
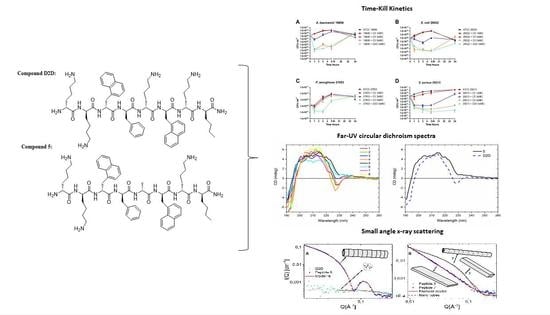
Structure-Activity Study of an All-d Antimicrobial Octapeptide D2D

 , , ,
, , ,  ,
,  ,
,  and
and
Abstract
1. Introduction
2. Results and Discussion
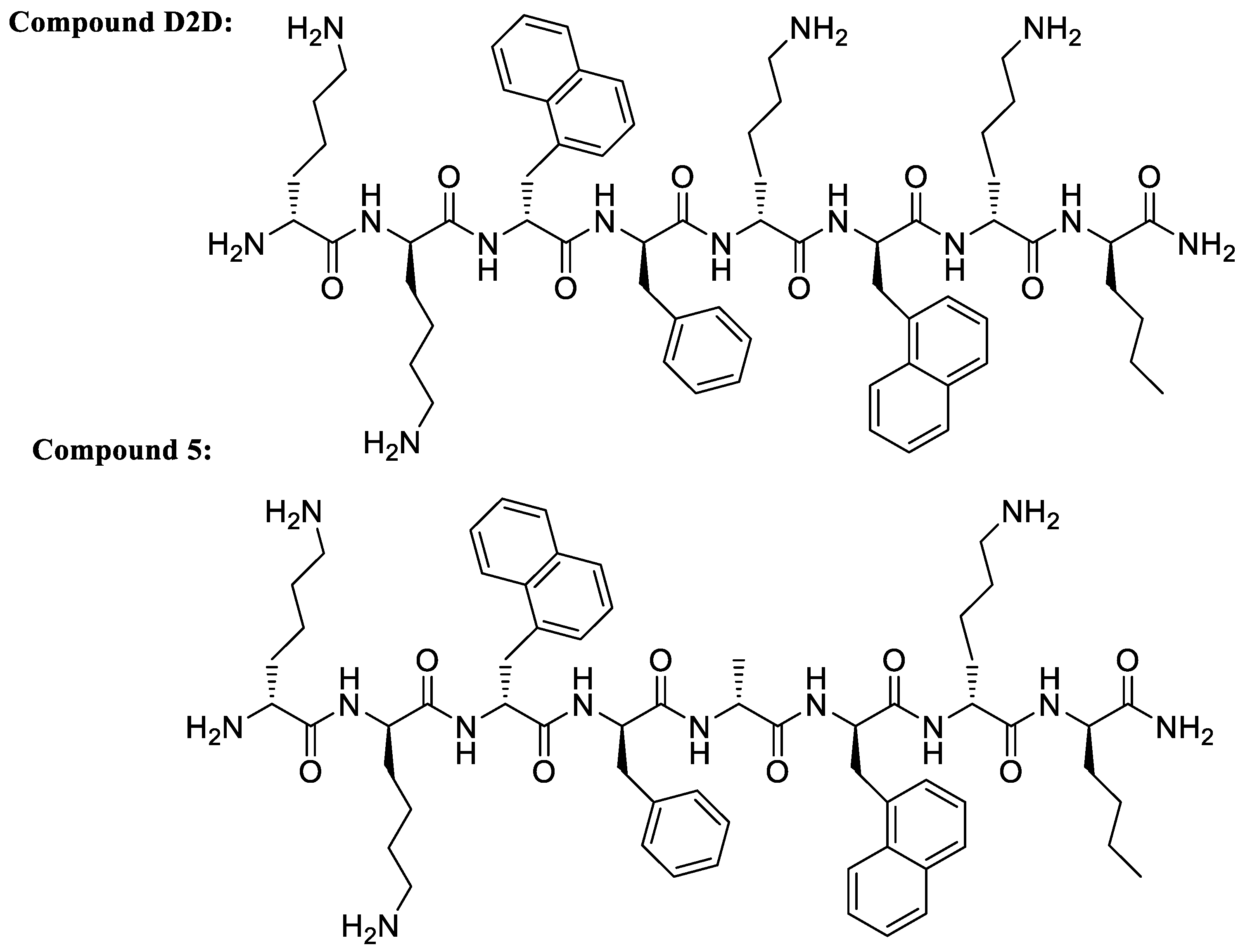
2.1. Alanine Scan of D2D
2.2. Investigation of Hydrophobicity at Position 5 and 7 Analogues
2.3. Manipulating Amphipaticity and Truncations (Compounds 25–32)
2.4. Manipulating Cationicity by Lys-Arg Substitutions (Compound 33–36)
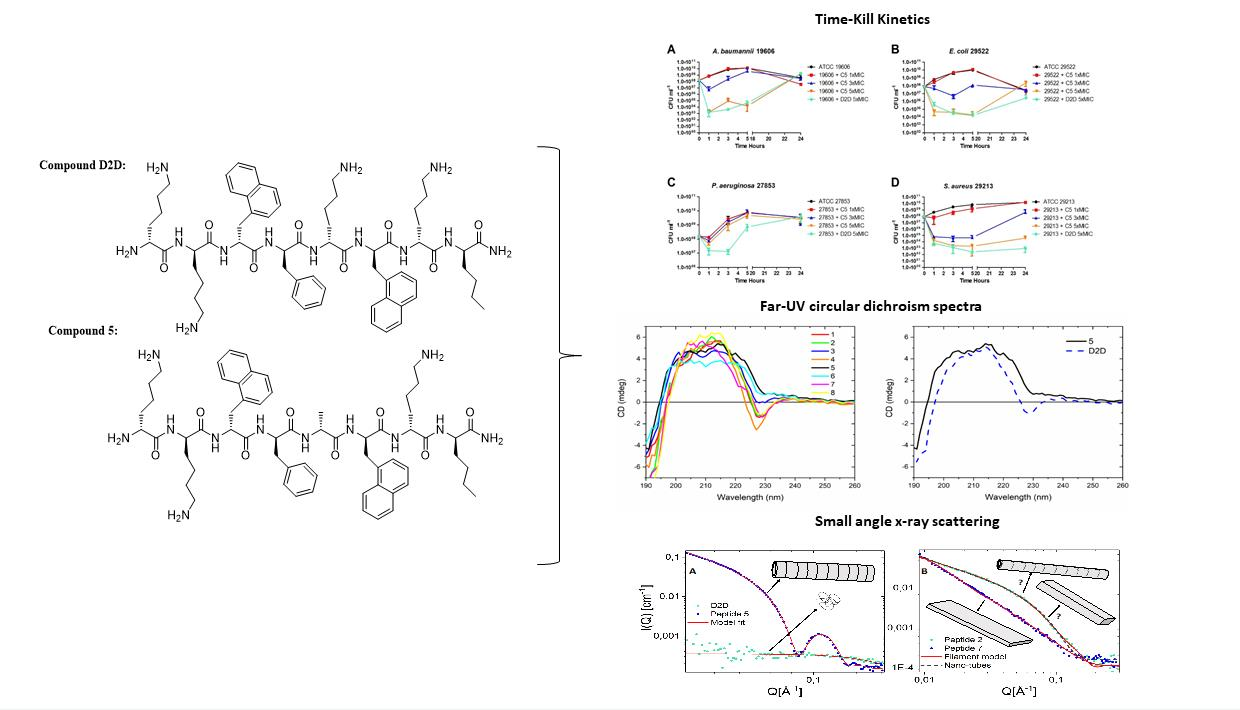
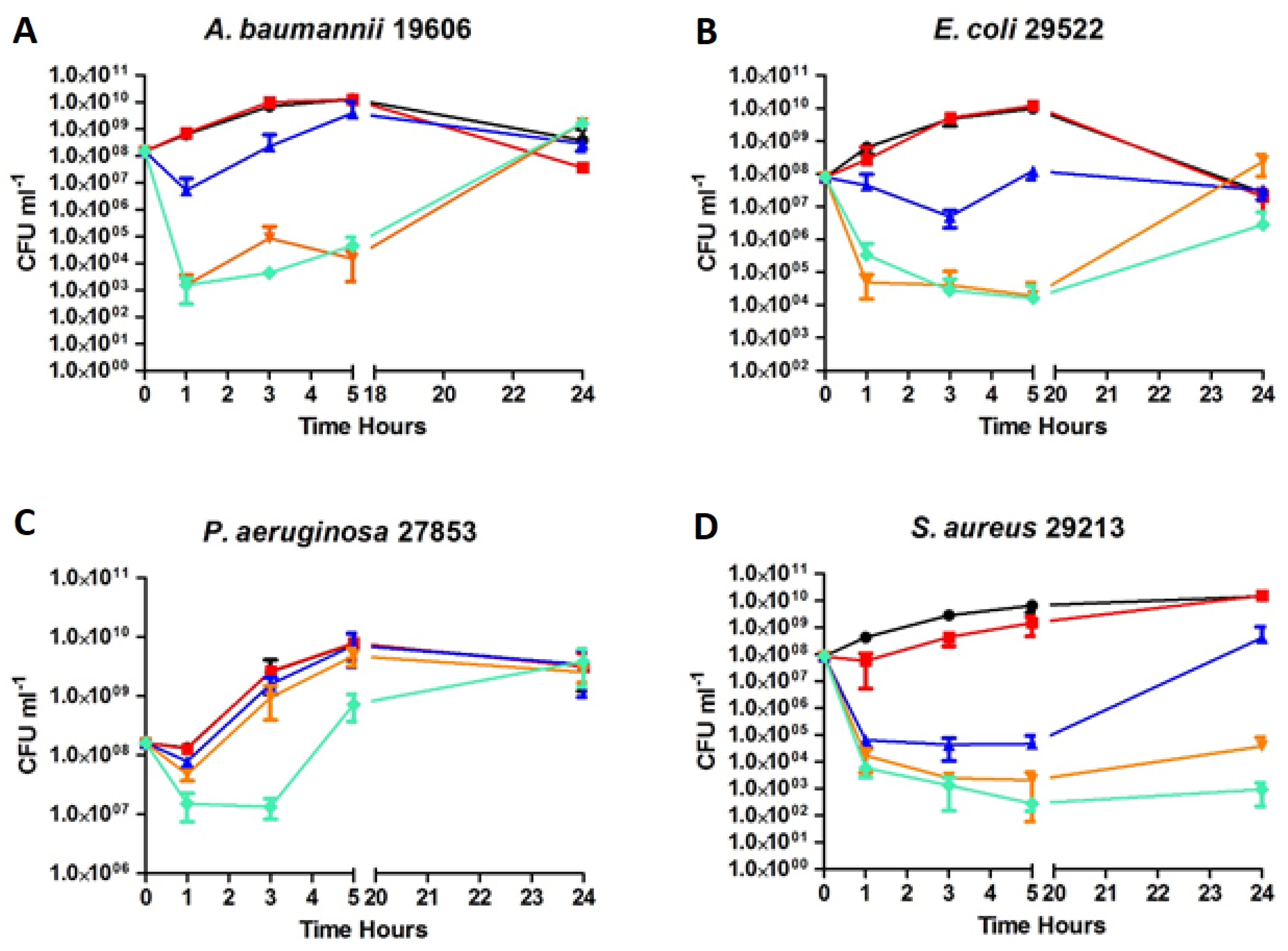
2.5. Time-kill Kinetics of D2D and Analogue 5
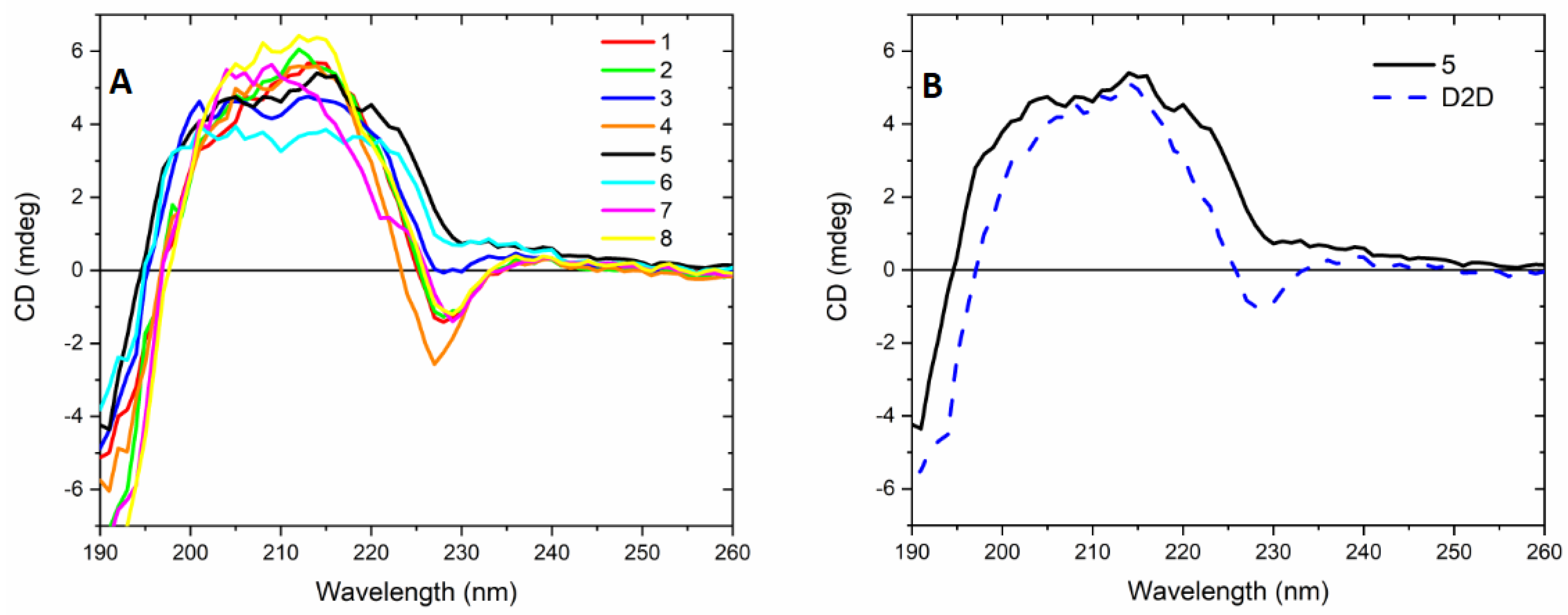
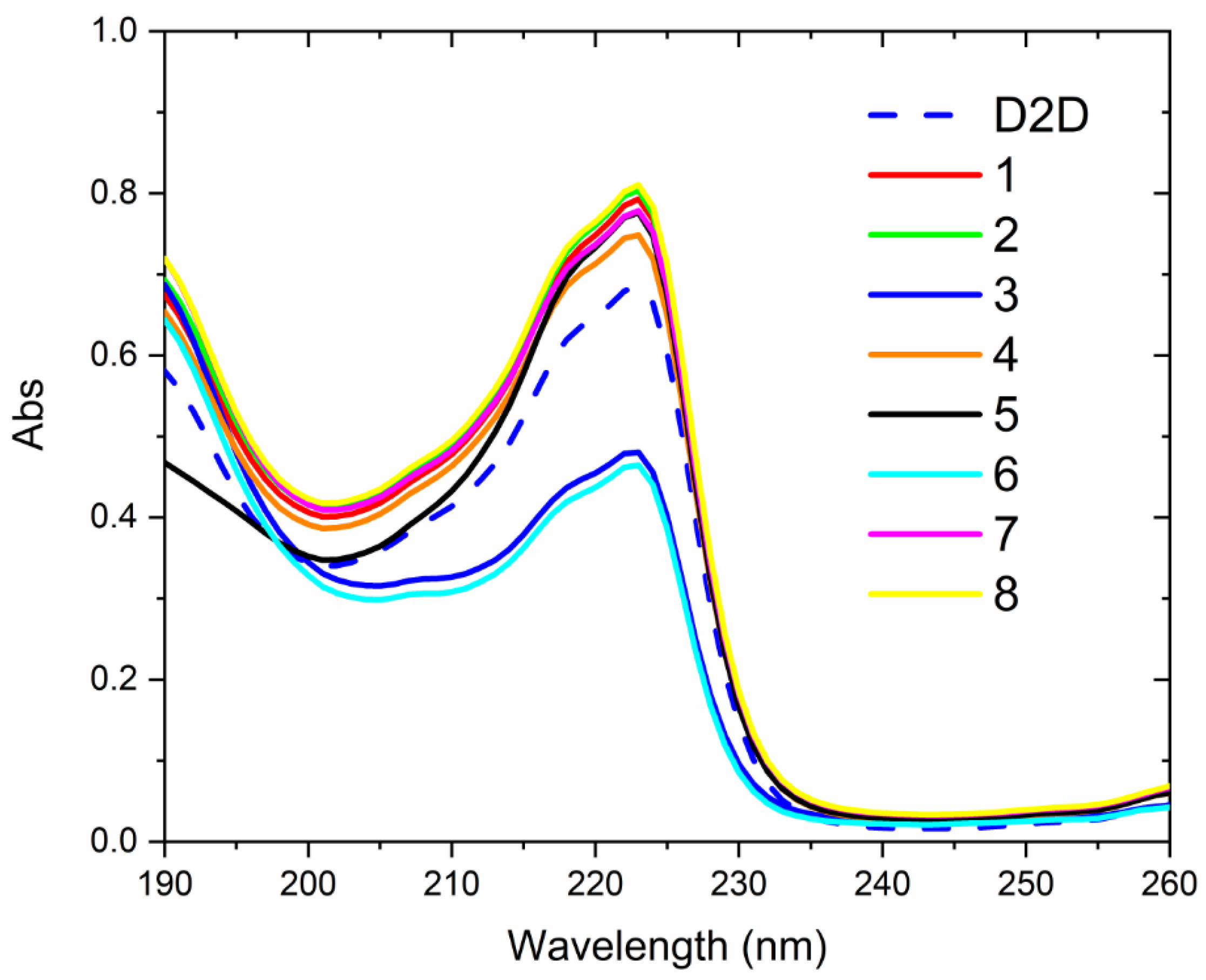
2.6. CD-Experiments
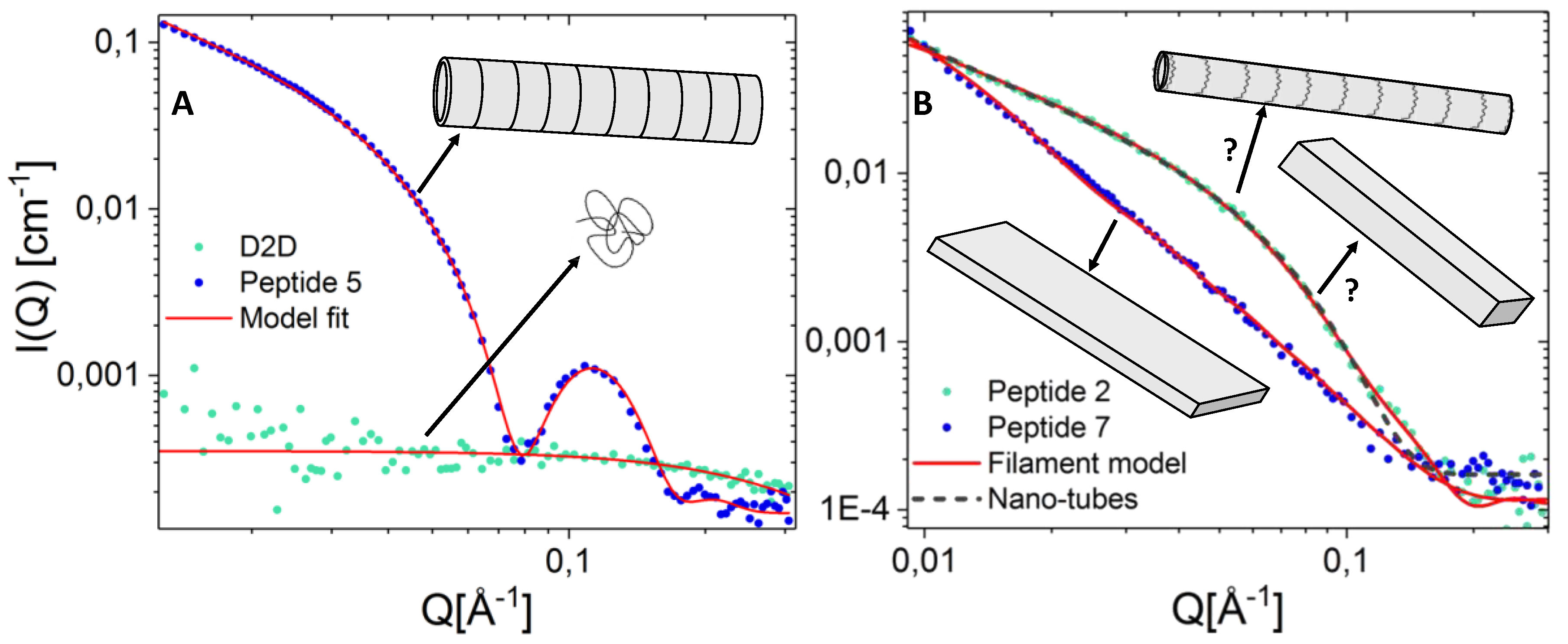
2.7. Peptide Self-Assembly Nanostructure Investigated by Small Angle X-ray Scattering
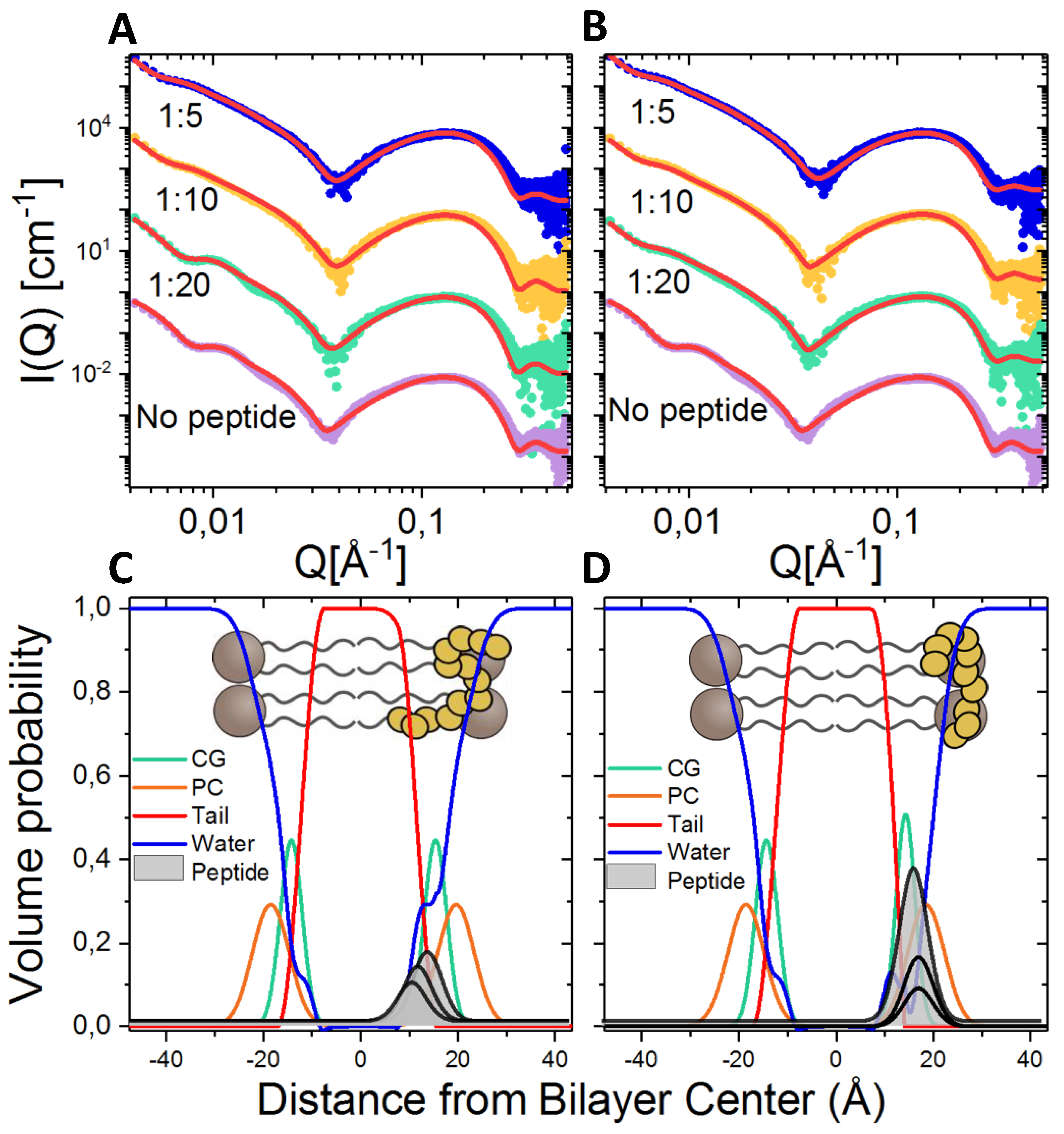
2.8. Lipid Interaction Probed by Small Angle X-ray Scattering
3. Materials and Methods
3.1. Synthesis of Peptides
3.2. Bacterial Strains
3.3. Antimicrobial Susceptibility Testing
3.4. Time Kill Curves
3.5. Hemolytic Activity
3.6. Circular Dichroism
3.7. Small Angle X-ray Scattering on Pure Peptides in Solution
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Konaklieva, M.I. Addressing Antimicrobial Resistance through New Medicinal and Synthetic Chemistry Strategies. SLAS DISCOVERY: Adv. Life Sci. R D 2018, 24, 419–439. [Google Scholar] [CrossRef]
