
Mimicking Strategy for Protein–Protein Interaction Inhibitor Discovery by Virtual Screening




Abstract
:1. Introduction
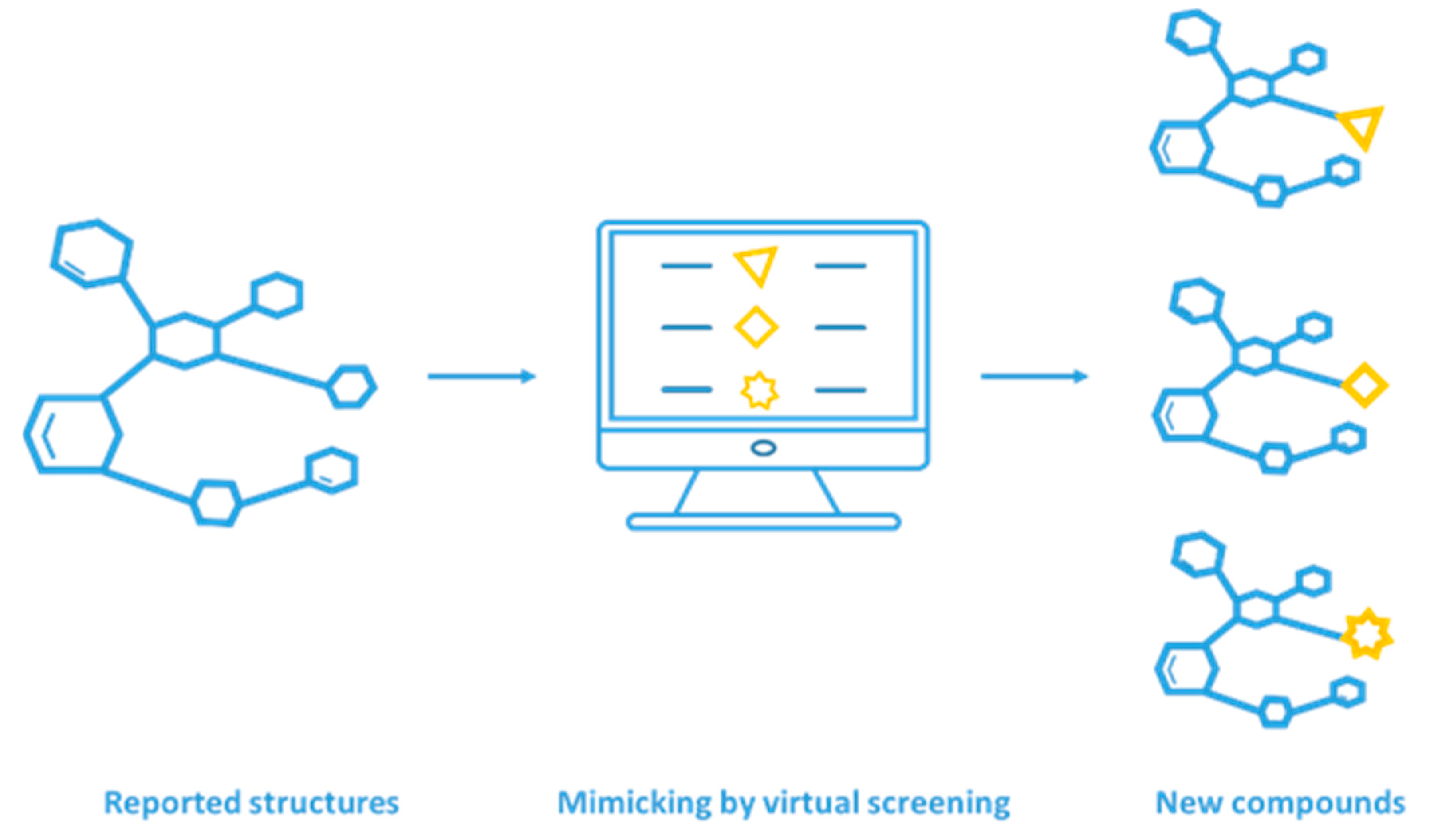
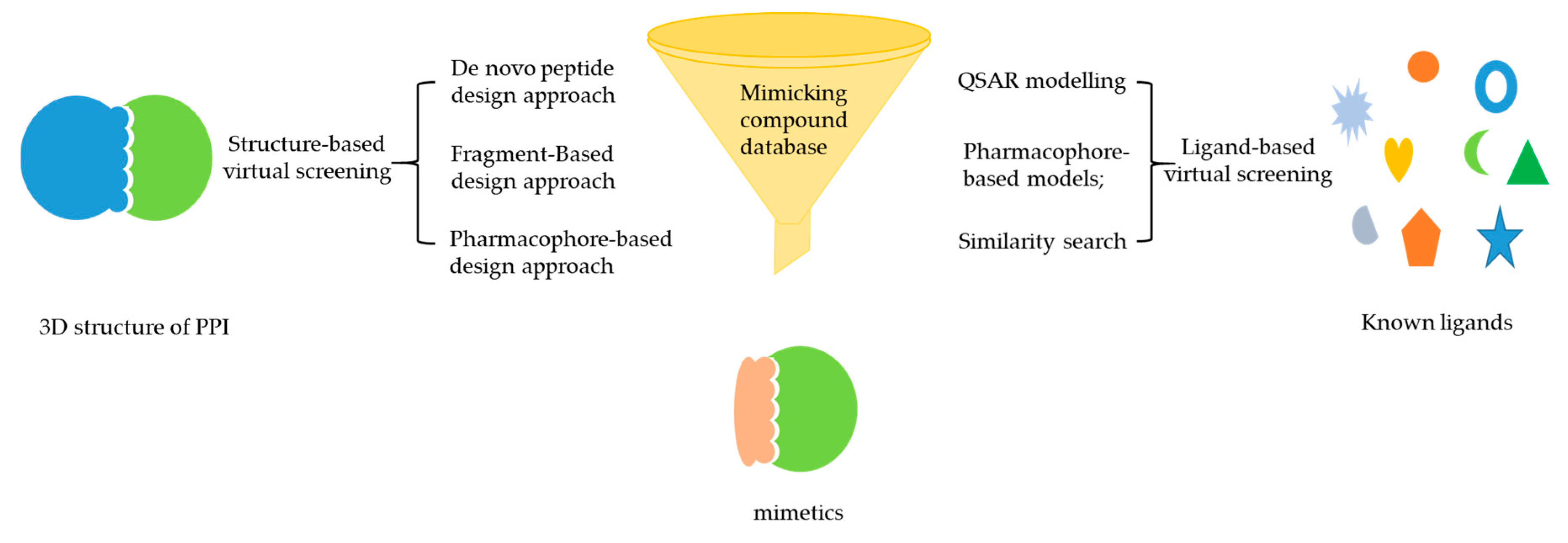
2. Integrating Mimicking and Virtual Screening Strategy for Protein–Protein Interaction Inhibitor Discovery
2.1. Virtual Screening for PPI Inhibitor Discovery
2.2. Structure-Based Mimicking Peptide Strategy for PPI Inhibitor Discovery
2.3. Integration of Mimicking Strategies with VS for PPI Inhibitor Discovery
2.3.1. De Novo Peptide Design Approach
2.3.2. Fragment-Based Design Approach
2.3.3. Pharmacophore-Based Design Approach
2.3.4. Integration of Mimicking Strategies with LBVS for PPI Inhibitor Discovery
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Scott, D.E.; Bayly, A.R.; Abell, C.; Skidmore, J. Small molecules, big targets: Drug discovery faces the protein–protein interaction challenge. Nat. Rev. Drug Discov. 2016, 15, 533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basile, L. Virtual screening in the search of new and potent anti-alzheimer agents. In Computational Modeling of Drugs against Alzheimer’s Disease; Springer: Totowa, NJ, USA, 2018; pp. 107–137. [Google Scholar]
- Lionta, E.; Spyrou, G.; Vassilatis, D.K.; Cournia, Z. Structure-based virtual screening for drug discovery: Principles, applications and recent advances. Curr. Top. Med. Chem. 2014, 14, 1923–1938. [Google Scholar] [CrossRef] [PubMed]
- Villoutreix, B.O.; Renault, N.; Lagorce, D.; Sperandio, O.; Montes, M.; Miteva, M.A. Free resources to assist structure-based virtual ligand screening experiments. Curr. Protein Pept. Sci. 2007, 8, 381–411. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zhou, H.; Zhu, H.; Leung, S.-W. Ligand-based virtual screening and inductive learning for identification of SIRT1 inhibitors in natural products. Sci. Rep. 2016, 6, 19312. [Google Scholar] [CrossRef]
- Yang, G.-J.; Ko, C.-N.; Zhong, H.-J.; Leung, C.-H.; Ma, D.-L. Structure-Based Discovery of a Selective KDM5A Inhibitor that Exhibits Anti-Cancer Activity via Inducing Cell Cycle Arrest and Senescence in Breast Cancer Cell Lines. Cancers 2019, 11, 92. [Google Scholar] [CrossRef] [Green Version]
