Author Contributions
Conceptualization, J.Y.K. and K.-J.P.; methodology, Y.-K.J. and S.Y.Y.; validation, S.Y.Y. and S.Y.K.; writing—original draft preparation, Y.-K.J., and K.-J.P.; writing—review and editing, Y.-K.J., J.Y.K and K.-J.P.; supervision, K.-J.P.
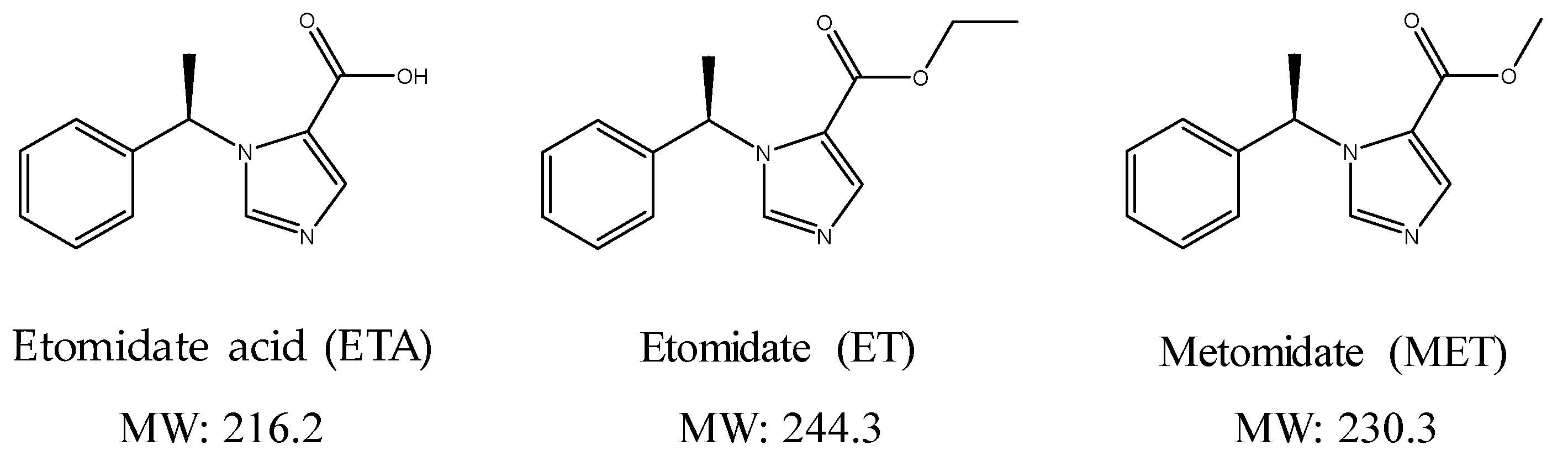
Figure 1.
Chemical structures and molecular weights of the analytes (etomidate acid, etomidate) and internal standard (metomidate).
Figure 1.
Chemical structures and molecular weights of the analytes (etomidate acid, etomidate) and internal standard (metomidate).
Figure 2.
Chromatograms for the separation of etomidate acid and etomidate using four analytical columns: (a) C18 column (X-bridge C18, 50 × 2.1 mm I.D., 3.5 μm, Waters Corporation), (b) multi-mode C18 column (Scherzo SM-C18, 30 × 2.0 mm I.D., 3.0 μm, Imtakt) (c) silica-based polar end-capped C18 column (Hypurity Aquastar , 50 × 2.1 mm I.D., 5.0 μm , Thermo Fisher Scientific), and (d) porous graphitic carbon column (Hypercarb, 50 × 2.1 mm I.D., 5.0 μm, Thermo Fisher Scientific). Solvent A (0.05 % formic acid in water) and solvent B (50%:50% acetonitrile:methanol) were used for analysis. The gradient elution included 15% B for 0.4 min, 85% B for 3 min to 5 min, and re-equilibration to 15% B for 7.5.
Figure 2.
