Novel Bispecific Aptamer Enhances Immune Cytotoxicity Against MUC1-Positive Tumor Cells by MUC1-CD16 Dual Targeting

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
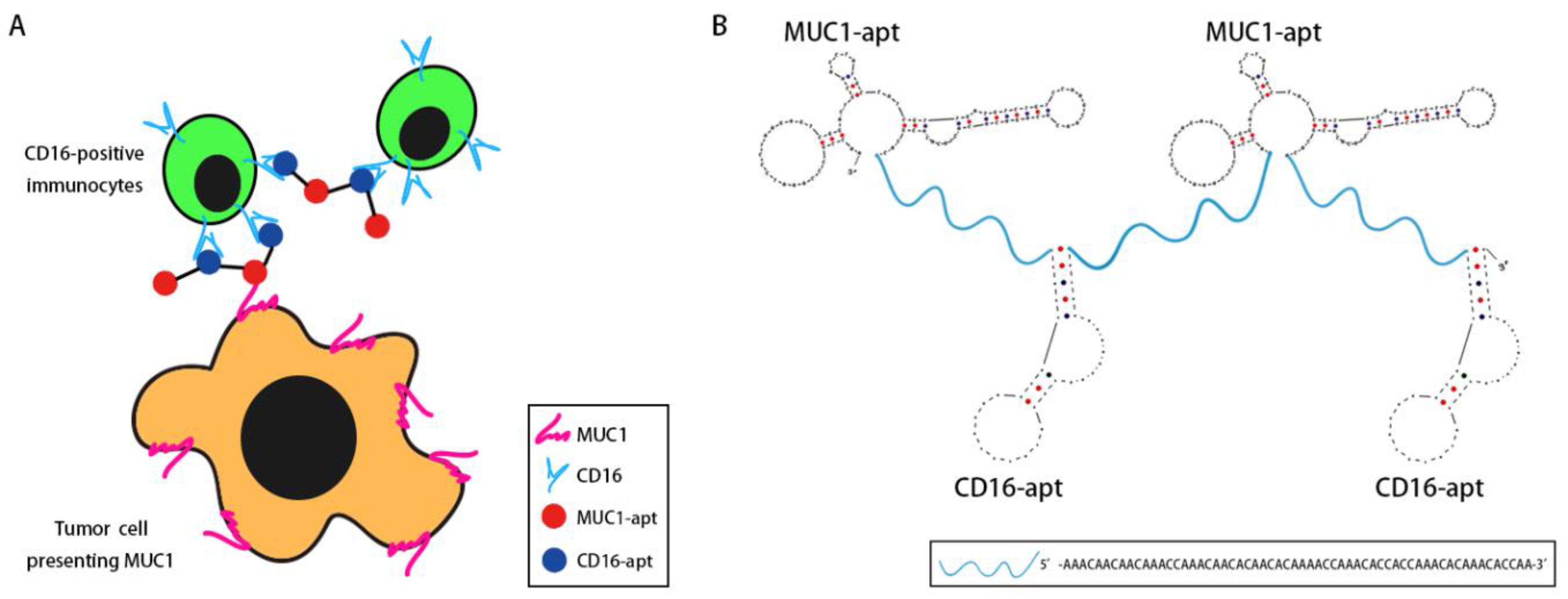
2.1. Design of BBiApt
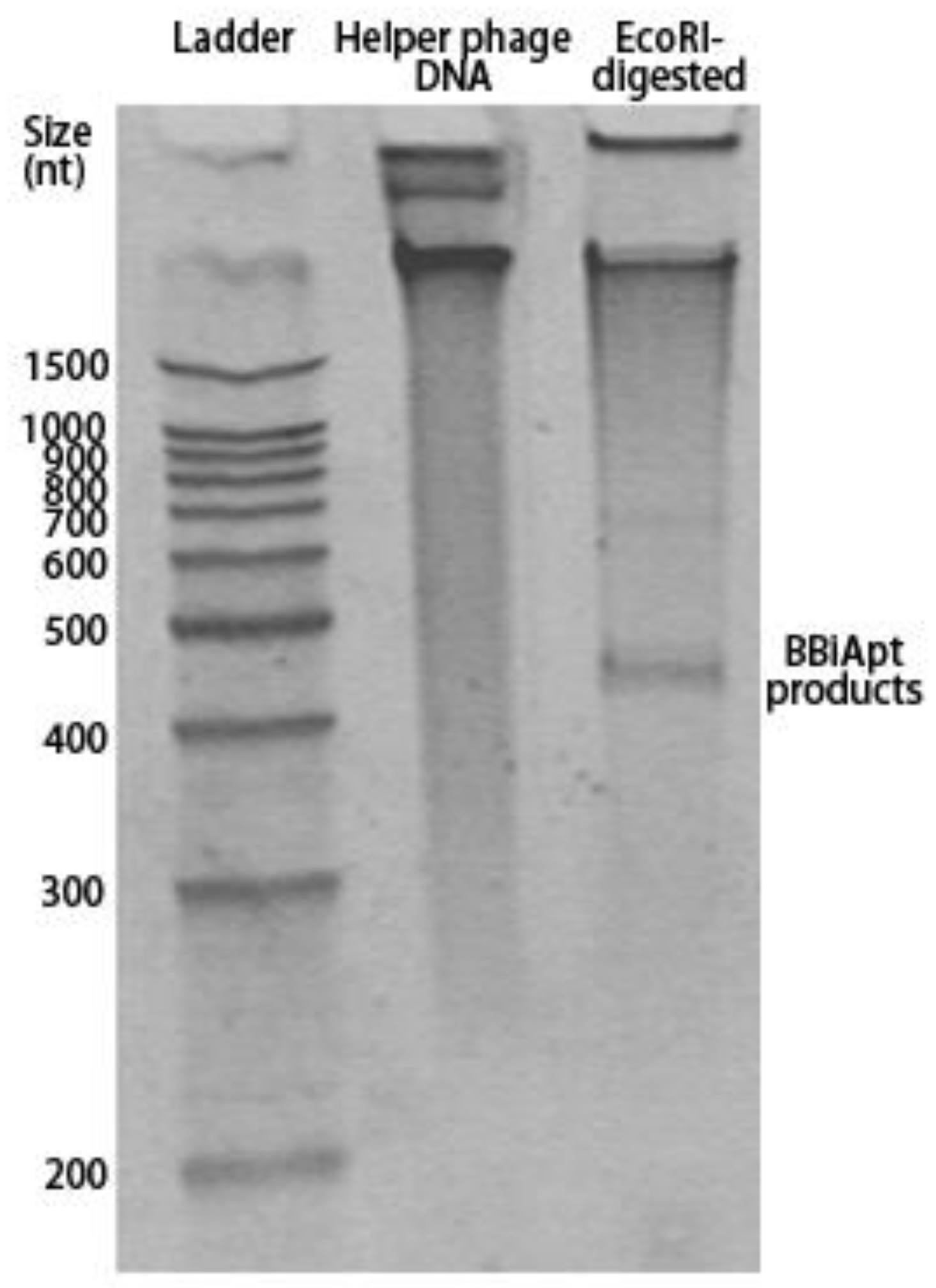
2.2. Production of BBiApt
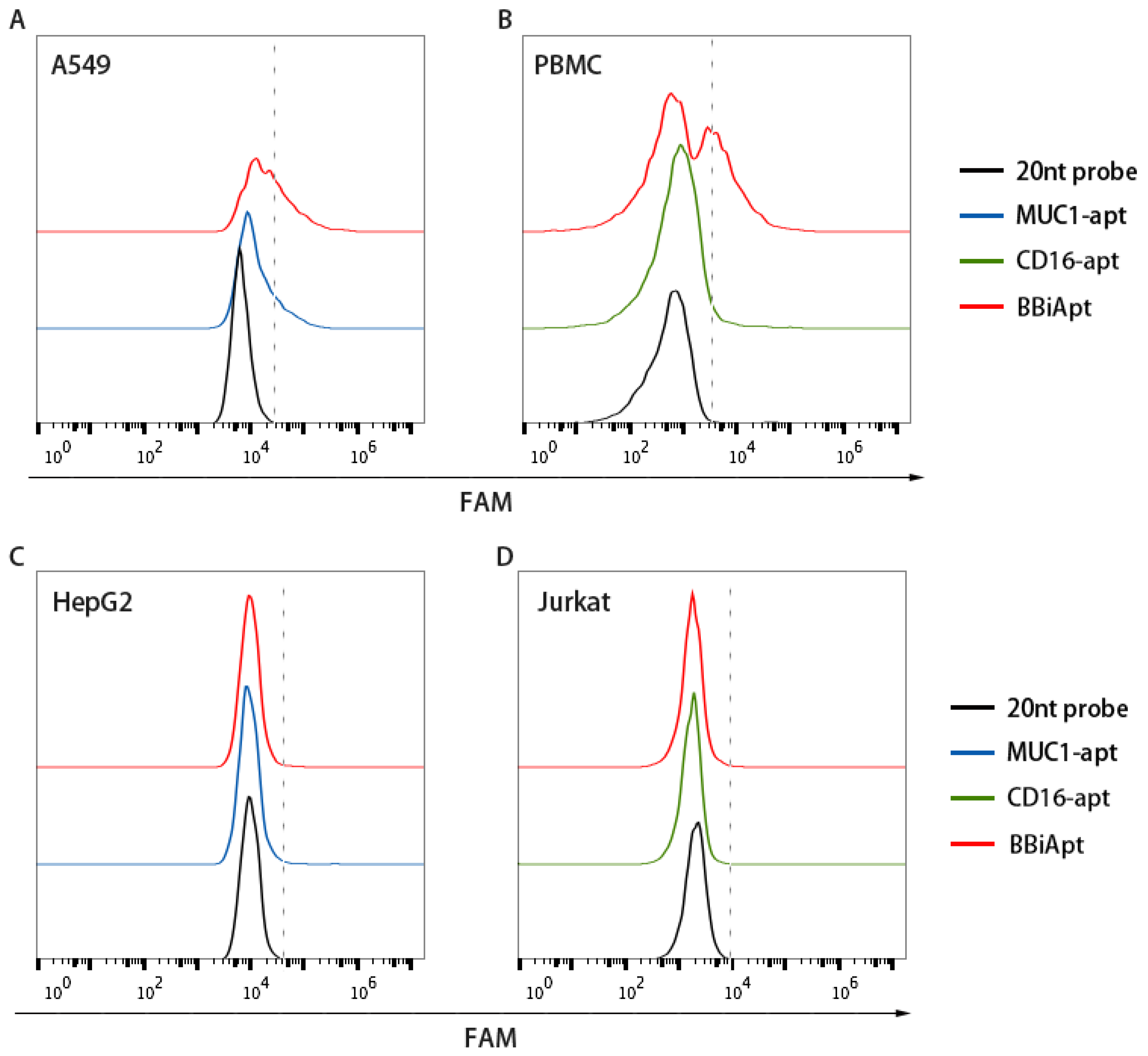
2.3. BBiApt‘s Binding to Target Cells
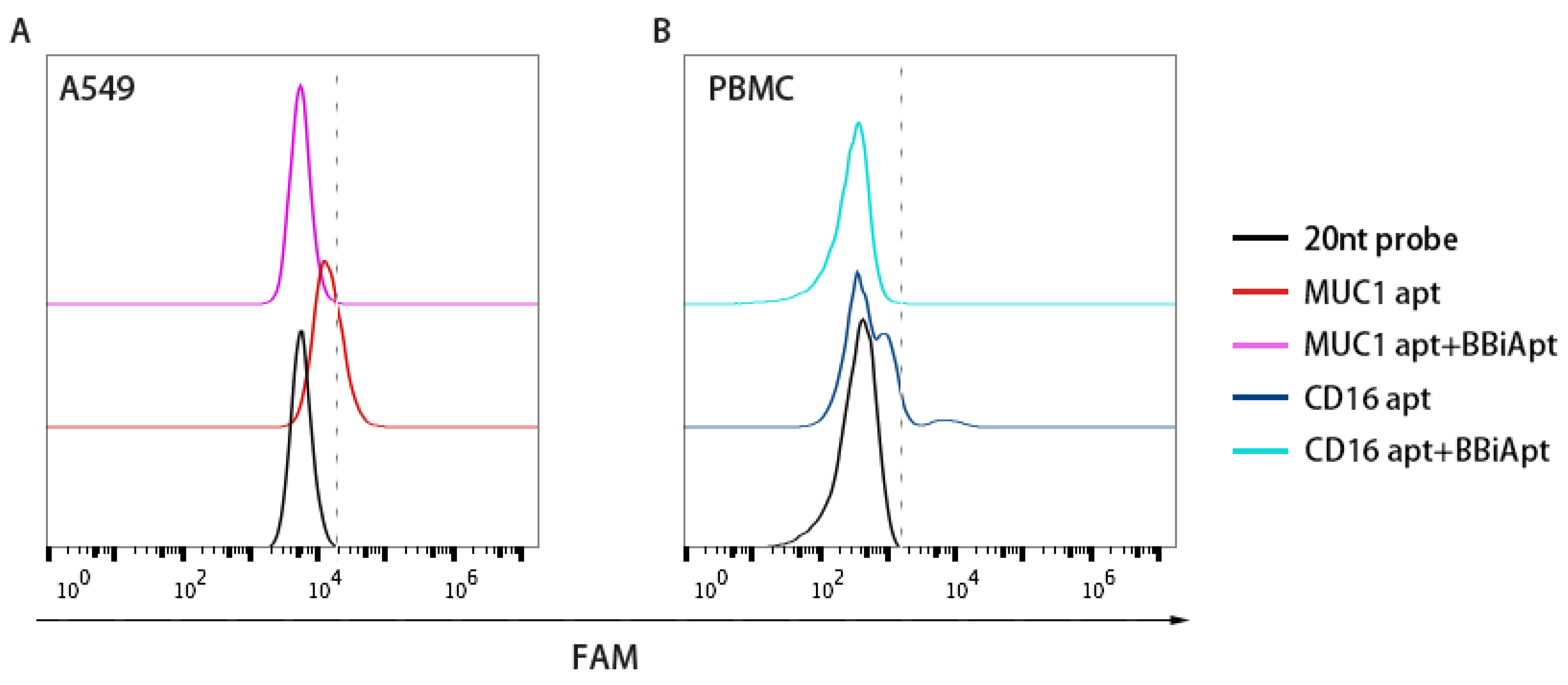
2.4. BBiApt Bound to the Same Targets as the Monovalent Aptamers
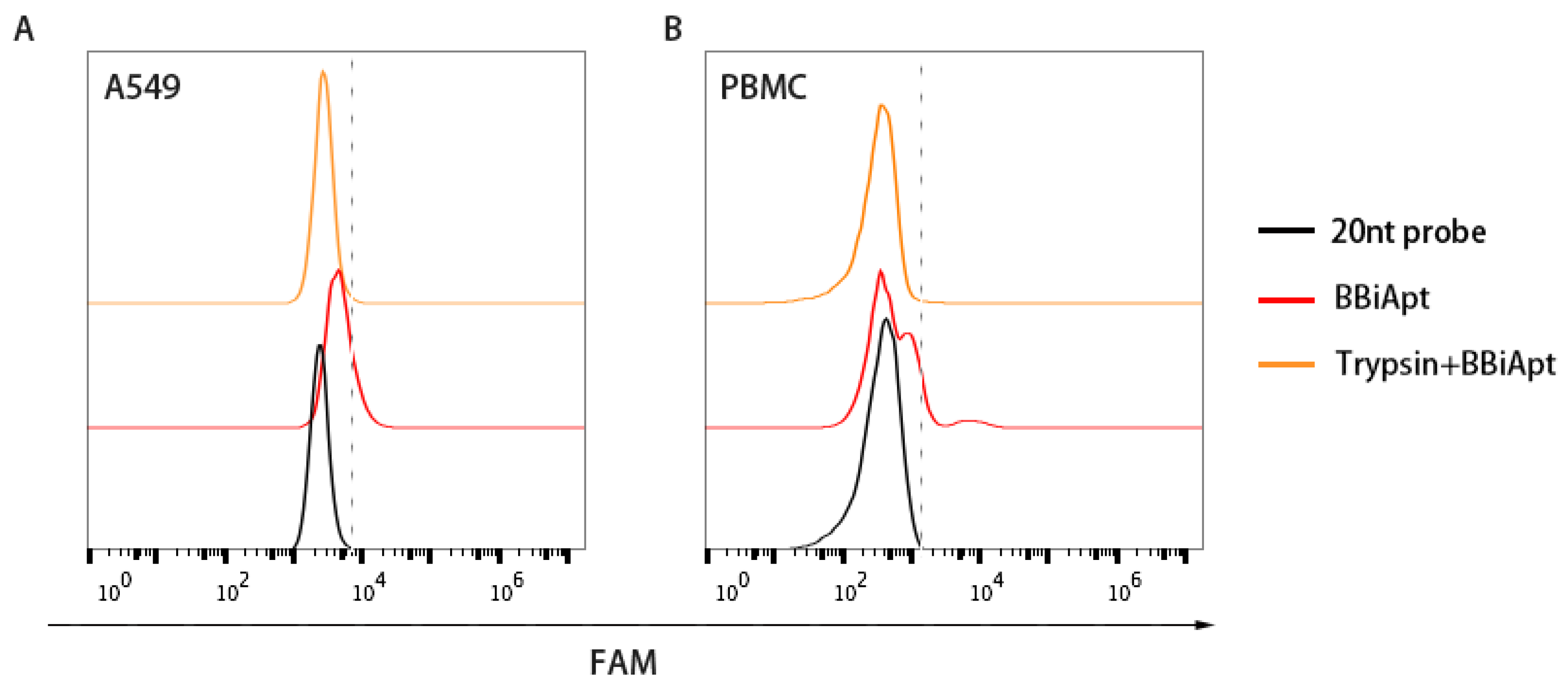
2.5. The Target Molecules of BBiApt were Membrane Proteins
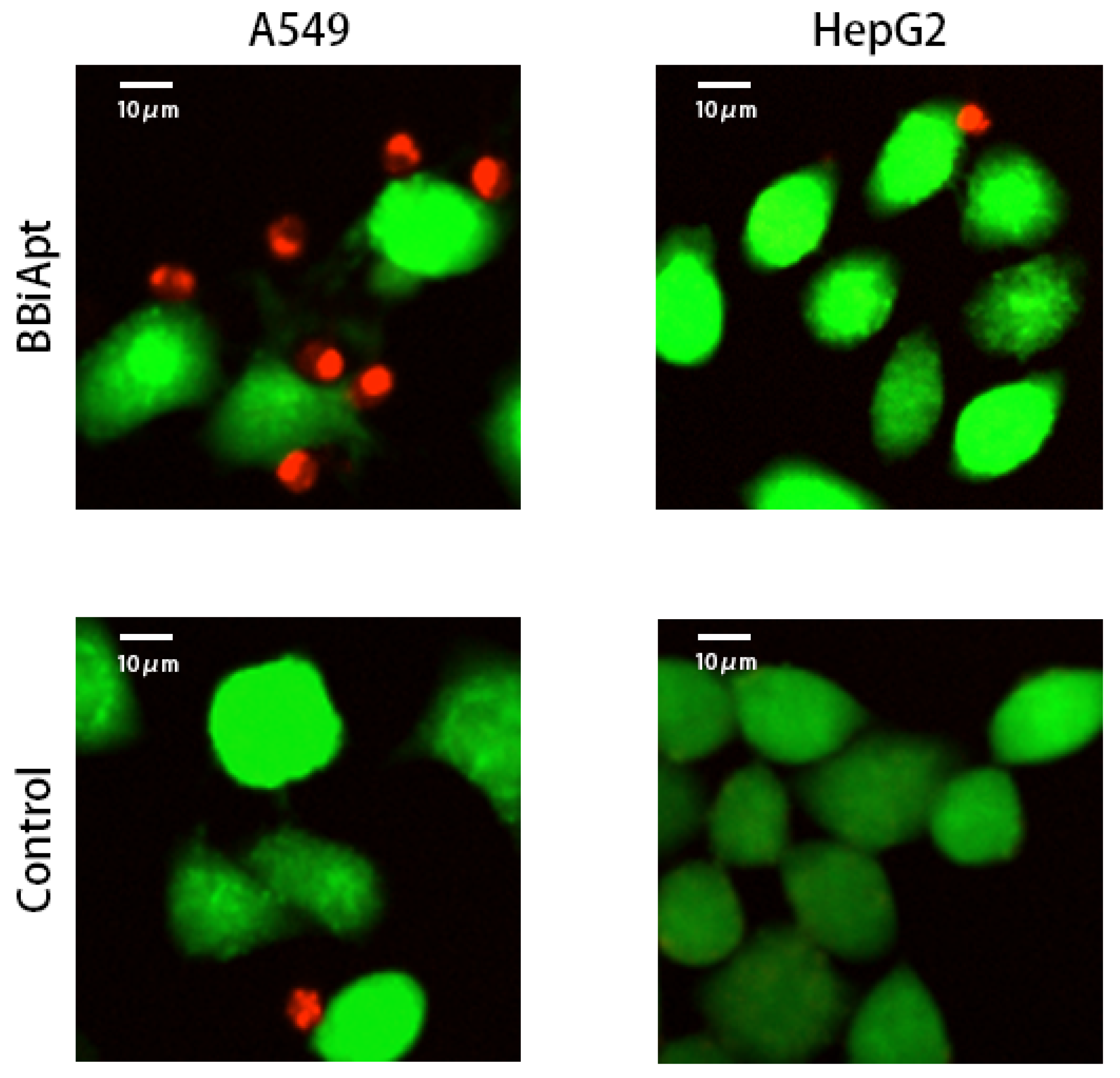
2.6. BBiApt Recruited CD16-positive Immunocytes Around A549 Tumor Cells
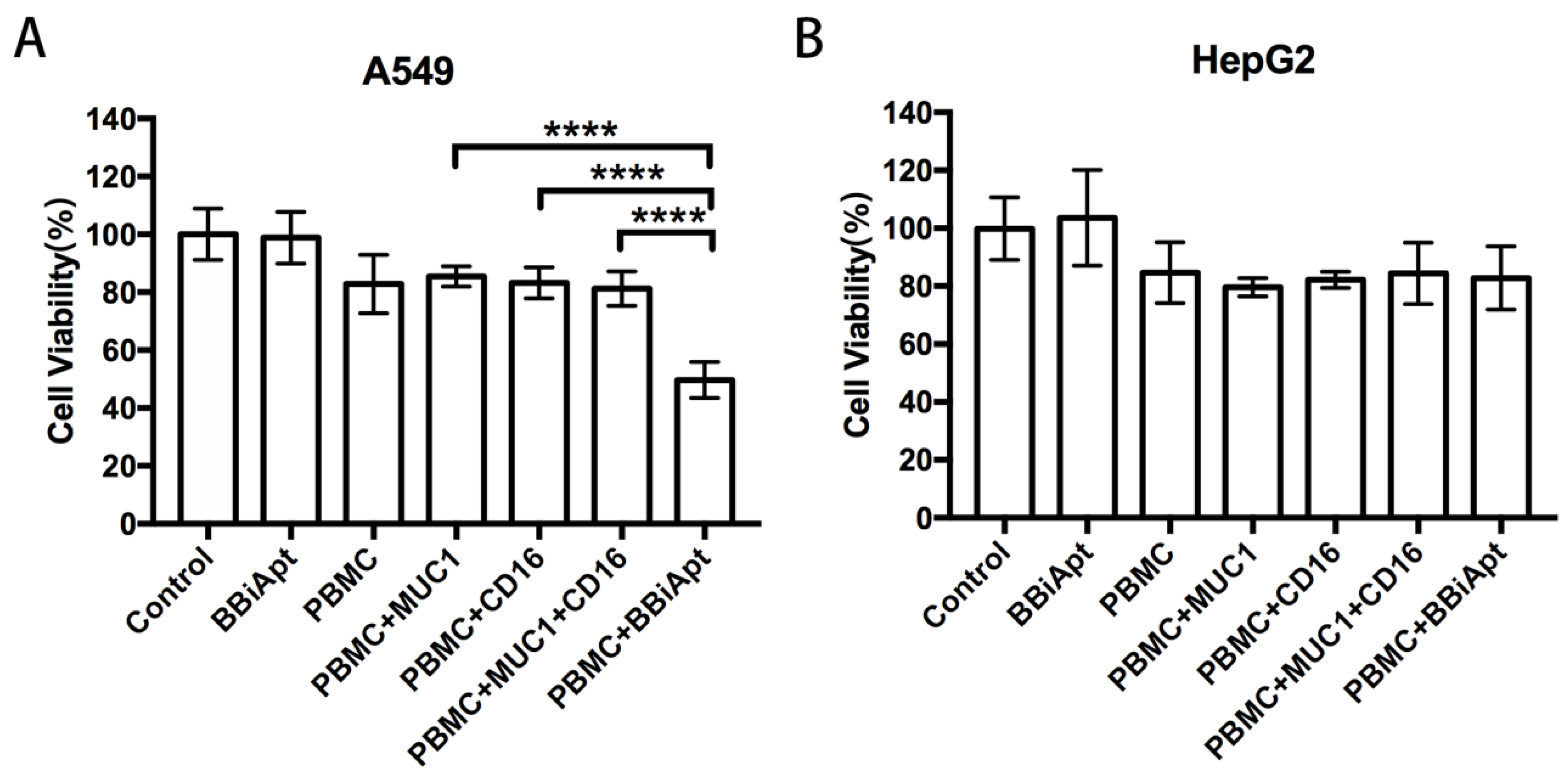
2.7. BBiApt Enhanced Cytotoxicity to MUC1-Positive Tumor Cells by CD16-Positive Immunocytes
3. Discussion
4. Materials and Methods
4.1. Generation of BBiApt
4.2. Cell Cultures
4.3. Cellular Binding Assays
4.4. Binding Competition Studies
4.5. Trypsin Digestion Experiment
4.6. Confocal Imaging Studies
4.7. In Vitro Cytotoxicity Assays
