Design, Synthesis, Anticancer Evaluation and Molecular Modeling of Novel Estrogen Derivatives

 ,
,  ,
,  ,
,  and
and
Abstract
1. Introduction
2. Results and Discussion
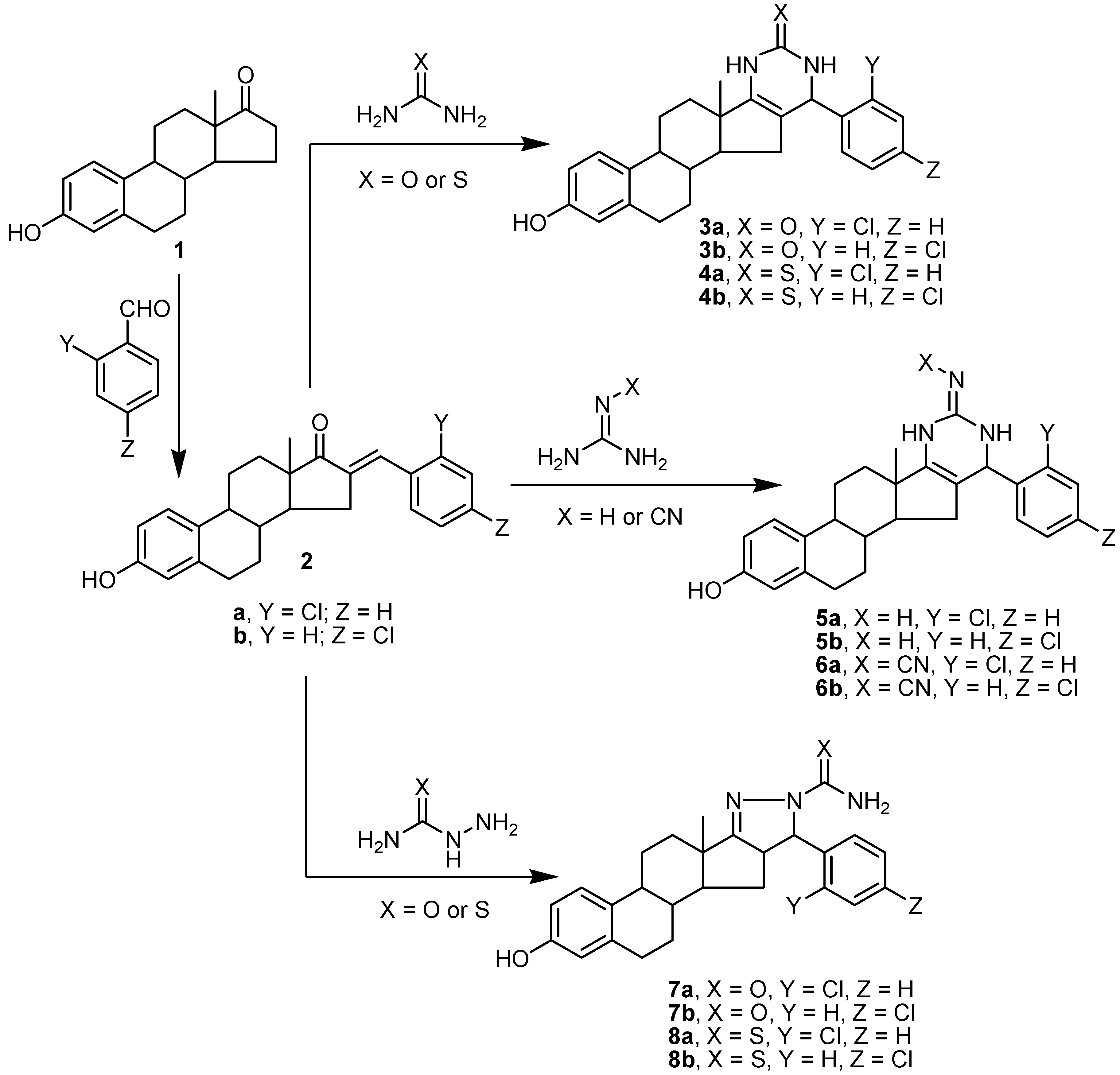
2.1. Chemistry
2.2. Biological Evaluation
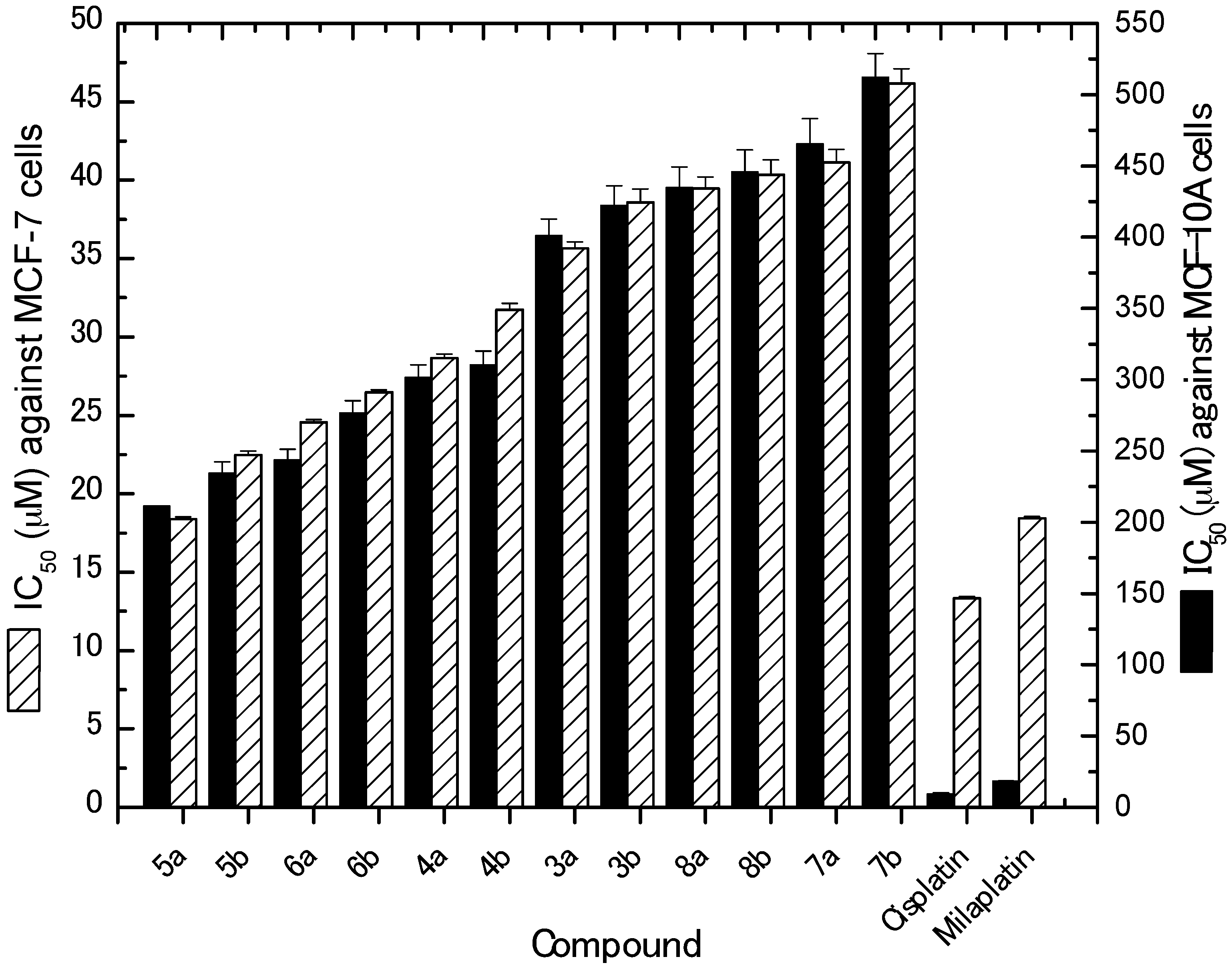
2.2.1. In Vitro Cytotoxic Activity against MCF-7 Cancer Cells
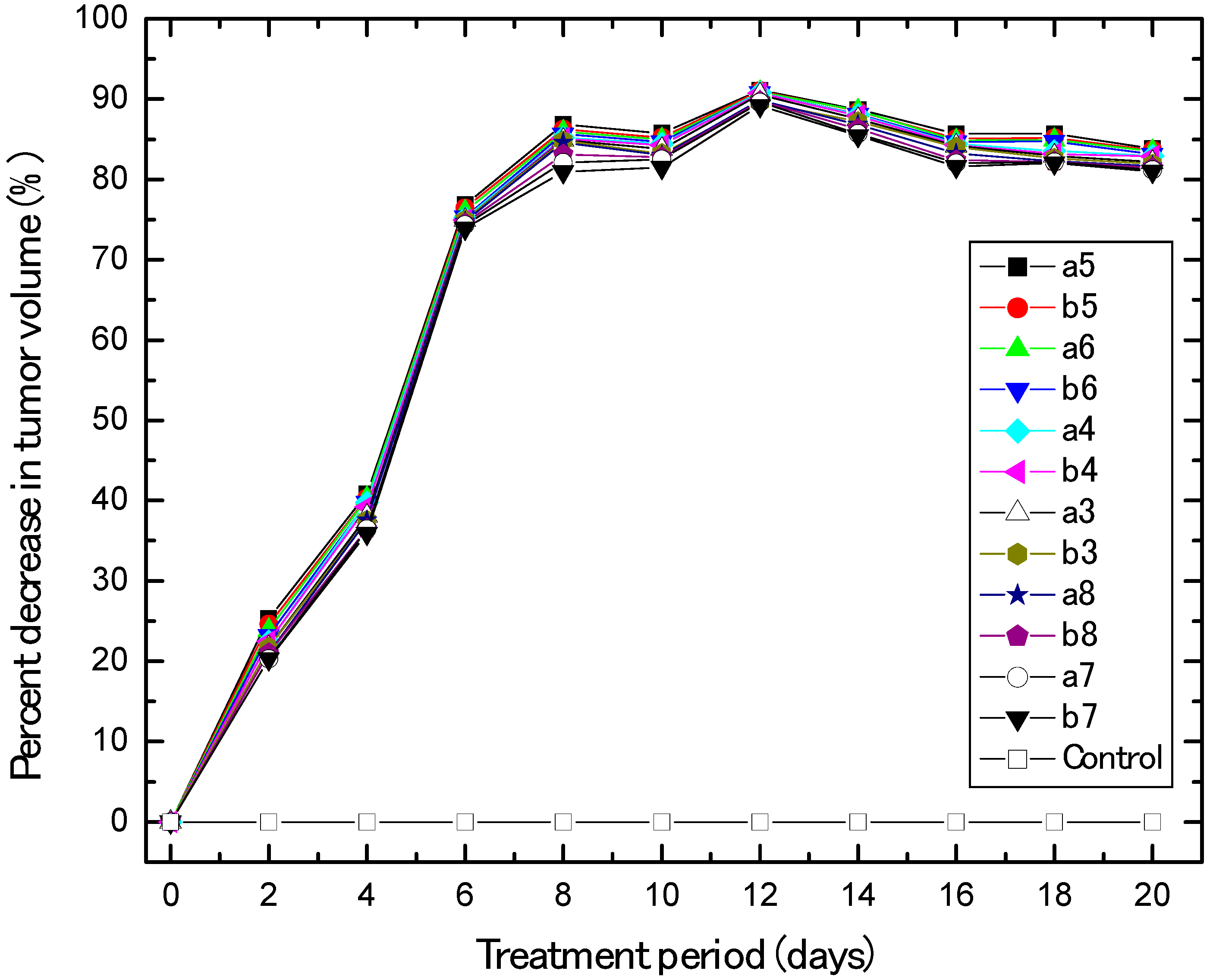
2.2.2. In Vivo Anti-Breast Cancer
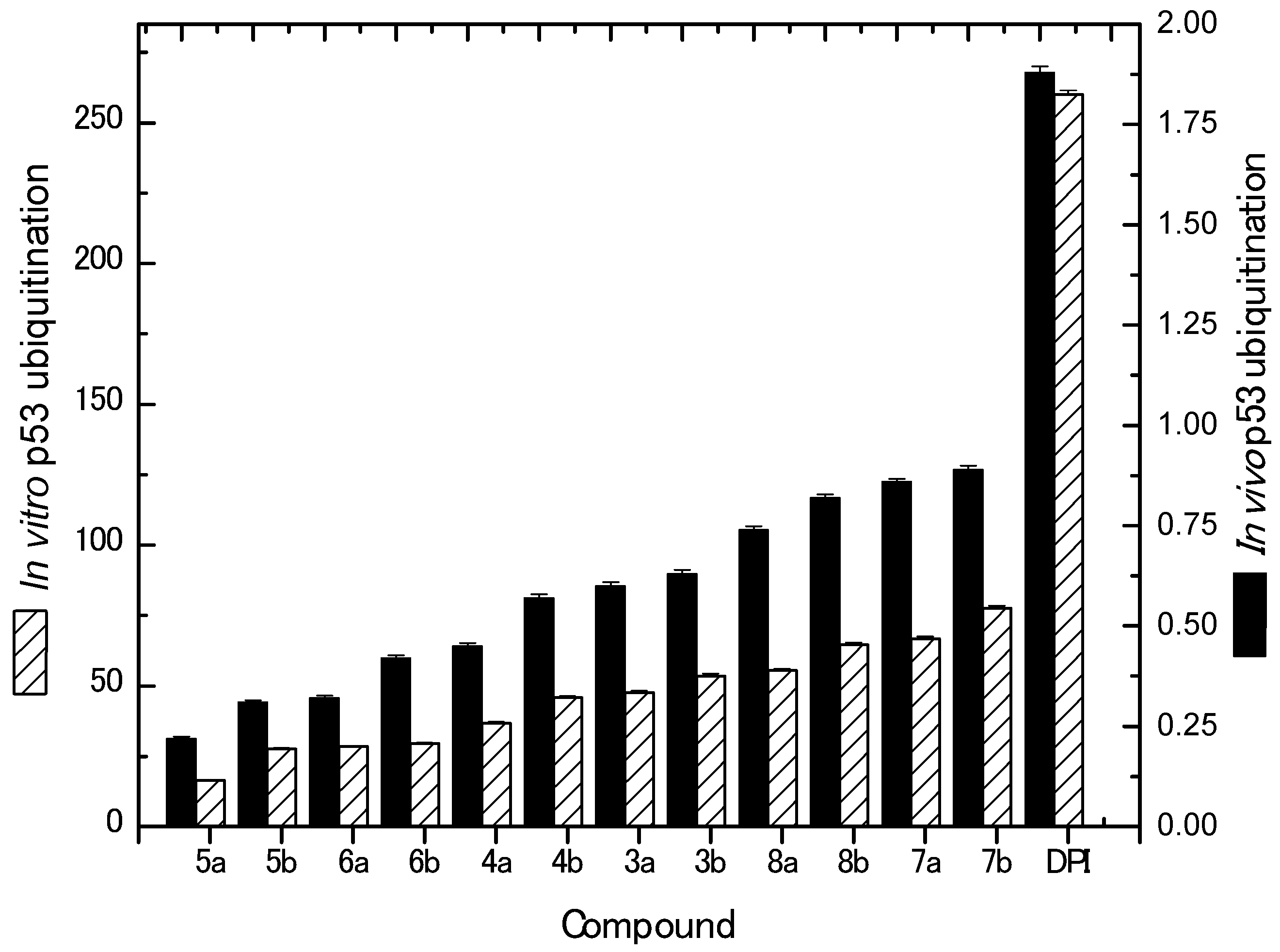
2.2.3. In Vivo and In Vitro Inhibition of p53 Ubiquitination Activities
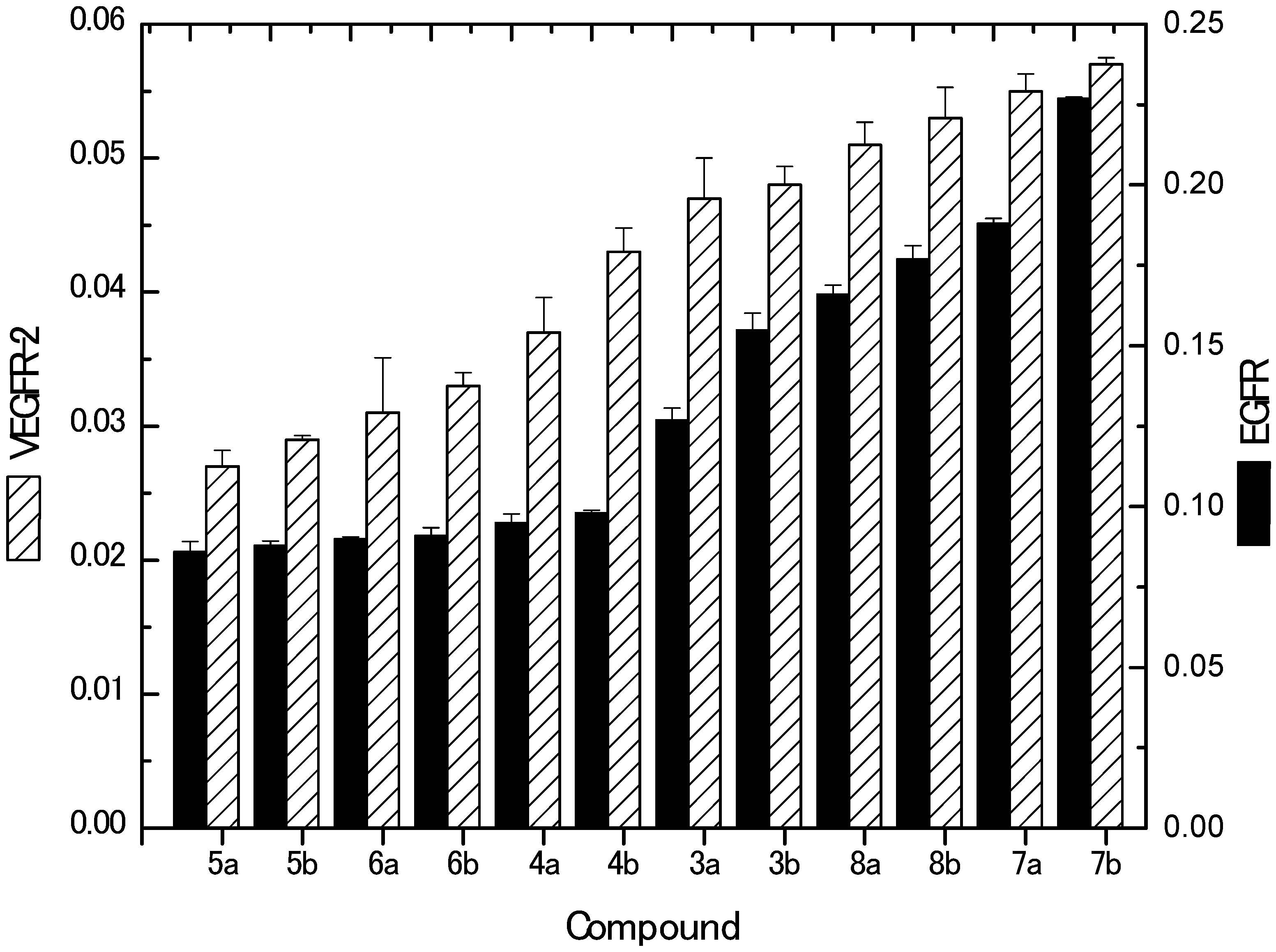
2.2.4. Inhibition of EGFR and VEGFR-2 Kinases
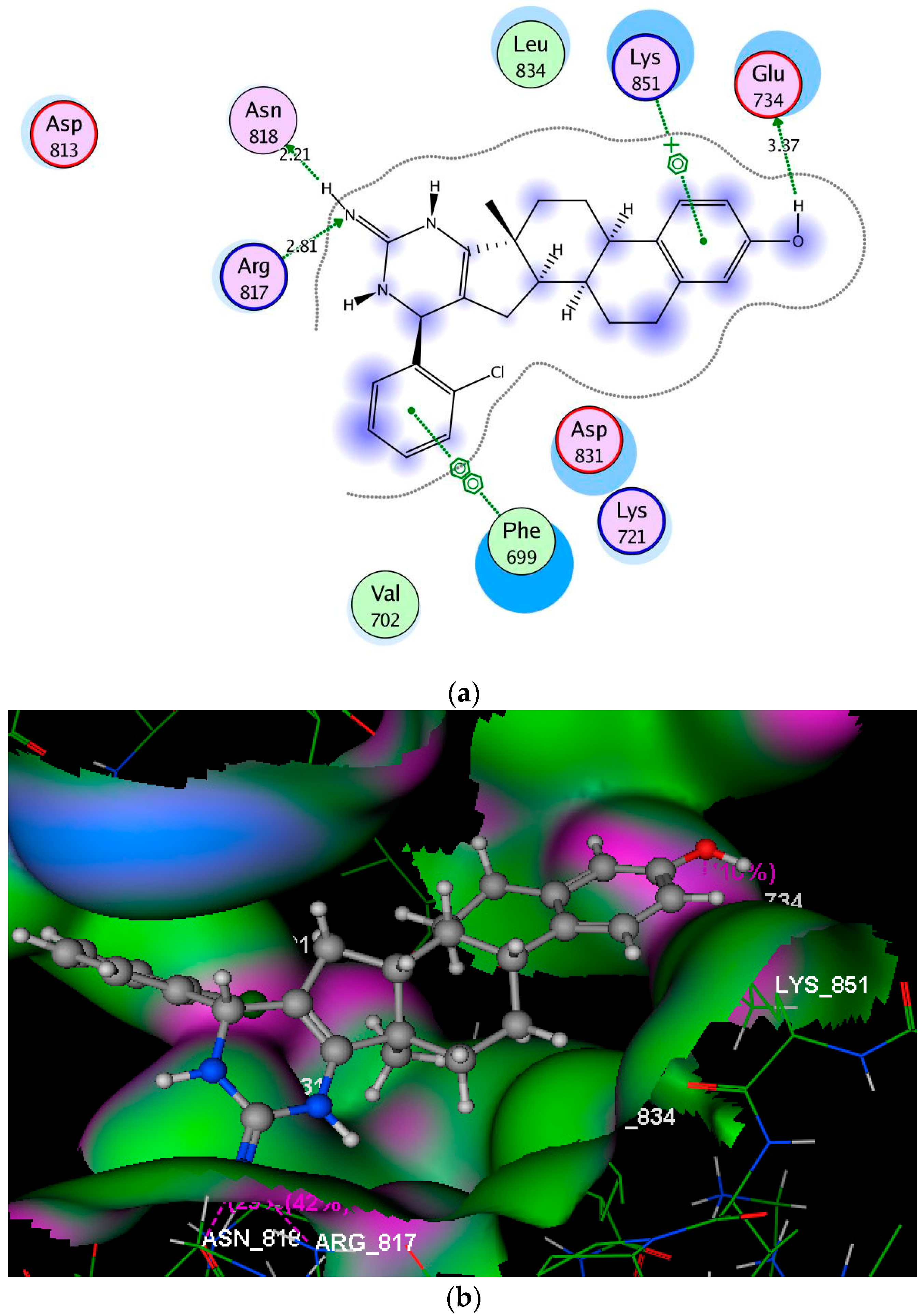
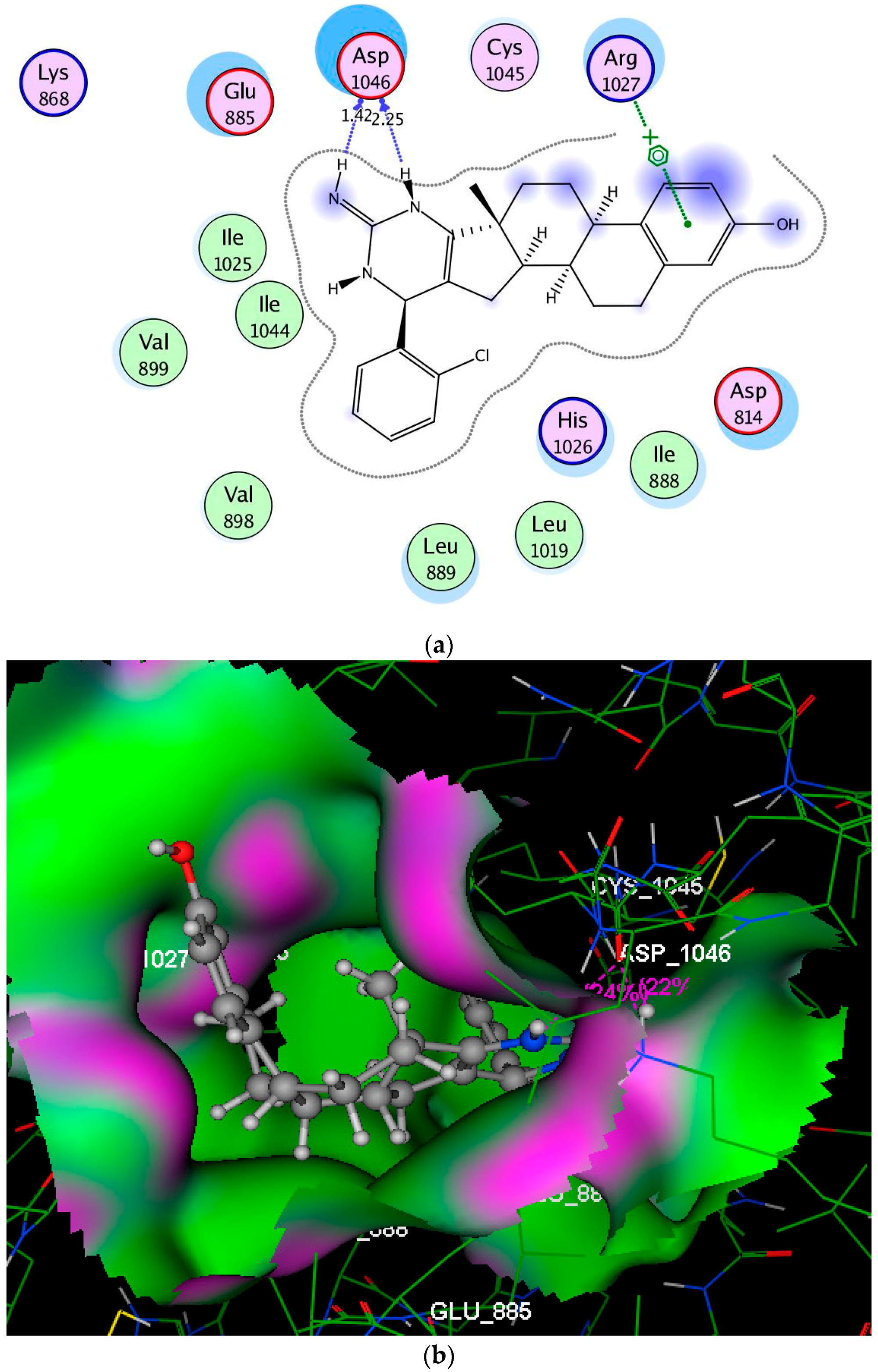
2.3. Molecular Modeling Studies
3. Materials and Methods
3.1. Chemistry
3.1.1. Synthesis of Aryl-estra-tetraien-[17,16-d]pyrimidinol Derivatives 3a,b and 4a,b
3.1.2. Synthesis of Imino-6′-aryl-estra-tetraien[17,16-d]pyrimidinool Derivatives 5a,b and 6a,b
3.1.3. Synthesis of Substituted Phenyl-estratrien[17,16-c]pyrazoline Derivatives 7a,b and 8a,b
3.2. Biological Evaluation
3.2.1. In Vitro Cytotoxic Activity against MCF-7 Cancer Cells
3.2.2. In Vivo Human Breast Cancer Xenograft Models and Animal Treatment
3.2.3. In Vitro p53 Ubiquitination Assay
3.2.4. In Vivo Ubiquitination of p53
3.2.5. Non-Fluorescent In Vitro Ubiquitination
3.2.6. Determination of EGFR and VEGFR-2 Kinase Activities
3.2.7. Statistical Analysis
3.3. Molecular Modeling Studies
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- WHO. Cancer: Factsheet No. 297; WHO: Geneva, Switzerland, February 2014. [Google Scholar]
- Yue, W.; Yager, J.D.; Wang, J.P.; Jupe, E.R.; Santen, R.J. Estrogen receptor-dependent and independent mechanisms of breast cancer carcinogenesis. Steroids 2013, 78, 161–170. [Google Scholar] [CrossRef]