- Tacconelli, E. Global priority list of antibiotic-resistant bacteria to guide research, discovery, and development of new antibiotics. World Health Organ. 2017, 27, 7. [Google Scholar]
- Mulani, M.S.; Kamble, E.E.; Kumkar, S.N.; Tawre, M.S.; Pardesi, K.R. Emerging Strategies to Combat ESKAPE Pathogens in the Era of Antimicrobial Resistance: A Review. Front. Microbiol. 2019, 10, 539. [Google Scholar] [CrossRef]
- Butler, M.S.; Blaskovich, M.A.T.; Cooper, M.A. Antibiotics in the clinical pipeline at the end of 2015. J Antibiot. 2017, 70, 3–24. [Google Scholar] [CrossRef]
- Mercer, D.K.; O’Neil, D.A. Peptides as the next generation of anti-infectives. Future Med. Chem. 2013, 5, 315–337. [Google Scholar] [CrossRef]
- Zasloff, M. Antimicrobial peptides of multicellular organisms. Nature 2002, 415, 389–395. [Google Scholar] [CrossRef]
- Greber, K.E.; Dawgul, M. Antimicrobial Peptides Under Clinical Trials. Curr. Top. Med. Chem. 2017, 17, 620–628. [Google Scholar] [CrossRef]
- Haney, E.F.; Mansour, S.C.; Hancock, R.E.W. Antimicrobial Peptides: An Introduction. In Antimicrobial Peptides: Methods and Protocols; Hansen, P.R., Ed.; Springer New York: New York, NY, USA, 2017; pp. 3–22. [Google Scholar]
- Fosgerau, K.; Hoffmann, T. Peptide therapeutics: Current status and future directions. Drug Discov. Today 2015, 20, 122–128. [Google Scholar] [CrossRef]
- Joo, S.H. Cyclic Peptides as Therapeutic Agents and Biochemical Tools. Biomol. Ther. 2012, 20, 19–26. [Google Scholar] [CrossRef]
