
Obtaining and Characterization of the PLA/Chitosan Foams with Antimicrobial Properties Achieved by the Emulsification Combined with the Dissolution of Chitosan by CO2 Saturation

 ,
,  , ,
, ,  ,
,
Abstract
1. Introduction
2. Results and Discussion
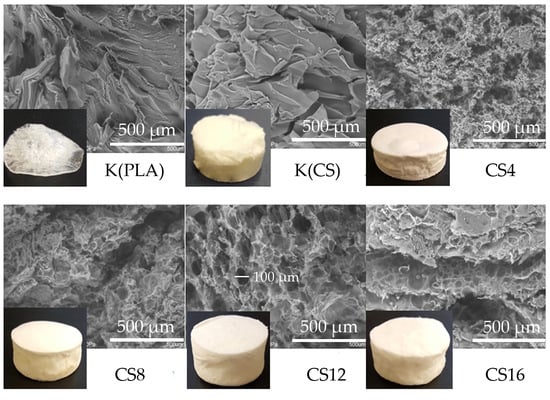
2.1. Structure and Physicochemical Properties
2.2. Swelling Capacity and Solubility
2.3. Thermal Properties
2.4. Antimicrobial Properties
2.5. Biocompatibility
3. Materials and Methods
3.1. Materials
3.2. Foams Preparation
3.3. Foams Characterization
3.3.1. Topography Evaluation
3.3.2. Porosity and Density Measurements
3.3.3. Fourier-transform Infrared Spectroscopy (FT-IR) Study
3.3.4. Swelling Capacity/Dissolution Evaluation
3.3.5. Mechanical Properties
3.3.6. Thermal Properties
3.3.7. Antimicrobial Properties
3.3.8. Biocompatibility Tests
Cell Culture
Sample Preparation
Cell Viability
3.3.9. Statistical Analyses
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Chanjuan, D.; Yonggang, L.V. Application of collagen scaffold in tissue engineering: Recent advances and new perspectives. Polymers 2016, 8, 42. [Google Scholar] [CrossRef]
- Wells, C.M.; Harris, M.; Choi, L.; Murali, V.P.; Delbuque Guerra, F.; Jennings, J.A. Muli-Responsive Drug Release from Smart Polymers. J. Funct. Biomater. 2019, 10, 34. [Google Scholar] [CrossRef] [PubMed]
- Tylingo, R.; Gorczyca, G.; Mania, S.; Szweda, P.; Milewski, S. Preparation and characterization of porous scaffolds from chitosan-collagen-gelatin composite. React. Funct. Polym. 2016, 103, 131–140. [Google Scholar] [CrossRef]
- Yao, Z.-A.; Chen, F.-J.; Cui, H.-L.; Lin, T.; Guo, N.; Wu, H.-G. Efficacy of chitosan and sodium alginate scaffolds for repair of spinal cord injury in rats. Neural. Regen. Res. 2018, 13, 502–509. [Google Scholar] [CrossRef] [PubMed]
- Gunatillake, P.A.; Adhikari, R. Biodegradable synthetic polymers for tissue engineering. Eur. Cell Mater 2003, 5, 1–16. [Google Scholar] [CrossRef]
- Munirah, S.; Kim, S.H.; Ruszymah, B.H.I.; Khang, G. The use of fibrin and poly (lactic-co-glycolic acid) hybrid scaffold for articular cartilage tissue engineering: An in vivo analysis. Eur. Cell Mater. 2008, 15, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Pal, A.K.; Das, A.; Katiyar, V. Chitosan from Muga silkworms (Antheraea assamensis) and its influence on thermal degradation behavior of poly(lactic acid) based biocomposite films. J. Appl. Polym. Sci. 2016, 133, 43710. [Google Scholar] [CrossRef]
- Dhar, P.; Tarafder, D.; Kumar, A.; Katiyar, V. Effect of cellulose nanocrystal polymorphs on mechanical, barrier and thermal properties of poly(lactic acid) based bionanocomposites. RSC Adv. 2015, 5, 60426–60440. [Google Scholar] [CrossRef]
- Borkotoky, S.S.; Dhar, P.; Katiyar, V. Biodegradable poly (lactic acid)/cellulose nanocrystals (CNCs) composite microcellular foam: Effect of nanofillers on foam cellular morphology, thermal and wettability behavior. Int. J. Biol. Macromol. 2018, 106, 433–446. [Google Scholar] [CrossRef]
- Zargar, V.; Asghari, M.; Dashti, A. A review on chitin and chitosan polymers: Structure, chemistry, solubility, derivatives, and applications. ChemBioEng Rev. 2015, 2, 1–24. [Google Scholar] [CrossRef]