- Izrailit, J.; Reedijk, M. Developmental pathways in breast cancer and breast tumor initiating cells: Therapeutic implications. Cancer Lett. 2012, 317, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Kumar, B.S.; Negi, A.S. Current status on development of steroids as anticancer agents. J. Steroid Biochem. Mol. Biol. 2013, 137, 242–270. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, V.; Lee, H. Synthesis and bio-evaluation of novel quinolino-stilbene derivatives as potential anticancer agents. Bioorg. Med. Chem. Lett. 2015, 23, 7629–7640. [Google Scholar] [CrossRef] [PubMed]
- Dancey, J.E.; Chen, H.X. Strategies for optimizing combinations of molecularly targeted anticancer agents. Nat. Rev. Drug Discov. 2006, 5, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Zhang, H.J.; Zhang, H.; Xu, C.; Zhao, J.; Zhu, H.L. Synthesis, molecular modeling, and biological evaluation of cinnamic acid metronidazole ester derivatives as novel anticancer agents. Bioorg. Med. Chem. 2010, 18, 4991–4996. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S. Review of epidermal growth factor receptor biology. Int. J. Radiat. Oncol. Biol. Phys. 2004, 59, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Colbert, L.S.; Fuller, M.; Zhang, Y.; Gonzalez-Perez, R.R. Vascular endothelial growth factor receptor-2 in breast cancer. Biochim. Biophys. Acta 2010, 1806, 108–121. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H.; Lane, D.P. p53 in health and disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Fang, S.; Jensen, J.P.; Ludwig, R.L.; Vousden, K.H.; Weissman, A.M. Mdm2 Is a RING Finger-dependent Ubiquitin Protein Ligase for Itself and p53. J. Biol. Chem. 2000, 275, 8945–8951. [Google Scholar] [CrossRef]