- Molchanova, N.; Hansen, P.R.; Franzyk, H. Advances in Development of Antimicrobial Peptidomimetics as Potential Drugs. Molecules 2017, 22, 1430. [Google Scholar] [CrossRef]
- Bessalle, R.; Kapitkovsky, A.; Gorea, A.; Shalit, I.; Fridkin, M. All-D-magainin-chirality, antimicrobial activity and proteolytic resistance. FEBS Lett. 1990, 274, 151–155. [Google Scholar]
- Zuckermann, R.N.; Kerr, J.M.; Kent, S.B.H.; Moos, W.H. Efficient method for the preparation of peptoids (oligo(N-substituted glycines)) by submonomer solid-phase synthesis. J. Am. Chem. Soc. 1992, 114, 10646–10647. [Google Scholar] [CrossRef]
- Schmitt, M.A.; Weisblum, B.; Gellman, S.H. Interplay among Folding, Sequence, and Lipophilicity in the Antibacterial and Hemolytic Activities of α/β-Peptides. J. Am. Chem. Soc. 2006, 129, 417–428. [Google Scholar] [CrossRef]
- Ryge, T.S.; Frimodt-Moller, N.; Hansen, P.R. Antimicrobial activities of twenty lysine-peptoid hybrids against clinically relevant bacteria and fungi. Chemotherapy 2008, 54, 152–156. [Google Scholar] [CrossRef]
- Molchanova, N.; Hansen, P.R.; Damborg, P.; Nielsen, H.M.; Franzyk, H. Lysine-Based α-peptide/β-peptoid peptidomimetics: Influence of hydrophobicity, fluorination and distribution of cationic charge on antimicrobial activity and cytotoxicity. ChemMedChem 2017, 20, 312–318. [Google Scholar] [CrossRef]
- Niu, Y.; Wu, H.; Li, Y.; Hu, Y.; Padhee, S.; Li, Q.; Cao, C.; Cai, J. AApeptides as a new class of antimicrobial agents. Org. Biomol. Chem. 2013, 11, 4283–4290. [Google Scholar] [CrossRef]