4.8. Statistics
Author Contributions
Funding
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Kassebaum, N.J.; Bertozzi-Villa, A.; Coggeshall, M.S.; Shackelford, K.A.; Steiner, C.; Heuton, K.R.; Gonzalez-Medina, D.; Barber, R.; Huynh, C.; Dicker, D.; et al. Global, regional, and national levels and causes of maternal mortality during 1990–2013: A systematic analysis for the Global Burden of Disease Study 2013. Lancet (Lond. Engl.) 2014, 384, 980–1004. [Google Scholar] [CrossRef]
- Khalil, D.N.; Smith, E.L.; Brentjens, R.J.; Wolchok, J.D. The future of cancer treatment: immunomodulation, CARs and combination immunotherapy. Nat. Rev. Clin. Oncol. 2016, 13, 273–290. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, L.; Forschner, A.; Loquai, C.; Goldinger, S.M.; Zimmer, L.; Ugurel, S.; Schmidgen, M.I.; Gutzmer, R.; Utikal, J.S.; Goppner, D.; et al. Cutaneous, gastrointestinal, hepatic, endocrine, and renal side-effects of anti-PD-1 therapy. Eur. J. Cancer (Oxf. Engl. 1990) 2016, 60, 190–209. [Google Scholar] [CrossRef] [PubMed]