- Kubbutat, M.H.; Jones, S.N.; Vousden, K.H. Regulation of p53 stability by Mdm2. Nature 1997, 387, 299–303. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.L.; Auchus, R.J. The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr. Rev. 2010, 32, 81–151. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.G.; Zeng, Q.; Tse, G.M. Estrogen and its receptors in cancer. Med. Res. Rev. 2008, 28, 954–974. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, N.R.; Abdelhalim, M.M.; Khadraway, Y.A.; Elmegeed, G.A.; Abdel-Salam, O.M.E. One-pot three-component synthesis of novel heterocyclic steroids as a central antioxidant and anti-inflammatory agents. Steroids 2012, 77, 1469–1476. [Google Scholar] [CrossRef] [PubMed]
- Burkhart, J.P.; Gates, C.A.; Laughlin, M.E.; Resvick, R.J.; Peet, N.P. Inhibition of steroid C17(20) lyase with C-17-heteroaryl steroids. Bioorg. Med. Chem. 1996, 4, 1411–1420. [Google Scholar] [CrossRef]
- Huang, L.H.; Zheng, Y.F.; Song, C.J.; Wang, Y.G.; Xie, Z.Y.; Lai, Y.W.; Lu, Y.Z.; Liu, H.M. Synthesis of novel D-ring fused 7′-aryl-androstano[17,16-d][1,2,4]triazolo[1,5-a]pyrimidines. Steroids 2011, 77, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.H.; Zheng, Y.F.; Lu, Y.Z.; Song, C.J.; Wang, Y.; Yu, G.B.; Liu, H.M. Synthesis and biological evaluation of novel steroidal[17,16-d41][1,2,4]triazolo[1,5-a]pyrimidines. Steroids 2012, 77, 710–715. [Google Scholar] [CrossRef]
- Cushman, M.; He, H.M.; Katzenellenbogen, J.A.; Varma, R.K.; Hamel, E.; Lin, C.M.; Ram, S.; Sachdeva, Y.P. Synthesis of analogs of 2-methoxyestradiol with enhanced inhibitory effects on tubulin polymerization and cancer cell growth. J. Med. Chem. 1997, 40, 2323–2334. [Google Scholar] [CrossRef]
- Möller, G.; Deluca, D.; Gege, C.; Rosinus, A.; Kowalik, D.; Peters, O.; Droescher, P.; Elger, W.; Adamski, J.; Hillisch, A. Structure-based design, synthesis and in vitro characterization of potent 17β-hydroxysteroid dehydrogenase type 1 inhibitors based on 2-substitutions of estrone and d-homo-estrone. Bioorg. Med. Chem. Lett. 2009, 19, 6740–6744. [Google Scholar] [CrossRef]