- Muzzarelli, R.A.A. Chitins and chitosans for the repair of wounded skin. Carbohydr. Polym. 2009, 76, 167–182. [Google Scholar] [CrossRef]
- Liu, X.; Ma, L.; Mao, Z.; Gao, C. Chitosan-Based Biomaterials for Tissue Repair and Regeneration. Adv. Polym. Sci. 2011, 244, 81–128. [Google Scholar] [CrossRef]
- Mania, S.; Tylingo, R.; Augustin, E.; Gucwa, K.; Szwacki, J.; Staroszczyk, H. Investigation of an elutable N-propylphosphonic acid chitosan derivative composition with a chitosan matrix prepared from carbonic acid solution. Carbohydr. Polym. 2018, 179, 196–206. [Google Scholar] [CrossRef] [PubMed]
- Rinaudo, M. Chitin and chitosan: Properties and applications. Prog. Polym. Sci. 2006, 31, 603–632. [Google Scholar] [CrossRef]
- Malinowska-Pańczyk, E.; Staroszczyk, H.; Gottfried, K.; Kołodziejska, I.; Wojtasz-Pająk, A. Antimicrobial properties of chitosan solutions, chitosan films and gelatin-chitosan films. Polimery 2015, 60, 735–740. [Google Scholar] [CrossRef]
- Movaffagh, J.; Fazly Bazzaz, B.S.; Yazdi, A.T.; Sajadi-Tabassi, A.; Azizzadeh, M.; Najafi, E.; Amiri, N.; Taghanaki, H.B.; Ebrahimzadeh, M.H.; Moradi, A. Wound Healing and Antimicrobial Effects of Chitosan-hydrogel/Honey Compounds in a Rat Full-thickness Wound Model. Wounds 2019, 31, 228–235. [Google Scholar]
- Phaechamud, T.; Charoenteeraboon, J. Antibacterial Activity and Drug Release of Chitosan Sponge Containing Doxycycline Hyclate. AAPS PharmSciTech. 2008, 9, 829–835. [Google Scholar] [CrossRef]
- Goy, R.C.; Morais, S.T.B.; Assis, O.B.G. Evaluation of the antimicrobial activity of chitosan and its quaternized derivative on E. coli and S. aureus growth. Rev. Bras. Farmacogn. 2016, 26, 122–127. [Google Scholar] [CrossRef]
- Sebti, I.; Martial-Gros, A.; Carnet-Pantiez, A.; Grelier, S.; Coma, V. Chitosan polymer as bioactive coating and film against Aspergillus niger contamination. J. Food Sci. 2005, 70, 100–104. [Google Scholar] [CrossRef]
- Raafat, D.; von Bargen, K.; Haas, A.; Sahl, H.G. Insights into the mode of action of chitosan as an antibacterial compound. Appl. Environmen. Microbiol. 2008, 74, 3764–3773. [Google Scholar] [CrossRef]
- Pal, A.K.; Katiyar, V. Thermal degradation behaviour of nanoamphiphilic chitosan dispersed poly (lactic acid) bionanocomposite films. Int. J. Biol. Macromol. 2017, 95, 1267–1279. [Google Scholar] [CrossRef] [PubMed]
- Haaparanta, A.M.; Järvinen, E.; Cengiz, I.F.; Ellä, V.; Kokkonen, H.T.; Kiviranta, I.; Kellomäki, M. Preparation and characterization of collagen/PLA, chitosan/PLA, and collagen/chitosan/PLA hybrid scaffolds for cartilage tissue engineering. J. Mater. Sci. Mater. Med. 2014, 25, 1129–1136. [Google Scholar] [CrossRef] [PubMed]
- Suryani; Agusnar, H.; Wirjosentono, B.; Rihayat, T.; Salisah, Z. Synthesis and characterization of poly(lactid acid)/chitosan nanocomposites based on renewable resources as biobased-material. J. Phys. Conf. 2018, 953, 012015. [Google Scholar] [CrossRef]
- Ali Raza, Z.; Anwar, F. Fabrication of poly(lactic acid) incorporated chitosan nanocomposites for enhanced functional polyester fabric. Polímeros 2018, 28, 120–124. [Google Scholar] [CrossRef]