Chromatograms for the separation of etomidate acid and etomidate using four analytical columns: (a) C18 column (X-bridge C18, 50 × 2.1 mm I.D., 3.5 μm, Waters Corporation), (b) multi-mode C18 column (Scherzo SM-C18, 30 × 2.0 mm I.D., 3.0 μm, Imtakt) (c) silica-based polar end-capped C18 column (Hypurity Aquastar , 50 × 2.1 mm I.D., 5.0 μm , Thermo Fisher Scientific), and (d) porous graphitic carbon column (Hypercarb, 50 × 2.1 mm I.D., 5.0 μm, Thermo Fisher Scientific). Solvent A (0.05 % formic acid in water) and solvent B (50%:50% acetonitrile:methanol) were used for analysis. The gradient elution included 15% B for 0.4 min, 85% B for 3 min to 5 min, and re-equilibration to 15% B for 7.5.
Figure 3.
Effect of the changing ratio of acetonitrile and methanol: (a) acetonitrile, (b) acetonitrile:methanol (v:v, 75:25), (c) acetonitrile:methanol (v:v, 50:50), (d) acetonitrile:methanol (v:v, 25:75), (e) methanol.
Figure 3.
Effect of the changing ratio of acetonitrile and methanol: (a) acetonitrile, (b) acetonitrile:methanol (v:v, 75:25), (c) acetonitrile:methanol (v:v, 50:50), (d) acetonitrile:methanol (v:v, 25:75), (e) methanol.
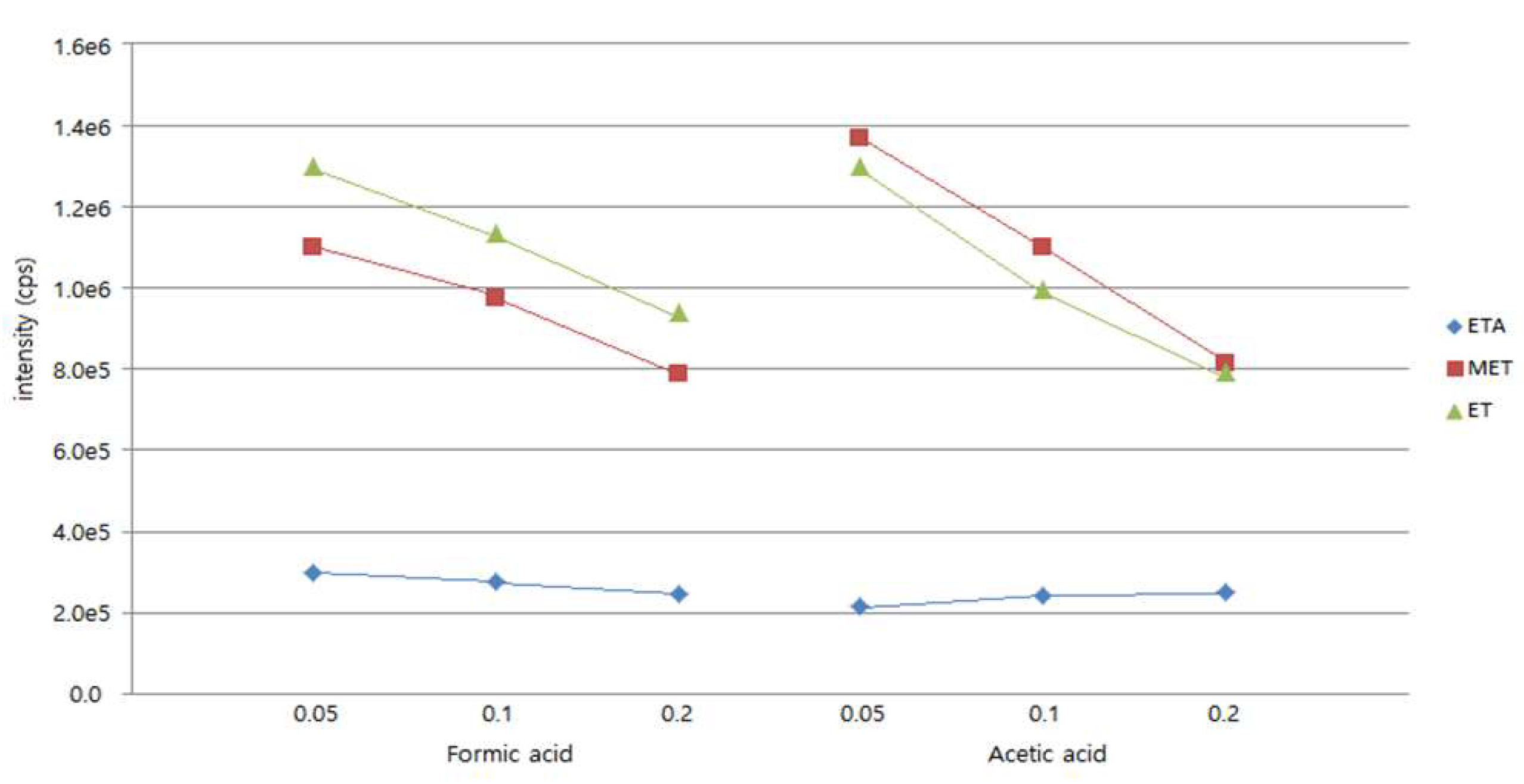
Figure 4.