- Fan, G.; Wang, Z.; Hao, M.; Li, J. Bispecific antibodies and their applications. J. Hematol. Oncol. 2015, 8, 130. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.; Stein, A.; Gokbuget, N.; Fielding, A.K.; Schuh, A.C.; Ribera, J.M.; Wei, A.; Dombret, H.; Foa, R.; Bassan, R.; et al. Blinatumomab versus Chemotherapy for Advanced Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2017, 376, 836–847. [Google Scholar] [CrossRef]
- Nath, S.; Mukherjee, P. MUC1: A multifaceted oncoprotein with a key role in cancer progression. Trends Mol. Med. 2014, 20, 332–342. [Google Scholar] [CrossRef] [PubMed]
- Apostolopoulos, V.; Stojanovska, L.; Gargosky, S.E. MUC1 (CD227): A multi-tasked molecule. Cell. Mol. Life Sci. 2015, 72, 4475–4500. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, J.A.; Masri, A.A.; Adriance, M.C.; Tessier, J.C.; Kotlarczyk, K.L.; Thompson, M.C.; Gendler, S.J. MUC1 overexpression results in mammary gland tumorigenesis and prolonged alveolar differentiation. Oncogene 2004, 23, 5739–5747. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Finn, O.J. MUC1 immunotherapy is here to stay. Expert Opin. Biol. Ther. 2013, 13, 35–49. [Google Scholar] [CrossRef] [PubMed]