- Greco, I.; Hansen, J.E.; Jana, B.; Molchanova, N.; Oddo, A.; Thulstrup, P.W.; Damborg, P.; Guardabassi, L.; Hansen, P.R. Structure–Activity Study, Characterization, and Mechanism of Action of an Antimicrobial Peptoid D2 and Its D- and L-Peptide Analogues. Molecules 2019, 24, 1121. [Google Scholar] [CrossRef]
- Greco, I.; Hummel, B.; Vasir, J.; Watts, J.; Koch, J.; Hansen, J.; Nielsen, H.; Damborg, P.; Hansen, P. In Vitro ADME Properties of Two Novel Antimicrobial Peptoid-Based Compounds as Potential Agents against Canine Pyoderma. Molecules 2018, 23, 630. [Google Scholar] [CrossRef]
- Staubitz, P.; Peschel, A.; Nieuwenhuizen, W.F.; Otto, M.; Götz, F.; Jung, G.; Jack, R.W. Structure-function relationships in the tryptophan-rich, antimicrobial peptide indolicidin. J. Pept. Sci. 2001, 7, 552–564. [Google Scholar] [CrossRef]
- Grieco, P.; Luca, V.; Auriemma, L.; Carotenuto, A.; Saviello, M.R.; Campiglia, P.; Barra, D.; Novellino, E.; Mangoni, M.L. Alanine scanning analysis and structure–function relationships of the frog-skin antimicrobial peptide temporin-1Ta. J. Pept. Sci. 2011, 17, 358–365. [Google Scholar] [CrossRef]
- Ifrah, D.; Doisy, X.; Ryge, T.; Hansen, P. Structure-activity relationship study of anoplin. J. Pept. Sci. 2005, 11, 113–121. [Google Scholar] [CrossRef]
- Manabe, T.; Kawasaki, K. D-form KLKLLLLLKLK-NH2 peptide exerts higher antimicrobial properties than its L-form counterpart via an association with bacterial cell wall components. Sci. Rep. 2017, 7, 43384. [Google Scholar] [CrossRef]