Effect of the concentration of charged additive.
Figure 4.
Effect of the concentration of charged additive.
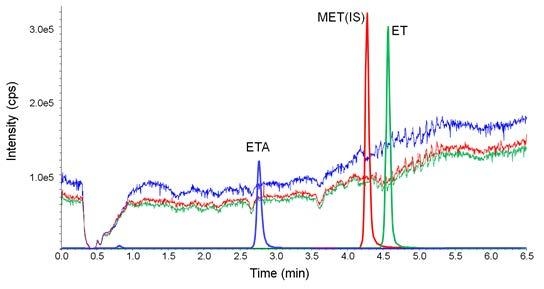
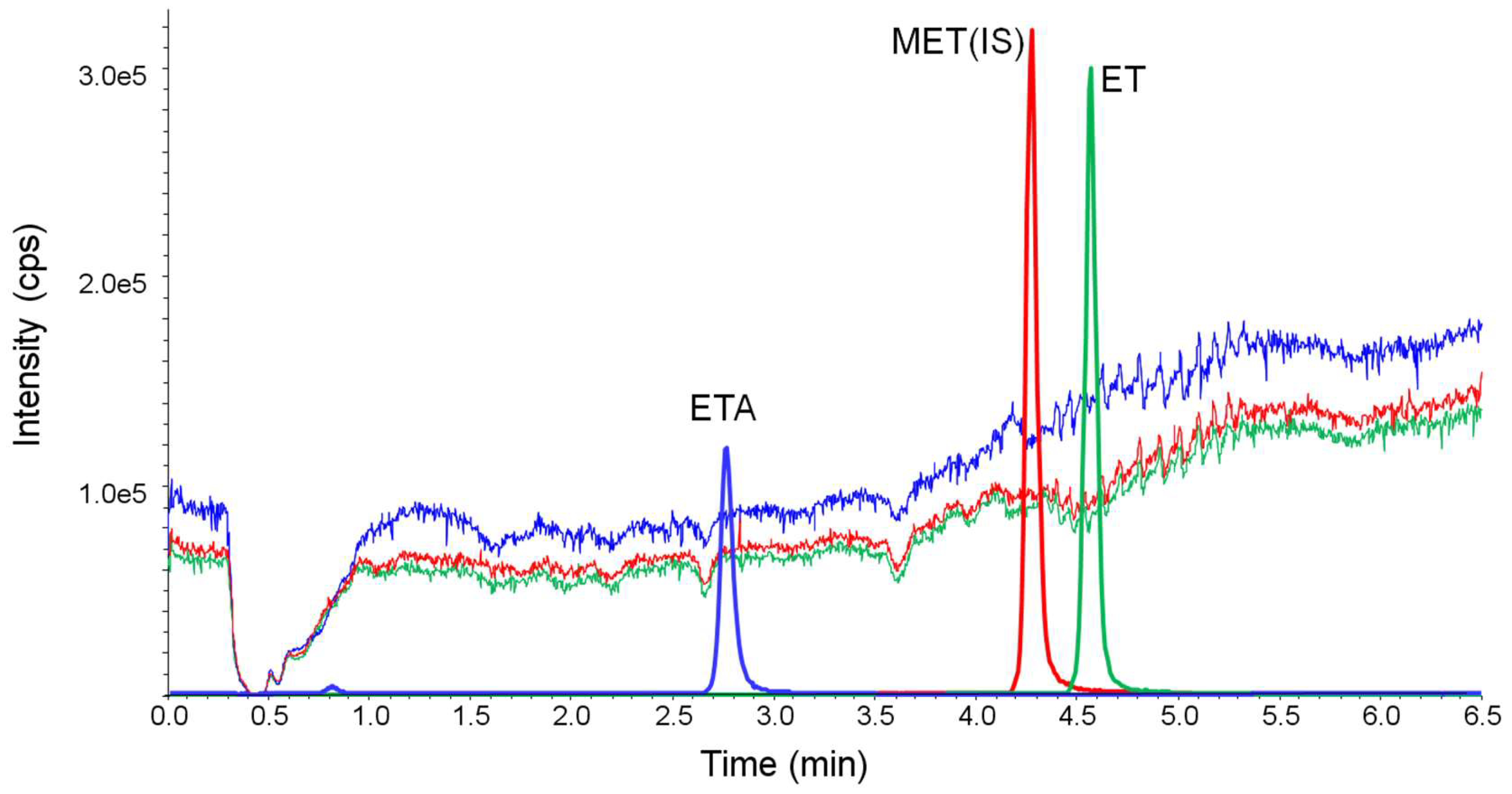
Figure 5.