- Wu, K.-J.; Zhong, H.-J.; Li, G.; Liu, C.; Wang, H.-M.D.; Ma, D.-L.; Leung, C.-H. Structure-based identification of a NEDD8-activating enzyme inhibitor via drug repurposing. Eur. J. Med. Chem. 2018, 143, 1021–1027. [Google Scholar] [CrossRef]
- Yang, C.; Wang, W.; Chen, L.; Liang, J.; Lin, S.; Lee, M.-Y.; Ma, D.-L.; Leung, C.-H. Discovery of a VHL and HIF1α interaction inhibitor with in vivo angiogenic activity via structure-based virtual screening. Chem. Commum. 2016, 52, 12837–12840. [Google Scholar] [CrossRef]
- Zhong, H.-J.; Lee, B.R.; Boyle, J.W.; Wang, W.; Ma, D.-L.; Chan, P.W.H.; Leung, C.-H. Structure-based screening and optimization of cytisine derivatives as inhibitors of the menin–MLL interaction. Chem. Commum. 2016, 52, 5788–5791. [Google Scholar] [CrossRef]
- Zhong, Z.; Liu, L.-J.; Dong, Z.-Q.; Lu, L.; Wang, M.; Leung, C.-H.; Ma, D.-L.; Wang, Y. Structure-based discovery of an immunomodulatory inhibitor of TLR1–TLR2 heterodimerization from a natural product-like database. Chem. Commum. 2015, 51, 11178–11181. [Google Scholar] [CrossRef] [Green Version]
- Leung, C.-H.; Zhang, J.-T.; Yang, G.-J.; Liu, H.; Han, Q.-B.; Ma, D.-L. Emerging Screening Approaches in the Development of Nrf2–Keap1 Protein–Protein Interaction Inhibitors. Int. J. Mol. Sci. 2019, 20, 4445. [Google Scholar] [CrossRef] [Green Version]
- Mullard, A. Protein–protein interaction inhibitors get into the groove. Nat. Rev. Drug Discov. 2012, 11, 173–175. [Google Scholar] [CrossRef] [PubMed]
- Mason, J.M. Design and development of peptides and peptide mimetics as antagonists for therapeutic intervention. Future Med. Chem. 2010, 2, 1813–1822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groß, A.; Möbius, K.; Haußner, C.; Donhauser, N.; Schmidt, B.; Eichler, J. Mimicking protein–protein interactions through peptide–peptide interactions: HIV-1 gp120 and CXCR4. Front Immunol. 2013, 4, 257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carry, J.