bilgin, A.A. 1-N-substituted thiocarbamoyl-3-phenyl-5-thienyl-2-pyrazolines: Synthesis and evaluation as MAO inhibitors. Arch. Pharm. 2003, 336, 362–371. [Google Scholar] [CrossRef]
- Pierini, M.; Carradori, S.; Menta, S.; Secci, D.; Cirilli, R. 3-(Phenyl-4-oxy)-5-phenyl-4,5-dihydro-(1H)-pyrazole: A fascinating molecular framework to study the enantioseparation ability of the amylose (3,5-dimethylphenylcarbamate) chiral stationary phase. Part II. Solvophobic effects in enantiorecognition process. J. Chromatogr. A 2017, 1499, 140–148. [Google Scholar] [CrossRef]
- Carradori, S.; Pierini, M.; Menta, S.; Secci, D.; Fioravanti, R.; Cirilli, R. 3-(Phenyl-4-oxy)-5-phenyl-4,5-dihydro-(1H)-pyrazole: A fascinating molecular framework to study the enantioseparation ability of the amylose (3,5-dimethylphenylcarbamate) chiral stationary phase. Part I. Structure-enantioselectivity relationships. J. Chromatogr. A 2016, 1467, 221–227. [Google Scholar] [CrossRef]
- Zhou, M.; Panchuk-Voloshina, N. A one-step fluorometric method for the continuous measurement of monoamine oxidase activity. Anal. Biochem. 1997, 253, 169–174. [Google Scholar] [CrossRef]
- Petzer, A.; Pienaar, A.; Petzer, J.P. The inhibition of monoamine oxidase by esomeprazole. Drug Res. 2013, 63, 462–467. [Google Scholar] [CrossRef]
- Nagesam, M.; Raju, K.M.; Raju, S. Synthesis of 4-hydroxy-, 4′-hydroxy- and 2-hydroxychalcone ethers. Acta Cienc. Indica Chem. 1984, 10, 165–169. [Google Scholar]
- Okaecwe, T.; Swanepoel, A.J.; Petzer, A.; Bergh, J.J.; Petzer, J.P. Inhibition of monoamine oxidase by 8-phenoxymethylcaffeine derivatives. Bioorg. Med. Chem. 2012, 20, 4336–4347. [Google Scholar] [CrossRef]
- Berman, H.M.; Battistuz, T.; Bhat, T.N.; Bluhm, W.F.; Bourne, P.E.; Burkhardt, K.; Feng, Z.; Gilliland, G.L.; Iype, L.; Jain, S.; et al. The protein data bank. Acta Crystallogr. Sect. D Biol. Crystallogr. 2002, 58, 899–907. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Poli, G.; Gelain, A.; Porta, F.; Asai, A.; Martinelli, A.; Tuccinardi, T. Identification of a new STAT3 dimerization inhibitor through a pharmacophore-based virtual screening approach. J. Enzyme Inhib. Med. Chem. 2016, 31, 1011–1017. [Google Scholar] [CrossRef]
- De Leo, M.; Huallpa, C.G.; Alvarado, B.; Granchi, C.; Poli, G.; De Tommasi, N.; Braca, A. New diterpenes from Salvia pseudorosmarinus and their activity as inhibitors of monoacylglycerol lipase (MAGL). Fitoterapia 2018, 130, 251–258. [Google Scholar] [CrossRef]
- Aghazadeh, T.M.; Baraldi, P.G.; Ruggiero, E.; Saponaro, G.; Baraldi, S.; Poli, G.; Tuccinardi, T.; Ravani, A.; Vincenzi, F.; Borea, P.A.; et al. Synthesis and structure activity relationship investigation of triazolo[1,5-a]pyrimidines as CB2 cannabinoid receptor inverse agonists. Eur. J. Med. Chem. 2016, 113, 11–27. [Google Scholar] [CrossRef]