Ion suppression studied by monitoring the multiple reaction monitoring (MRM) chromatograms during post-column infusion of 30 ng/min of etomidate acid (ETA), 2 ng/min of metomidate (MET, internal standard, IS) and ET, and subsequent injection of the diluted urine sample.
Figure 5.
Ion suppression studied by monitoring the multiple reaction monitoring (MRM) chromatograms during post-column infusion of 30 ng/min of etomidate acid (ETA), 2 ng/min of metomidate (MET, internal standard, IS) and ET, and subsequent injection of the diluted urine sample.
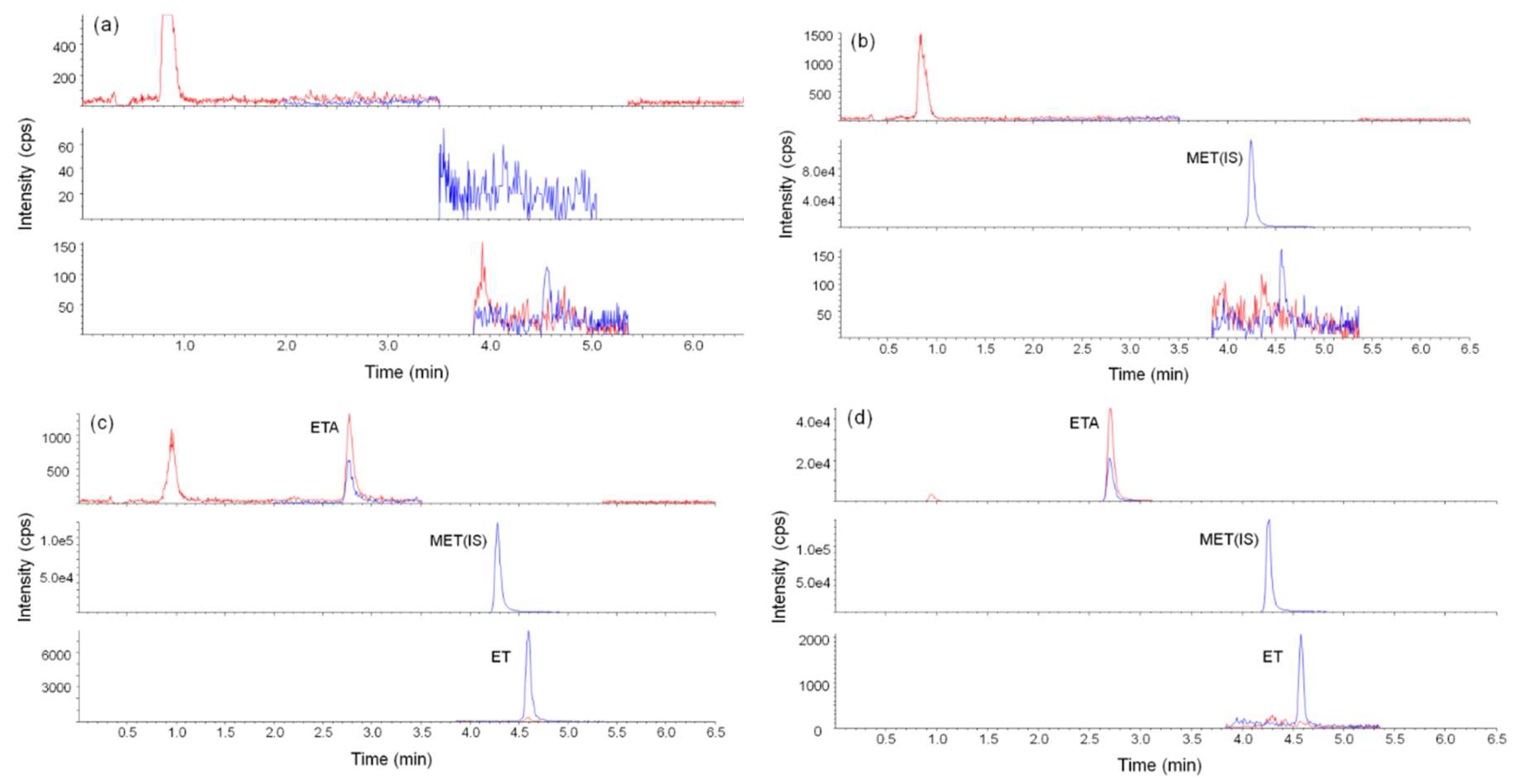
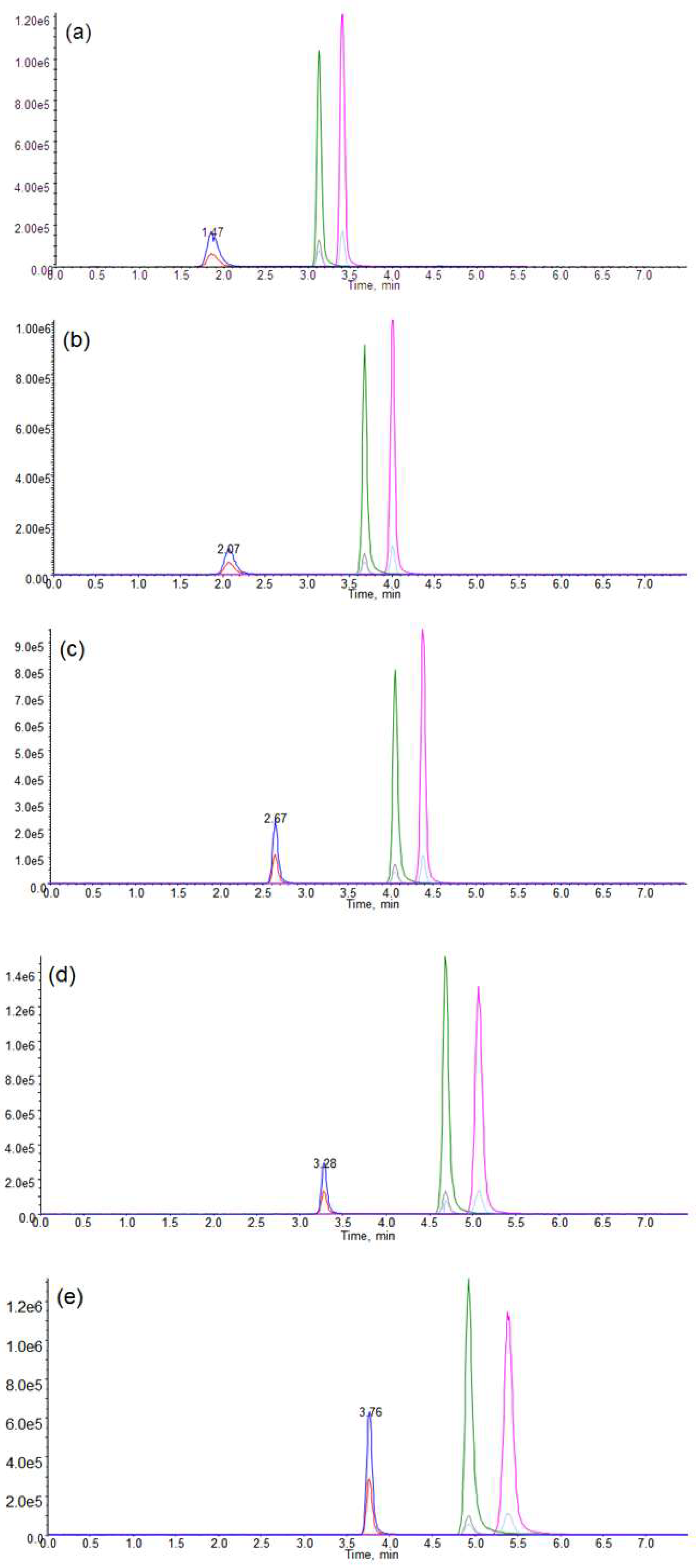
Figure 6.