- Hijazi, N.; Le Moigne, N.; Rodier, E.; Letourneau, J.J.; Sauceau, M.; Fages, J.; Guibal, E.; Vincent, T.; Benezet, J.C. Development of nanostructured film based on PLA and chitosan nanoparticles generated by supercritical CO2 assisted processes. In Proceedings of the ECCM16-16th European Conference on Composite Materials, Seville, Spain, 22–26 June 2014. [Google Scholar]
- Kazimierczak, P.; Palka, K.; Przekora, A. Development and Optimization of the Novel Fabrication Method of Highly Macroporous Chitosan/Agarose/Nanohydroxyapatite Bone Scaffold for Potential Regenerative Medicine Applications. Biomolecules 2019, 9, 434. [Google Scholar] [CrossRef]
- Bryśkiewicz, A.; Zieleniewska, M.; Przyjemska, K.; Chojnacki, P.; Ryszkowska, J. Modification of flexible polyurethane foams by the addition of natural origin fillers. Polym. Degrad. Stabil. 2016, 132, 32–40. [Google Scholar] [CrossRef]
- Lin, Y.; Hsieh, F.; Huff, H.E. Water-blown flexible polyurethane foam extended with biomass materials. J. Appl. Polym. Sci. 1997, 695–703. [Google Scholar] [CrossRef]
- Auras, R.; Harte, B.; Selke, S. An Overview of Polylactides as Packaging Materials. Macromol. Biosci. 2004, 4, 835–864. [Google Scholar] [CrossRef]
- Mathias, J.-D.; Tessier-Doyen, N.; Michaud, P. Development of a Chitosan-Based Biofoam: Application to the Processing of a Porous Ceramic Material. Int. J. Mol. Sci. 2011, 12, 1175–1186. [Google Scholar] [CrossRef]
- Karageorgiou, V.; Kaplan, D. Porosity of 3D biomaterial scaffolds and osteogenesis. Biomaterials 2005, 26, 5474–5491. [Google Scholar] [CrossRef]
- Gorczyca, G.; Tylingo, R.; Szweda, P.; Augustin, E.; Sadowska, M.; Milewski, S. Preparation and characterization of genipin cross-linked porous chitosan–collagen–gelatin scaffolds using chitosan–CO2 solution. Carbohyd. Polym. 2014, 102, 901–911. [Google Scholar] [CrossRef] [PubMed]
- Guarino, V.; Causa, F.; Ambrosio, L. Porosity and mechanical properties relationship in PCL porous scaffold. J. Appl. Biomater. 2007, 5, 149–157. [Google Scholar] [CrossRef]
- Mohanty, A.K.; Misra, M.; Drzal, L.T. Natural Fibers, Biopolymers, and Biocomposites; CRC Press: Boca Raton, FL, USA, 2005; pp. 2–31. [Google Scholar]
- Popa, E.E.; Rapa, M.; Popa, O.; Mustatea, G.; Popa, V.I.; Mitelut, A.C.; Popa, M.E. Polylactic Acid/Cellulose Fibres Based Composites for Food Packaging Applications. Mater. Plast. 2017, 54, 673–677. [Google Scholar]
- Daver, F.; Marcian Lee, K.P.; Brandt, M.; Shanks, R. Cork–PLA composite filaments for fused deposition modelling. Compos. Sci. Technol. 2018, 168, 230–237. [Google Scholar] [CrossRef]
- Chieng, B.; Ibrahim, N.; Yunus, W.; Hussein, M. Poly(lactic acid)/Poly(ethylene glycol) Polymer Nanocomposites: Effects of Graphene Nanoplatelets. Polymers 2013, 6, 93–104. [Google Scholar] [CrossRef]
- Khairuddin; Pramono, E.; Utomo, S.B.; Wulandari, V.; Zahrotul, W.A.; Clegg, F. FTIR studies on the effect of concentration of polyethylene glycol on polimerization of Shellac. J. Phys.: Conf. Ser. 2016, 776, 012053. [Google Scholar] [CrossRef]