- Oddo, A.; Thomsen, T.T.; Kjelstrup, S.; Gorey, C.; Franzyk, H.; Frimodt-Møller, N.; Løbner-Olesen, A.; Hansen, P.R. An all-D amphipathic undecapeptide shows promising activity against colistin-resistant strains of Acinetobacter baumannii and a dual mode of action. Antimicrob. Agents Chemother. 2016, 60, 592–599. [Google Scholar] [CrossRef]
- Schiffer, M.; Edmunson, A.B. Use of helical wheels to represent the structures of proteins and to identify segments with helical potential. Biophys. J. 1967, 7, 121–135. [Google Scholar] [CrossRef]
- Nielsen, S.L.; Frimodt-Moller, N.; Kragelund, B.B.; Hansen, P.R. Structure activity study of the antibacterial peptide fallaxin. Protein Sci. 2007, 16, 1969–1976. [Google Scholar] [CrossRef]
- Deslouches, B.; Hasek, M.L.; Craigo, J.K.; Steckbeck, J.D.; Montelaro, R.C. Comparative functional properties of engineered cationic antimicrobial peptides consisting exclusively of tryptophan and either lysine or arginine. J. Med. Microbiol. 2016, 65, 554–565. [Google Scholar] [CrossRef]
- Greenfield, N.J. Using circular dichroism spectra to estimate protein secondary structure. Nat. Protoc. 2006, 1, 2876–2890. [Google Scholar] [CrossRef]
- Nielsen, J.E.; Bjørnestad, V.A.; Lund, R. Resolving the structural interactions between antimicrobial peptides and lipid membranes using small-angle scattering methods: The case of indolicidin. Soft Matter 2018, 14, 8750–8763. [Google Scholar] [CrossRef]