- Ayan, D.; Roy, J.; Maltais, R.; Poirier, D. Impact of estradiol structural modifications (18-methyl and/or 17-hydroxy inversion of configuration) on the in vitro and in vivo estrogenic activity. J. Steroid Biochem. Mol. Biol. 2011, 127, 324–330. [Google Scholar] [CrossRef]
- Schönecker, B.; Lange, C.; Kötteritzsch, M.; Gunther, W.; Weston, J.; Anders, E.; Görls, H. Conformational design for 13-alpha-steroids. J. Org. Chem. 2000, 65, 5487–5497. [Google Scholar] [CrossRef] [PubMed]
- Jovanović-Santa, S.; Petrović, J.; Andrić, S.; Kovačević, R.; Đurendić, E.; Sakač, M.; Lazar, D.; Stanković, S. Synthesis, structure, and screening of estrogenic and antiestrogenic activity of new 3,17-substituted-16,17-seco-estratriene derivatives. Bioorg. Chem. 2003, 31, 475–484. [Google Scholar] [CrossRef]
- Amr, A.E.; El-Naggar, M.; Al-Omar, M.A.; Elsayed, E.A.; Abdalla, M.M. In vitro and in vivo anti-breast cancer activities of some synthesized pyrazolinyl-estran-17-one candidates. Molecules 2018, 23, 1572. [Google Scholar] [CrossRef] [PubMed]
- Mohareb, R.M.; Samir, E.M.; Halim, P.A. Synthesis, and anti-proliferative, Pim-1 kinase inhibitors and molecular docking of thiophenes derived from estrone. Bioorg. Chem. 2018. [Google Scholar] [CrossRef] [PubMed]
- Amr, A.E.; Abo-Ghalia, M.H.; Abdalah, M.M. Synthesis of new (Nα-dipicolinoyl)-bis-l-valyl-l-phenylalanyl linear and macrocyclic bridged peptides as anti-inflammatory agents. Arch. Pharm. 2007, 340, 304–309. [Google Scholar] [CrossRef] [PubMed]
- Al-Omar, M.A.; Amr, A.E. Synthesis of some new pyridine-2,6-carboxamide-derived schiff bases as potential antimicrobial agents. Molecules 2010, 15, 4711–4721. [Google Scholar] [CrossRef] [PubMed]
- Amr, A.E.; Abdel-Latif, N.A.; Abdalla, M.M. Synthesis of some new testosterone derivatives fused with substituted pyrazoline ring as promising 5α-reductase inhibitors. Acta Pharm. 2006, 56, 203–218. [Google Scholar]