- Tiğh, R.S.; Karakecili, A. In vitro characterization of chitosan scaffolds: Influence of composition and deacethylation degree. J. Mater. Sci. Mater. Med. 2007, 18, 1665–1674. [Google Scholar] [CrossRef]
- Dimida, S.; Demitri, C.; De Benedicti, V.M.; Scalera, F.; Gervaso, F.; Sannino, A. Genipin-cross-linked chitosan-based hydrogels: Reaction kinetics and structure-related characteristics. J. Appl. Polym. Sci. 2015, 132, 42256. [Google Scholar] [CrossRef]
- Xiang, Q.; Ren, Y.; Wang, X. New advances in the biodegradation of Poly(lactic) acid. Int. Biodeter. Biodegr. 2017, 117, 215–223. [Google Scholar] [CrossRef]
- Carrasco, F.; Pagès, P.; Gámez-Pérez, J.; Santana, O.O.; Maspoch, M.L. Processing of poly(lactic acid): Characterization of chemical structure, thermal stability and mechanical properties. Polym. Degrad. Stabil. 2010, 95, 116–125. [Google Scholar] [CrossRef]
- Mohapatra, A.K.; Mohanty, S.; Nayak, S.K. Effect of PEG on PLA/PEG Blend and Its Nanocomposites: A Study of Thermo-Mechanical and Morphological Characterization. Polym. Compos. 2013, 35, 283–293. [Google Scholar] [CrossRef]
- Bijarimi, M.; Ahmad, S.; Rasid, R.; Khushairi, M.A.; Zakir, M. Poly (lactic acid)/Poly (ethylene glycol) blends: Mechanical, thermal and morphological properties. AIP Conf. Proc. 2016, 1727, 020002. [Google Scholar] [CrossRef]
- Sungsanit, K.; Kao, N.; Bhattacharya, S.N. Properties of linear poly(lactic acid)/polyethylene glycol blends. Polym. Eng. Sci. 2011, 52, 108–116. [Google Scholar] [CrossRef]
- Noootsuwan, N.; Wattanathana, W.; Jongrungruangchok, S.; Veranitisagul, C.; Koonsaeng, N.; Laobuthee, A. Development of novel hybrid materials from polylactic acid and nano-silver coated carbon black with distinct antimicrobial and electrical properties. J. Polym. Res. 2018, 25, 90. [Google Scholar] [CrossRef]
- Damian, L.; Paţchia, S. Method for Testing the Antimicrobial Character of the Materials and Their Fitting to the Scope. Bull. Transylv. Univ. Braşov. 2014, 7, 37–44. [Google Scholar] [CrossRef]
- Rapacz-Kmita, A.; Pierchała, M.K.; Tomas-Trybuś, A.; Szaraniec, B.; Karwot, J. The wettability, mechanical and antimicrobial properties of polylactide/montmorillonite nanocomposite films. Acta Bioeng. Biomech. 2017, 19, 25–33. [Google Scholar] [CrossRef]
- Turalija, M.; Bischof, S.; Budimir, A.; Gaan, S. Antimicrobial PLA films from environment. Friendly additives. Compos. Part B-Eng. 2016, 102, 94–99. [Google Scholar] [CrossRef]
- Pariente, J.L.; Kim, B.S.; Atala, A. In vitro biocompatibility assessment ofnaturally derived and synthetic biomaterials using normal human urothelial cells. J. Biomed. Mater. Res. 2001, 55, 33–39. [Google Scholar] [CrossRef]
- Zhang, R.Y.; Ma, P.X. Poly(α-hydroxyl acids)/hydroxyapatite porous composites for bone-tissue engineering. I. Preparation and morphology. J. Biomed. Mater. Res. 1999, 44, 446–455. [Google Scholar] [CrossRef]
- Hoyer, B.; Bernhardt, A.; Heinemann, S.; Stachel, I.; Meyer, M.; Gelinsky, M. Biomimetically mineralized salmon collagen scaffolds for application in bone tissue engineering. Biomacromolecules 2012, 13, 1059–1066. [Google Scholar] [CrossRef]