- Nielsen, J.E.; Lind, T.K.; Lone, A.; Gerelli, Y.; Hansen, P.R.; Jenssen, H.; Cárdenas, M.; Lund, R. A biophysical study of the interactions between the antimicrobial peptide indolicidin and lipid model systems. BBA Biomembr. 2019, 1861, 1355–1364. [Google Scholar] [CrossRef]
- Greco, I.; Emborg, A.P.; Jana, B.; Molchanova, N.; Oddo, A.; Damborg, P.; Guardabassi, L.; Hansen, P.R. Characterization, mechanism of action and optimization of activity of a novel peptide-peptoid hybrid against bacterial pathogens involved in canine skin infections. Sci. Rep.-UK 2019, 9, 3679. [Google Scholar] [CrossRef]
- Wiegand, I.; Hilpert, K.; Hancock, R.E.W. Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat. Protoc. 2008, 3, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Oddo, A.; Hansen, P.R. Hemolytic Activity of Antimicrobial Peptides. Methods Mol. Biol. 2017, 1548, 427–435. [Google Scholar] [PubMed]
- Sun, J.; Jiang, X.; Lund, R.; Downing, K.H.; Balsara, N.P.; Zuckermann, R.N. Self-assembly of crystalline nanotubes from monodisperse amphiphilic diblock copolypeptoid tiles. Proc. Natl. Acad. Sci. USA 2016, 113, 3954. [Google Scholar] [CrossRef] [PubMed]
- Pernot, P.; Round, A.; Barrett, R.; De Maria Antolinos, A.; Gobbo, A.; Gordon, E.; Huet, J.; Kieffer, J.; Lentini, M.; Mattenet, M.; et al. Upgraded ESRF BM29 beamline for SAXS on macromolecules in solution. J. Synchrotron Radiat. 2013, 20, 660–664. [Google Scholar] [CrossRef] [PubMed]