- Shigdar, S.; Qiao, L.; Zhou, S.F.; Xiang, D.; Wang, T.; Li, Y.; Lim, L.Y.; Kong, L.; Li, L.; Duan, W. RNA aptamers targeting cancer stem cell marker CD133. Cancer Lett. 2013, 330, 84–95. [Google Scholar] [CrossRef]
- Zhou, J.; Rossi, J. Aptamers as targeted therapeutics: current potential and challenges. Nat. Rev. Drug Discov. 2017, 16, 181–202. [Google Scholar] [CrossRef] [PubMed]
- Kaur, H.; Yung, L.Y. Probing high affinity sequences of DNA aptamer against VEGF165. PLoS ONE 2012, 7, e31196. [Google Scholar] [CrossRef] [PubMed]
- Vorobyeva, M.; Vorobjev, P.; Venyaminova, A. Multivalent Aptamers: Versatile Tools for Diagnostic and Therapeutic Applications. Molecules (Basel) 2016, 21, 1613. [Google Scholar] [CrossRef]
- Hu, Y.; Duan, J.; Zhan, Q.; Wang, F.; Lu, X.; Yang, X.D. Novel MUC1 aptamer selectively delivers cytotoxic agent to cancer cells in vitro. PLoS ONE 2012, 7, e31970. [Google Scholar] [CrossRef]