- Abdel-Wahab, B.F.; Mohamed, S.F.; Amr, A.E.; Abdalla, M.M. Synthesis and reactions of thiosemicarbazides, triazoles, and Schiff bases as antihypertensive α-blocking agents. Monatshefte fur Chemie 2008, 139, 1083–1090. [Google Scholar] [CrossRef]
- Amr, A.E.; Abdulla, M.M. Synthesis and anti-inflammatory activities of new cyanopyrane derivatives fused with steroidal nuclei. Arch. Pharm. 2006, 339, 88–95. [Google Scholar] [CrossRef]
- Khalifa, N.M.; Al-Omar, M.A.; Amr, A.E.; Haiba, M.E. HIV-1 and HSV-1 virus activities of some new polycyclic nucleoside pyrene candidates. Int. J. Biol. Macromol. 2013, 54, 51–56. [Google Scholar] [CrossRef]
- Abdalla, M.M.; Al-Omar, M.A.; Bhat, M.A.; Amr, A.E.; Al-Mohizea, A.M. Steroidal pyrazolines evaluated as aromatase and quinone reductase-2 inhibitors for chemoprevention of cancer. Int. J. Biol. Macromol. 2012, 50, 1127–1132. [Google Scholar] [CrossRef] [PubMed]
- Maninder, M.; Jindal, D.P.; Leclercq, G.; Borras, M. Synthesis and biological activity of 16-arylidene derivatives of estrone and estrone methyl ether. Ind. J. Chem. 2003, 42B, 166–172. [Google Scholar]
- Gavaskar, D.; Suresh Babu, A.R.; Raghunathan, R.; Dharani, M.; Balasubramanian, S. An expedient sequential one-pot four component synthesis of novel steroidal spiro-pyrrolidine heterocycles in ionic liquid. Steroids 2016, 109, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Jeyachandran, V.; Vivek Kumar, S.; Ranjith Kumar, R. Synthesis of novel 16-spiro steroids: 7-(Aryl)tetrahydro-1H-pyrrolo[1,2-c][1,3]thiazolo-estrone-hybrid heterocycles. Steroids 2014, 82, 29–37. [Google Scholar] [CrossRef]