-C.; Garcia-Echeverria, C. Inhibitors of the p53/hdm2 protein–protein interaction—path to the clinic. Bioorg. Med. Chem. Lett. 2013, 23, 2480–2485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, K.; Lu, Y.; Nikolovska-Coleska, Z.; Qiu, S.; Ding, Y.; Gao, W.; Stuckey, J.; Krajewski, K.; Roller, P.P.; Tomita, Y. Structure-based design of potent non-peptide MDM2 inhibitors. J. Am. Chem. Soc. 2005, 127, 10130–10131. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Chen, J.; Elenbaas, B.; Levine, A.J. Several hydrophobic amino acids in the p53 amino-terminal domain are required for transcriptional activation, binding to mdm-2 and the adenovirus 5 E1B 55-kD protein. Genes Dev. 1994, 8, 1235–1246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray-Coquard, I.; Blay, J.-Y.; Italiano, A.; Le Cesne, A.; Penel, N.; Zhi, J.; Heil, F.; Rueger, R.; Graves, B.; Ding, M. Effect of the MDM2 antagonist RG7112 on the P53 pathway in patients with MDM2-amplified, well-differentiated or dedifferentiated liposarcoma: An exploratory proof-of-mechanism study. Lancet Oncol. 2012, 13, 1133–1140. [Google Scholar] [CrossRef]
- Rew, Y.; Sun, D.; Gonzalez-Lopez De Turiso, F.; Bartberger, M.D.; Beck, H.P.; Canon, J.; Chen, A.; Chow, D.; Deignan, J.; Fox, B.M. Structure-based design of novel inhibitors of the MDM2–p53 interaction. J. Med. Chem. 2012, 55, 4936–4954. [Google Scholar] [CrossRef]
- Vassilev, L.T.; Vu, B.T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004, 303, 844–848. [Google Scholar] [CrossRef] [Green Version]
- Opydo-Chanek, M.; Gonzalo, O.; Marzo, I. Multifaceted anticancer activity of BH3 mimetics: Current evidence and future prospects. Biochem. Pharm. 2017, 136, 12–23. [Google Scholar] [CrossRef]
- Petros, A.M.; Dinges, J.; Augeri, D.J.; Baumeister, S.A.; Betebenner, D.A.; Bures, M.G.; Elmore, S.W.; Hajduk, P.J.; Joseph, M.K.; Landis, S.K. Discovery of a potent inhibitor of the antiapoptotic protein Bcl-xL from NMR and parallel synthesis. J. Med. Chem. 2006, 49, 656–663. [Google Scholar] [CrossRef] [PubMed]
- Park, C.-M.; Oie, T.; Petros, A.M.; Zhang, H.; Nimmer, P.M.; Henry, R.F.; Elmore, S.W. Design, synthesis, and computational studies of inhibitors of Bcl-XL. J. Am. Chem. Soc. 2006, 128, 16206–16212. [Google Scholar] [CrossRef] [PubMed]