- Boltz, A.; Piater, B.; Toleikis, L.; Guenther, R.; Kolmar, H.; Hock, B. Bi-specific aptamers mediating tumor cell lysis. J. Biol. Chem. 2011, 286, 21896–21905. [Google Scholar] [CrossRef]
- Ducani, C.; Kaul, C.; Moche, M.; Shih, W.M.; Hogberg, B. Enzymatic production of ‘monoclonal stoichiometric’ single-stranded DNA oligonucleotides. Nat. Methods 2013, 10, 647–652. [Google Scholar] [CrossRef]
- Mousavi, T.; Poormoghim, H.; Moradi, M.; Tajik, N.; Shahsavar, F.; Soofi, M. Phenotypic study of natural killer cell subsets in ankylosing spondylitis patients. Iran. J. Allergy Asthma Immunol. 2009, 8, 193–198. [Google Scholar]
- Ansari, A.W.; Meyer-Olson, D.; Schmidt, R.E. Selective expansion of pro-inflammatory chemokine CCL2-loaded CD14+CD16+ monocytes subset in HIV-infected therapy naive individuals. J. Allergy Clin. Immunol. 2013, 33, 302–306. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.Q.; Bapat, A.S.; Rayanade, R.J.; Dagtas, A.S.; Hoffmann, M.K. Interleukin-10 induces macrophage apoptosis and expression of CD16 (FcgammaRIII) whose engagement