- Round, A.; Felisaz, F.; Fodinger, L.; Gobbo, A.; Huet, J.; Villard, C.; Blanchet, C.E.; Pernot, P.; McSweeney, S.; Roessle, M.; et al. BioSAXS Sample Changer: A robotic sample changer for rapid and reliable high-throughput X-ray solution scattering experiments. Acta. Crystallogr. D 2015, 71, 67–75. [Google Scholar] [CrossRef] [PubMed]
- De Maria Antolinos, A.; Pernot, P.; Brennich, M.E.; Kieffer, J.; Bowler, M.W.; Delageniere, S.; Ohlsson, S.; Malbet Monaco, S.; Ashton, A.; Franke, D.; et al. ISPyB for BioSAXS, the gateway to user autonomy in solution scattering experiments. Acta. Crystallogr. D 2015, 71, 76–85. [Google Scholar] [CrossRef]
Sample Availability: Not available. |


 ), 3 × MIC (
), 3 × MIC ( ), 5 × MIC (
), 5 × MIC ( ) (MIC = 4 µg/mL), D2D at 5 × MIC (
) (MIC = 4 µg/mL), D2D at 5 × MIC ( ) and non-treated cells (
) and non-treated cells ( ). Sampling of viable cells was performed at time points 0, 1, 3, 5 and 24 h (X-axis). Viable cell counts (CFU ml−1, Y-axis), was done by spot plating of washed cells.
), 3 × MIC (), 5 × MIC () (MIC = 4 µg/mL), D2D at 5 × MIC () and non-treated cells (). Sampling of viable cells was performed at time points 0, 1, 3, 5 and 24 h (X-axis). Viable cell counts (CFU ml−1, Y-axis), was done by spot plating of washed cells.
). Sampling of viable cells was performed at time points 0, 1, 3, 5 and 24 h (X-axis). Viable cell counts (CFU ml−1, Y-axis), was done by spot plating of washed cells.