- Case, D.A.; Berryman, J.T.; Betz, R.M.; Cerutti, D.S.; Darden, T.A.; Duke, R.E.; Giese, T.J.; Gohlke, H.; Goetz, A.W.; Homeyer, N.; et al. AMBER, Version 16; University of California: San Francisco, CA, USA, 2016. [Google Scholar]
Sample Availability: Samples of the compounds are available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Structure | IC50 (µM ± SD) a | SI b | |
|---|---|---|---|---|
| hMAO-A | hMAO-B | |||
| (S)-P1 |  | 46.6 ± 2.62 | >100 | <0.47 |
| (R)-P1 |  | 50.6 ± 2.67 | >100 | <0.51 |
| (S)-P2 |  | 32.3 ± 4.46 | >100 | <0.32 |
| (R)-P2 |  | 33.9 ± 1.29 | >100 | <0.34 |
| (S)-P3 |  | 9.59 ± 0.83 | 42.5 ± 2.61 | 0.23 |
| (R)-P3 |  | 8.00 ± 0.83 | 2.77 ± 0.39 | 2.89 |
| (S)-P4 |  | 39.2 ± 2.15 | 5.03 ± 0.88 | 7.78 |
| (R)-P4 |  | 3.63 ± 0.50 | 0.38 ± 0.06 | 9.55 |
| (S)-P5 |  | 55.2 ± 2.72 | 2.79 ± 0.87 | 19.77 |
| (R)-P5 |  | 3.03 ± 0.60 | 0.44 ± 0.028 | 6.89 |
| P6 |  | >100 | 54.1 ± 7.54 | >1.85 |
| P7 |  | 39.3 ± 5.94 | 14.5 ± 2.78 | 2.72 |
| P8 |  | >100 | 2.29 ± 0.42 | >43.67 |
| P9 |  | 9.13 ± 0.34 | 27.3 ± 0.57 | 0.33 |
| P10 |  | 86.8 ± 14.7 | 3.22 ± 0.48 | 26.96 |
| P11 |  | >100 | 12.2 ± 0.71 | >8.19 |
| P12 |  | 80.6 ± 10.0 | >100 | <0.81 |
| P13 |  | 5.74 ± 1.66 | 3.11 ± 0.66 | 1.85 |
| P14 |  | 4.13 ± 0.26 | 1.08 ± 0.05 | 3.82 |
| P15 |  | 12.1 ± 0.14 | 11.0 ± 3.56 | 1.11 |
| P16 |  | >100 | >100 | \ |
| P17 |  | >100 | >100 | \ |
| P18 |  | >100 | >100 | \ |
| Toloxatone | 3.92 ± 0.15 c | - | - | |
| Lazabemide | - | 0.091 ± 0.15 c | - | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guglielmi, P.; Carradori, S.; Poli, G.; Secci, D.; Cirilli, R.; Rotondi, G.; Chimenti, P.; Petzer, A.; Petzer, J.P. Design, Synthesis, Docking Studies and Monoamine Oxidase Inhibition of a Small Library of 1-acetyl- and 1-thiocarbamoyl-3,5-diphenyl-4,5-dihydro-(1H)-pyrazoles. Molecules 2019, 24, 484. https://doi.org/10.3390/molecules24030484
Guglielmi P, Carradori S, Poli G, Secci D, Cirilli R, Rotondi G, Chimenti P, Petzer A, Petzer JP. Design, Synthesis, Docking Studies and Monoamine Oxidase Inhibition of a Small Library of 1-acetyl- and 1-thiocarbamoyl-3,5-diphenyl-4,5-dihydro-(1H)-pyrazoles. Molecules. 2019; 24(3):484. https://doi.org/10.3390/molecules24030484
Chicago/Turabian StyleGuglielmi, Paolo, Simone Carradori, Giulio Poli, Daniela Secci, Roberto Cirilli, Giulia Rotondi, Paola Chimenti, Anél Petzer, and Jacobus P. Petzer. 2019. "Design, Synthesis, Docking Studies and Monoamine Oxidase Inhibition of a Small Library of 1-acetyl- and 1-thiocarbamoyl-3,5-diphenyl-4,5-dihydro-(1H)-pyrazoles" Molecules 24, no. 3: 484. https://doi.org/10.3390/molecules24030484
APA StyleGuglielmi, P., Carradori, S., Poli, G., Secci, D., Cirilli, R., Rotondi, G., Chimenti, P., Petzer, A., & Petzer, J. P. (2019). Design, Synthesis, Docking Studies and Monoamine Oxidase Inhibition of a Small Library of 1-acetyl- and 1-thiocarbamoyl-3,5-diphenyl-4,5-dihydro-(1H)-pyrazoles. Molecules, 24(3), 484. https://doi.org/10.3390/molecules24030484