blocks the cell death programme and facilitates differentiation. Immunology 2001, 102, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liu, H.; Sefah, K.; Liu, B.; Pu, Y.; Van Simaeys, D.; Tan, W. Selection of aptamers specific for adipose tissue. PLoS ONE 2012, 7, e37789. [Google Scholar] [CrossRef]
- Wang, Y.; Luo, Y.; Bing, T.; Chen, Z.; Lu, M.; Zhang, N.; Shangguan, D.; Gao, X. DNA aptamer evolved by cell-SELEX for recognition of prostate cancer. PLoS ONE 2014, 9, e100243. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.W.; Kochenderfer, J.N.; Stetler-Stevenson, M.; Cui, Y.K.; Delbrook, C.; Feldman, S.A.; Fry, T.J.; Orentas, R.; Sabatino, M.; Shah, N.N.; et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet (Lond. Engl.) 2015, 385, 517–528. [Google Scholar] [CrossRef]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef]
- Nabavinia, M.S.; Gholoobi, A.; Charbgoo, F.; Nabavinia, M.; Ramezani, M.; Abnous, K. Anti-MUC1 aptamer: A potential opportunity for cancer treatment. Med. Res. Rev. 2017, 37, 1518–1539. [Google Scholar] [CrossRef]
- Xiang, D.; Zheng, C.; Zhou, S.F.; Qiao, S.; Tran, P.H.; Pu, C.; Li, Y.; Kong, L.; Kouzani, A.Z.; Lin, J.; et al. Superior Performance of Aptamer in Tumor Penetration over Antibody: Implication of Aptamer-Based Theranostics in Solid Tumors. Theranostics 2015, 5, 1083–1097. [Google Scholar] [CrossRef]
- Nunez-Prado, N.; Compte, M.; Harwood, S.; Alvarez-Mendez, A.; Lykkemark, S.; Sanz, L.; Alvarez-Vallina, L. The coming of age of engineered multivalent antibodies. Drug Discov. Today 2015, 20, 588–594. [Google Scholar] [CrossRef]
- Pastor, F.; Kolonias, D.; McNamara, J.O., 2nd; Gilboa, E. Targeting 4-1BB costimulation to disseminated tumor lesions with bi-specific oligonucleotide aptamers. Mol. Ther. 2011, 19, 1878–1886. [Google Scholar] [CrossRef]
- Soldevilla, M.M.; Villanueva, H.; Casares, N.; Lasarte, J.J.; Bendandi, M.; Inoges, S.; Lopez-Diaz de Cerio, A.; Pastor, F. MRP1-CD28 bi-specific oligonucleotide aptamers: target costimulation to drug-resistant melanoma cancer stem cells. Oncotarget 2016, 7, 23182–23196. [Google Scholar] [CrossRef] [PubMed]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Z.; Hu, Y.; An, Y.; Duan, J.; Li, X.; Yang, X.