), 3 × MIC (), 5 × MIC () (MIC = 4 µg/mL), D2D at 5 × MIC () and non-treated cells (). Sampling of viable cells was performed at time points 0, 1, 3, 5 and 24 h (X-axis). Viable cell counts (CFU ml−1, Y-axis), was done by spot plating of washed cells.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Minimum Inhibitory Concentration [a] | ||||||
|---|---|---|---|---|---|---|
| No. | Sequence [b] | E.C. [c] | S.A. [d] | P.A. [e] | A.B. [f] | %H [g] |
| D2D | kk(1-nal)fk(1-nal)k(nle)-NH2 | 8 | 8 | 4 | 4 | 23 |
| 1 | ak(1-nal)fk(1-nal)k(nle)-NH2 | 8 | 2 | 8 | 4 | 41 |
| 2 | ka(1-nal)fk(1-nal)k(nle)-NH2 | 16 | 8 | 16 | 16 | 59 |
| 3 | kkafk(1-nal)k(nle)-NH2 | >128 | 64 | 16 | 16 | 2 |
| 4 | kk(1-nal)ak(1-nal)k(nle)-NH2 | >128 | 128 | 32 | 64 | 22 |
| 5 | kk(1-nal)fa(1-nal)k(nle)-NH2 | 4 | 4 | 4 | 4 | 47 |
| 6 | kk(1-nal)fkak(nle)-NH2 | >128 | >128 | >128 | >128 | 10 |
| 7 | kk(1-nal)fk(1-nal)a(nle)-NH2 | 8 | 8 | 8 | 4 | 67 |
| 8 | kk(1-nal)fk(1-nal)ka-NH2 | 64 | 64 | 16 | 64 | 5 |
| 9 | kk(1-nal)f(1-nal)(1-nal)k(nle)-NH2 | 64 | 4 | 128 | 32 | 20 |
| 10 | kk(1-nal)f(dap)(1-nal)k(nle)-NH2 | 8 | 16 | 16 | 16 | 28 |
| 11 | kk(1-nal)f(nle)(1-nal)k(nle)-NH2 | 16 | 4 | 64 | 64 | 33 |
| 12 | kk(1-nal)ff(1-nal)k(nle)-NH2 | 16 | 4 | 32 | 16 | 74 |
| 13 | kk(1-nal)fs(1-nal)k(nle)-NH2 | 4 | 8 | 32 | 8 | 29 |
| 14 | kk(1-nal)ft(1-nal)k(nle)-NH2 | 8 | 8 | 32 | 8 | 28 |
| 15 | kk(1-nal)fy(1-nal)k(nle)-NH2 | 8 | 2 | 16 | 8 | 40 |
| 16 | kk(1-nal)fv(1-nal)k(nle)-NH2 | 32 | 4 | 64 | 64 | 54 |
| 17 | kk(1-nal)fk(1-nal)(1-nal)(nle)-NH2 | 16 | 2 | 32 | 4 | 89 |
| 18 | kk(1-nal)fk(1-nal)(dap)(nle)-NH2 | 8 | 16 | 16 | 16 | 21 |
| 19 | kk(1-nal)fk(1-nal)(nle)(nle)-NH2 | 8 | 4 | 32 | 8 | 56 |
| 20 | kk(1-nal)fk(1-nal)f(nle)-NH2 | 8 | 4 | 32 | 4 | 91 |
| 21 | kk(1-nal)fk(1-nal)s(nle)-NH2 | 8 | 16 | 64 | 16 | 85 |
| 22 | kk(1-nal)fk(1-nal)t(nle)-NH2 | 8 | 8 | 32 | 8 | 66 |
| 23 | kk(1-nal)fk(1-nal)y(nle)-NH2 | 8 | 4 | 64 | 8 | 74 |
| 24 | kk(1-nal)fk(1-nal)v(nle)-NH2 | 8 | 4 | 32 | 8 | 50 |
| 25 | kk(1-nal)fkk(1-nal)(nle)-NH2 | 128 | 64 | 32 | 32 | 50 |
| 26 | kk(1-nal)fk(1-nal)(nle)k-NH2 | 128 | 64 | 32 | 32 | 18 |
| 27 | k(1-nal)fk(1-nal)k(nle)-NH2 | 8 | 8 | 32 | 16 | 49 |
| 28 | (1-nal)fk(1-nal)k(nle)-NH2 | >128 | >128 | >128 | >128 | 62 |
| 29 | fk(1-nal)k(nle)-NH2 | >128 | >128 | 128 | >128 | 12 |
| 30 | kk(1-nal)fk(1-nal)k-NH2 | >128 | >128 | 128 | >128 | 11 |
| 31 | kk(1-nal)fk(1-nal)-NH2 | >128 | >128 | 128 | >128 | 20 |
| 32 | kk(1-nal)fk-NH2 | >128 | >128 | >128 | >128 | 5 |
| 33 | rk(1-nal)fk(1-nal)k(nle)-NH2 | 8 | 8 | 16 | 16 | 60 |
| 34 | kr(1-nal)fk(1-nal)k(nle)-NH2 | 8 | 4 | 16 | 16 | 35 |
| 35 | kk(1-nal)fr(1-nal)k(nle)-NH2 | 8 | 4 | 16 | 16 | 49 |
| 36 | kk(1-nal)fk(1-nal)r(nle)-NH2 | 8 | 2 | 16 | 8 | 51 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lone, A.; Thomsen, T.T.; Nielsen, J.E.; Thulstrup, P.W.; Klitgaard, R.N.; Løbner-Olesen, A.; Lund, R.; Jenssen, H.; Hansen, P.R. Structure-Activity Study of an All-d Antimicrobial Octapeptide D2D. Molecules 2019, 24, 4571. https://doi.org/10.3390/molecules24244571
Lone A, Thomsen TT, Nielsen JE, Thulstrup PW, Klitgaard RN, Løbner-Olesen A, Lund R, Jenssen H, Hansen PR. Structure-Activity Study of an All-d Antimicrobial Octapeptide D2D. Molecules. 2019; 24(24):4571. https://doi.org/10.3390/molecules24244571
Chicago/Turabian StyleLone, Abdullah, Thomas T. Thomsen, Josefine Eilsø Nielsen, Peter W. Thulstrup, Rasmus N. Klitgaard, Anders Løbner-Olesen, Reidar Lund, Håvard Jenssen, and Paul R. Hansen. 2019. "Structure-Activity Study of an All-d Antimicrobial Octapeptide D2D" Molecules 24, no. 24: 4571. https://doi.org/10.3390/molecules24244571
APA StyleLone, A., Thomsen, T. T., Nielsen, J. E., Thulstrup, P. W., Klitgaard, R. N., Løbner-Olesen, A., Lund, R., Jenssen, H., & Hansen, P. R. (2019). Structure-Activity Study of an All-d Antimicrobial Octapeptide D2D. Molecules, 24(24), 4571. https://doi.org/10.3390/molecules24244571