- Day, J.M.; Foster, P.A.; Chander, S.K.; Tutill, H.J.; Parsons, M.F.C.; Allan, G.M.; Lawrence, H.R.; Vicker, N.; Potter, B.V.L.; Reed, M.J.; et al. Inhibition of estrone-dependent tumor growth in vivo by the 17β-HSD1 inhibitor, 2-ethyl-16β-m-pyridylmethylamidomethyl-estrone (2-EtE1-F). Breast Cancer Res. Treat. 2006, 100, S197. Available online: https://insights.ovid.com/breast-cancer-research-treatment/bcart/2006/12/001/inhibitionestrone-dependent-tumor-growth-vivo-17/585/00001803 (accessed on 28 September 2013).
- Day, J.M.; Foster, P.A.; Tutill, H.J.; Schmidlin, F.; Sharland, C.M.; Hargrave, J.D.; Vicker, N.; Potter, B.V.L.; Reed, M.J.; Purohit, A. STX2171, a 17β-hydroxysteroid dehydrogenase type 3 inhibitor, is efficacious in vivo in a novel hormone-dependent prostate cancer model. Endocr. Relat. Cancer 2013, 20, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Dai, M.-S.; Lu, H. Inhibition of MDM2-mediated p53 Ubiquitination and Degradation by Ribosomal Protein L5. J. Biol. Chem. 2004, 279, 44475–44482. [Google Scholar] [CrossRef]
- Amr, A.E.; Abo-Ghalia, M.H.; Moustafa, G.; Al-Omar, M.A.; Nossier, E.S.; Elsayed, E.A. Design, synthesis and docking studies of novel macrocyclic pentapeptides as anticancer multi-targeted kinase inhibitors. Molecules 2018, 23, 2416. [Google Scholar] [CrossRef]
- Elzahabi, H.S.A.; Nossier, E.S.; Khalifa, N.M.; Alasfoury, R.A.; El-Manawat, M.A. Anticancer evaluation and molecular modeling of multi-targeted kinase inhibitors based pyrido[2,3-d]pyrimidine scaffold. J. Enzy. Inh. Med. Chem. 2018, 33, 546–557. [Google Scholar] [CrossRef]
- Elsayed, E.A.; Sharaf-Eldin, M.A.; Wadaan, M. In vitro evaluation of cytotoxic activities of essential oil from Moringa oleifera seeds on HeLa, HepG2, MCF-7, CACO-2 and L929 cell lines. Asian Pac. J. Cancer Prev. 2015, 16, 4671–4675. [Google Scholar] [CrossRef]
- Elsayed, E.A.; Farooq, M.; Dailin, D.; El-Enshasy, H.A.; Othman, N.Z.; Malek, R.; Danial, E.; Wadaan, M. In vitro and in vivo biological screening of kefiran polysaccharide produced by Lactobacillus kefiranofaciens. Biomed. Res. 2017, 28, 594–600. [Google Scholar]