-D. Novel Bispecific Aptamer Enhances Immune Cytotoxicity Against MUC1-Positive Tumor Cells by MUC1-CD16 Dual Targeting. Molecules 2019, 24, 478. https://doi.org/10.3390/molecules24030478
Li Z, Hu Y, An Y, Duan J, Li X, Yang X-D. Novel Bispecific Aptamer Enhances Immune Cytotoxicity Against MUC1-Positive Tumor Cells by MUC1-CD16 Dual Targeting. Molecules. 2019; 24(3):478. https://doi.org/10.3390/molecules24030478
Chicago/Turabian StyleLi, Zhaoyi, Yan Hu, Yacong An, Jinhong Duan, Xundou Li, and Xian-Da Yang. 2019. "Novel Bispecific Aptamer Enhances Immune Cytotoxicity Against MUC1-Positive Tumor Cells by MUC1-CD16 Dual Targeting" Molecules 24, no. 3: 478. https://doi.org/10.3390/molecules24030478
APA StyleLi, Z., Hu, Y., An, Y., Duan, J., Li, X., & Yang, X.-D. (2019). Novel Bispecific Aptamer Enhances Immune Cytotoxicity Against MUC1-Positive Tumor Cells by MUC1-CD16 Dual Targeting. Molecules, 24(3), 478. https://doi.org/10.3390/molecules24030478