- Bi, L.; Cao, Z.; Hu, Y.; Song, Y.; Yu, L.; Yang, B.; Mu, J.; Huang, Z.; Han, Y. Effects of different cross-linking conditions on the properties of genipin-cross-linked chitosan/collagen scaffolds for cartilage tissue engineering. J. Mater. Sci. Mater. Med. 2011, 22, 51–62. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the PLA/CS/PEG foams are available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Density [kg/m3] | Porosity [%] |
|---|---|---|
| PLA | 1250 ± 6 a | Nd. |
| CS | 49 ± 7 b | 80.4 ± 2.2 A |
| CS4 | 168 ± 8 c | 53.1 ± 2.0 B |
| CS8 | 92 ± 10 d | 60.5 ± 3.1 C |
| CS12 | 69 ± 9 d | 73.9 ± 3.4 D |
| CS16 | 55 ± 11 b | 85.3 ± 2.7 A |
| Sample | Tg [°C] | Tc [°C] | Tm [°C] |
|---|---|---|---|
| PLA | 65.7 | - | 156.3 |
| CS4 | 41.9 | 77.6 | 149.8 |
| CS8 | 39.7 | 82.1 | 150.5 |
| CS12 | 39.0 | 82.4 | 148.6 |
| CS16 | 39.6 | 84.1 | 149.2 |
| Sample | Time of Interaction [h] | Average Number of Cells [cfu/mL] | |
|---|---|---|---|
| S. Aureus | E. Coli | ||
| K(-) | 0 | 1.16 × 105 | 2.36 × 105 |
| K(-) | 24 | 3.12 × 107 | 1.54 × 107 |
| PLA | 24 | 3.31 × 107 | 2.00 × 107 |
| CS | 24 | 1.02 × 102 | 1.23 × 103 |
| CS4 | 24 | 8.45 × 105 | 2.20 × 105 |
| CS8 | 24 | 3.25 × 105 | 1.16 × 105 |
| CS12 | 24 | 1.03 × 105 | 5.85 × 104 |
| CS16 | 24 | 3.56 × 104 | 1.28 × 104 |
| Sample | Concentration of Polymer in Foam [% w/w] | ||
|---|---|---|---|
| PLA | Chitosan | PEG | |
| PLA | 100 | 0 | 0 |
| CS | 0 | 100 | 0 |
| CS4 | 73 | 4 | 23 |
| CS8 | 69 | 8 | 23 |
| CS12 | 65 | 12 | 23 |
| CS16 | 61 | 16 | 23 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mania, S.; Partyka, K.; Pilch, J.; Augustin, E.; Cieślik, M.; Ryl, J.; Jinn, J.-R.; Wang, Y.-J.; Michałowska, A.; Tylingo, R. Obtaining and Characterization of the PLA/Chitosan Foams with Antimicrobial Properties Achieved by the Emulsification Combined with the Dissolution of Chitosan by CO2 Saturation. Molecules 2019, 24, 4532. https://doi.org/10.3390/molecules24244532
Mania S, Partyka K, Pilch J, Augustin E, Cieślik M, Ryl J, Jinn J-R, Wang Y-J, Michałowska A, Tylingo R. Obtaining and Characterization of the PLA/Chitosan Foams with Antimicrobial Properties Achieved by the Emulsification Combined with the Dissolution of Chitosan by CO2 Saturation. Molecules. 2019; 24(24):4532. https://doi.org/10.3390/molecules24244532
Chicago/Turabian StyleMania, Szymon, Karolina Partyka, Joanna Pilch, Ewa Augustin, Mateusz Cieślik, Jacek Ryl, Jia-Rong Jinn, Ya-Jane Wang, Anna Michałowska, and Robert Tylingo. 2019. "Obtaining and Characterization of the PLA/Chitosan Foams with Antimicrobial Properties Achieved by the Emulsification Combined with the Dissolution of Chitosan by CO2 Saturation" Molecules 24, no. 24: 4532. https://doi.org/10.3390/molecules24244532
APA StyleMania, S., Partyka, K., Pilch, J., Augustin, E., Cieślik, M., Ryl, J., Jinn, J.-R., Wang, Y.-J., Michałowska, A., & Tylingo, R. (2019). Obtaining and Characterization of the PLA/Chitosan Foams with Antimicrobial Properties Achieved by the Emulsification Combined with the Dissolution of Chitosan by CO2 Saturation. Molecules, 24(24), 4532. https://doi.org/10.3390/molecules24244532