- Wang, H.; Yu, D.; Agrawal, S.; Zhang, R. Experimental therapy of human prostate cancer by inhibiting MDM2 expression with novel mixed-backbone antisense oligonucleotides: In Vitro and in vivo activities and mechanisms. Prostate 2003, 54, 194–205. [Google Scholar] [CrossRef] [PubMed]
- Gomha, S.M.; Abdel-aziz, H.M. An efficient synthesis of functionalized 2-(heteroaryl)-3H-benzo[f]chromen-3-ones and antibacterial evaluation. J. Chem. Res. 2013, 2, 298–303. [Google Scholar] [CrossRef]
- Breitmaier, E. Structure Elucidation by NMR in Organic Chemistry, A Practical Guide; John Wiley: Chichester, UK, 1993; p. 27. [Google Scholar]
Sample Availability: Samples of the compounds are available from the authors. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound No. | IC50 (Mean ± SEM) (µM) | |
|---|---|---|
| MCF-7 | MCF-10A | |
| 3a | 35.66 ± 0.40 | 401.23 ± 11.56 |
| 3b | 38.57 ± 0.87 | 422.33 ± 13.76 |
| 4a | 28.64 ± 0.28 | 301.54 ± 8.86 |
| 4b | 31.75 ± 0.39 | 310.56 ± 9.59 |
| 5a | 18.38 ± 0.13 | 211.23 ± 7.53 |
| 5b | 22.48 ± 0.24 | 234.56 ± 7.76 |
| 6a | 24.57 ± 0.16 | 243.54 ± 7.78 |
| 6b | 26.46 ± 0.17 | 276.87 ± 8.23 |
| 7a | 41.12 ± 0.84 | 465.77 ± 17.34 |
| 7b | 46.17 ± 0.93 | 512.34 ± 16.65 |
| 8a | 39.46 ± 0.75 | 434.65 ± 14.57 |
| 8b | 40.35 ± 0.95 | 445.67 ± 15.43 |
| Cisplatin | 13.34 ± 0.11 | 9.55 ± 0.54 |
| Milaplatin | 18.43 ± 0.13 | 18.30 ± 0.34 |
| Compound No. | IC50 (Mean ± SEM) (µM) | |
|---|---|---|
| In Vitro p53 Ubiquitination | In Vivo p53 Ubiquitination | |
| 3a | 47.65 ± 0.60 | 0.60 ± 0.0086 |
| 3b | 53.56 ± 0.78 | 0.63 ± 0.0095 |
| 4a | 36.64 ± 0.58 | 0.45 ± 0.0085 |
| 4b | 45.76 ± 0.49 | 0.57 ± 0.0097 |
| 5a | 16.45 ± 0.23 | 0.22 ± 0.0043 |
| 5b | 27.56 ± 0.34 | 0.31 ± 0.0053 |
| 6a | 28.47 ± 0.25 | 0.32 ± 0.0063 |
| 6b | 29.55 ± 0.36 | 0.42 ± 0.0074 |
| 7a | 66.65 ± 0.86 | 0.86 ± 0.0068 |
| 7b | 77.56 ± 0.97 | 0.89 ± 0.0099 |