Representative liquid chromatography–tandem mass spectrometry (LC–MS/MS) chromatograms for the (a) blank (b) blank with metomidate (internal standard (IS)) (c) blank with IS, etomidate and etomidate acid, and (d) etomidate-positive urine sample.
Figure 6.
Representative liquid chromatography–tandem mass spectrometry (LC–MS/MS) chromatograms for the (a) blank (b) blank with metomidate (internal standard (IS)) (c) blank with IS, etomidate and etomidate acid, and (d) etomidate-positive urine sample.
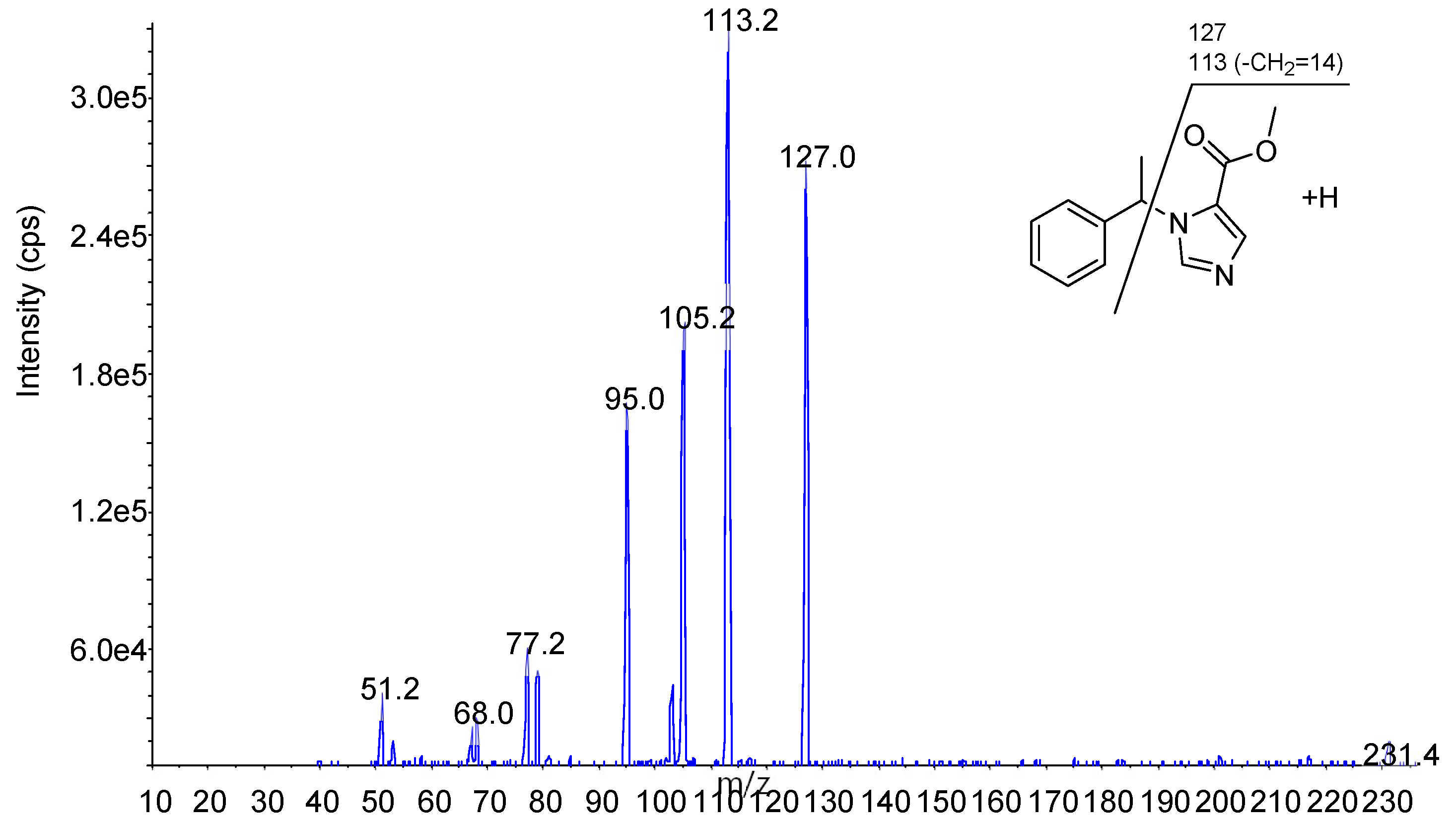
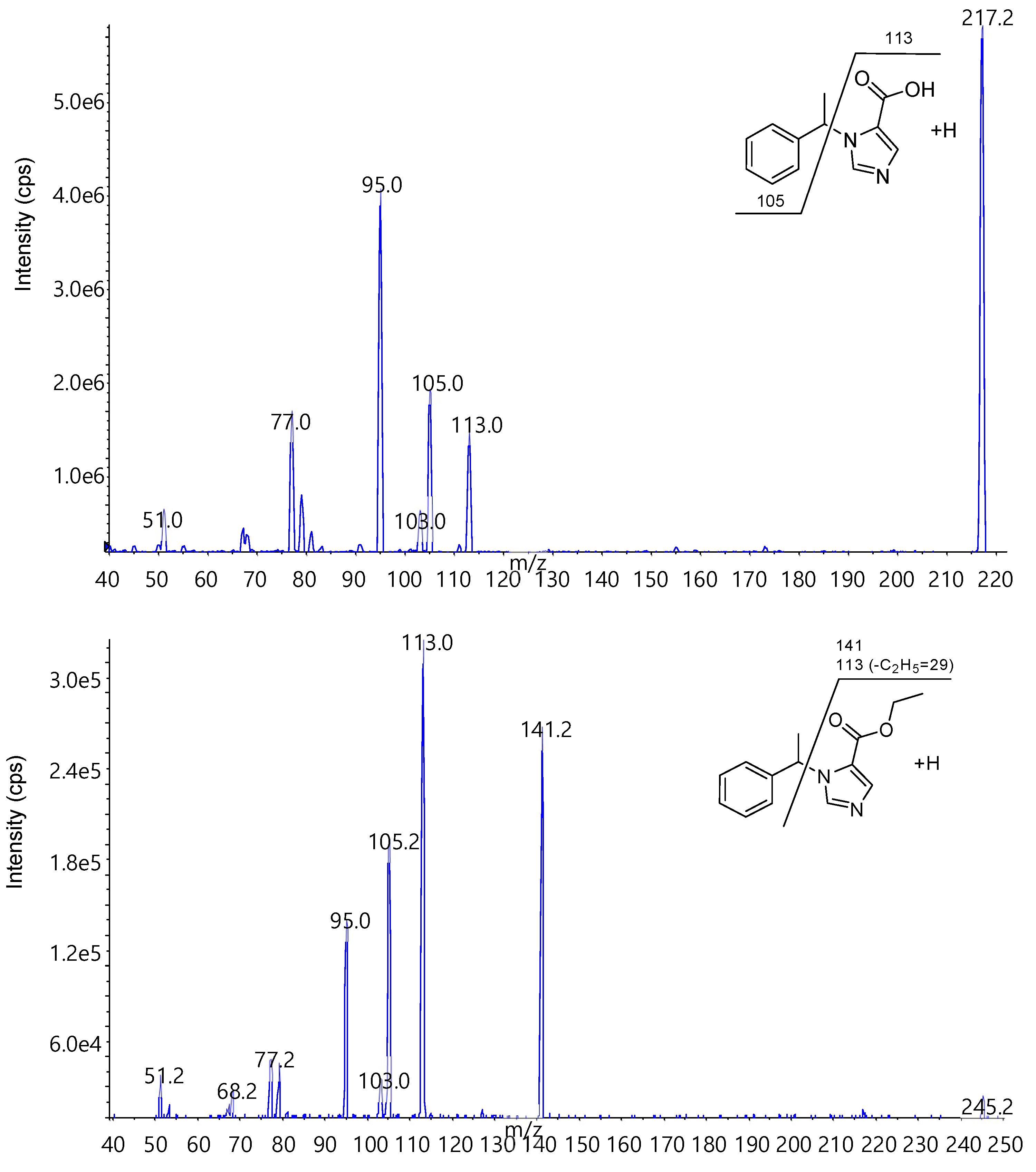
Figure 7.
Precursor and product ion of each compound.
Figure 7.
Precursor and product ion of each compound.
Table 1.
Gradient program condition for the separation of analytes.
Table 1.