| 8a | 55.45 ± 0.69 | 0.74 ± 0.0087 |
| 8b | 64.56 ± 0.78 | 0.82 ± 0.0078 |
| Diphenyl Imidazole | 260 ± 0.02 | 1.88 ± 0.0047 |
| Compound No. | IC50 (Mean ± SEM) (µM) | |
|---|---|---|
| EGFR | VEGFR-2 | |
| 3a | 0.127 ± 0.0037 | 0.047 ± 0.003 |
| 3b | 0.155 ± 0.0051 | 0.048 ± 0.0014 |
| 4a | 0.095 ± 0.0028 | 0.037 ± 0.0026 |
| 4b | 0.098 ± 0.0009 | 0.043 ± 0.0018 |
| 5a | 0.086 ± 0.0032 | 0.027 ± 0.0012 |
| 5b | 0.088 ± 0.0013 | 0.029 ± 0.0003 |
| 6a | 0.090 ± 0.0006 | 0.031 ± 0.0041 |
| 6b | 0.091 ± 0.0025 | 0.033 ± 0.0010 |
| 7a | 0.188 ± 0.0016 | 0.055 ± 0.0013 |
| 7b | 0.227 ± 0.0004 | 0.057 ± 0.0005 |
| 8a | 0.166 ± 0.0028 | 0.051 ± 0.0017 |
| 8b | 0.177 ± 0.0041 | 0.053 ± 0.0023 |
| Delphinidin | 6.27 ± 0.00076 | 5.09 ± 0.0012 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amr, A.E.-G.E.; Elsayed, E.A.; Al-Omar, M.A.; Badr Eldin, H.O.; Nossier, E.S.; Abdallah, M.M. Design, Synthesis, Anticancer Evaluation and Molecular Modeling of Novel Estrogen Derivatives. Molecules 2019, 24, 416. https://doi.org/10.3390/molecules24030416
Amr AE-GE, Elsayed EA, Al-Omar MA, Badr Eldin HO, Nossier ES, Abdallah MM. Design, Synthesis, Anticancer Evaluation and Molecular Modeling of Novel Estrogen Derivatives. Molecules. 2019; 24(3):416. https://doi.org/10.3390/molecules24030416
Chicago/Turabian StyleAmr, Abd El-Galil E., Elsayed A. Elsayed, Mohamed A. Al-Omar, Hanan O. Badr Eldin, Eman S. Nossier, and Mohamed M. Abdallah. 2019. "Design, Synthesis, Anticancer Evaluation and Molecular Modeling of Novel Estrogen Derivatives" Molecules 24, no. 3: 416. https://doi.org/10.3390/molecules24030416
APA StyleAmr, A. E.-G. E., Elsayed, E. A., Al-Omar, M. A., Badr Eldin, H. O., Nossier, E. S., & Abdallah, M. M. (2019). Design, Synthesis, Anticancer Evaluation and Molecular Modeling of Novel Estrogen Derivatives. Molecules, 24(3), 416. https://doi.org/10.3390/molecules24030416