Gradient program condition for the separation of analytes.
| Total Time (min) | Flow Rate (μL/min) | Mobile Phase |
|---|
| Solvent A (%) a | Solvent B (%) b |
|---|
| 1 | 400 | 85 | 15 |
| 0.4 | 400 | 85 | 15 |
| 3 | 450 | 15 | 85 |
| 5 | 450 | 15 | 85 |
| 5.1 | 400 | 85 | 15 |
| 7.5 | 400 | 85 | 15 |
Table 2.
Validation parameters for the calibration curves of ETS and ET.
Table 2.
Validation parameters for the calibration curves of ETS and ET.
| Analyte | Concentration Range (ng/mL) | Slope (mean ± SD) | y-Intercept (mean) | Linearity a (R2) | LOD b (ng/mL) | LLOQ c (ng/mL) |
|---|
| Etomidate acid | 1.0–300.0 | 0.0124 ± 0.0008 | 0.0004 | 0.9958 | 0.03 | 1.0 |
| Etomidate | 0.4–120.0 | 0.1148 ± 0.0098 | 0.0051 | 0.9963 | 0.01 | 0.4 |
Table 3.
Intra- and inter-day precision and accuracy.
Table 3.
Intra- and inter-day precision and accuracy.
| Analyte | Nominal Concentration (ng/mL) | Intra-Day (n = 18) | Inter-Day (n = 24) |
|---|
| Precision a (% CV) | Accuracy b (% bias) | Precision (% CV) | Accuracy (% bias) |
|---|
| Etomidate acid | 1.0 | 10.2 | 2.9 | 8.4 | −6.4 |
| | 3.0 | 4.7 | 2.4 | 6.0 | −4.1 |
| | 30.0 | 2.5 | −0.2 | 2.3 | −1.3 |
| | 90.0 | 1.7 | −7.4 | 2.7 | −7.0 |
| Etomidate | 0.4 | 6.3 | −9.9 | 4.1 | −5.5 |
| | 1.2 | 3.3 | −3.7 | 3.3 | 0.6 |
| | 12.0 | 2.0 | −4.0 | 4.0 | 1.3 |
| | 36.0 | 4.2 | −8.6 | 3.0 | −6.1 |
Table 4.
Stability test results for bench-top stability, long-term stability, and auto-sampler stability.
Table 4.
Stability test results for bench-top stability, long-term stability, and auto-sampler stability.
| Compound | Concentration (ng/mL) | Bench-Top Stability (%) | Long-Term Stability (%) | Auto-Sampler Stability (%) |
|---|
| (Room Temperature for 6 h) | (4 °C for 7 days) | (4 °C for 14 days) | (4 °C for 12 h) |
|---|
| Etomidate acid | 3.0 | −11.0 | −13.3 | −20.9 | −12.5 |
| | 90.0 | −10.5 | −8.3 | −11.8 | −9.3 |
| Etomidate | 1.2 | −0.5 | −7.5 | −9.7 | −7.0 |
| | 36.0 | −4.7 | −5.3 | −5.7 | −4.5 |
Table 5.
Urinary concentrations (ng/mL) of etomidate acid and etomidate for authentic samples.
Table 5.
Urinary concentrations (ng/mL) of etomidate acid and etomidate for authentic samples.
| Sample# | Etomidate Acid Concentration (ng/mL) | Etomidate Concentration (ng/mL) |
|---|
| # 1 | 23.79 | < LLOQ |
| # 2 | 31.38 | < LLOQ |
Table 6.
Retention times, MRM transitions and compound dependent parameters for LC–MS/MS analysis of the analytes and internal standard (IS).
Table 6.
Retention times, MRM transitions and compound dependent parameters for LC–MS/MS analysis of the analytes and internal standard (IS).
| Analyte | RT a (min) | MRM Transition | DP b | EP c | CEP d | CE e | CXP f |
|---|
| Q1 | Q3 |
|---|
| Etomidate acid | 2.75 | 217.1 | 113.1 | 24 | 8.5 | 12 | 18 | 6 |
| 105.2 | 24 | 8.5 | 12 | 37 | 6 |
| Metomidate (IS) | 4.29 | 231.2 | 127.1 | 26 | 4 | 12 | 15 | 4 |
| 113.2 | 26 | 4 | 12 | 31 | 4 |
| Etomidate | 4.6 | 245.2 | 113.2 | 26 | 4.5 | 12 | 27 | 4 |
| 141.2 | 26 | 4.5 | 12 | 15 | 6 |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}