- Oltersdorf, T.; Elmore, S.W.; Shoemaker, A.R.; Armstrong, R.C.; Augeri, D.J.; Belli, B.A.; Bruncko, M.; Deckwerth, T.L.; Dinges, J.; Hajduk, P.J. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 2005, 435, 677. [Google Scholar] [CrossRef] [PubMed]
- Park, C.-M.; Bruncko, M.; Adickes, J.; Bauch, J.; Ding, H.; Kunzer, A.; Marsh, K.C.; Nimmer, P.; Shoemaker, A.R.; Song, X. Discovery of an orally bioavailable small molecule inhibitor of prosurvival B-cell lymphoma 2 proteins. J. Med. Chem. 2008, 51, 6902–6915. [Google Scholar] [CrossRef] [PubMed]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202. [Google Scholar] [CrossRef] [PubMed]
- Cekay, M.J.; Roesler, S.; Frank, T.; Knuth, A.-K.; Eckhardt, I.; Fulda, S. Smac mimetics and type II interferon synergistically induce necroptosis in various cancer cell lines. Cancer Lett. 2017, 410, 228–237. [Google Scholar] [CrossRef]
- Bai, L.; Smith, D.C.; Wang, S. Small-molecule SMAC mimetics as new cancer therapeutics. Pharmacol. Ther. 2014, 144, 82–95. [Google Scholar] [CrossRef] [Green Version]
- Derakhshan, A.; Chen, Z.; Van Waes, C. Therapeutic small molecules target inhibitor of apoptosis proteins in cancers with deregulation of extrinsic and intrinsic cell death pathways. Clin. Cancer Res. 2017, 23, 1379–1387. [Google Scholar] [CrossRef] [Green Version]
- Modell, A.E.; Blosser, S.L.; Arora, P.S. Systematic targeting of protein–protein interactions. Trends Pharm. Sci. 2016, 37, 702–713. [Google Scholar] [CrossRef] [Green Version]
- Pelay-Gimeno, M.; Glas, A.; Koch, O.; Grossmann, T.N. Structure-based design of inhibitors of protein–protein interactions: Mimicking peptide binding epitopes. Angew Chem. Int. 2015, 54, 8896–8927. [Google Scholar] [CrossRef]
- Wu, K.-J.; Liu, X.; Wong, S.-Y.; Zhou, Y.; Ma, D.-L.; Leung, C.-H. Synthesis and Evaluation of Dibenzothiophene Analogues as Pin1 Inhibitors for Cervical Cancer Therapy. ACS Omega 2019, 4, 9228–9234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Tse, A.K.; Li, P.; Ma, Q.; Xiang, S.; Nicosia, S.V.; Seto, E.; Zhang, X.; Bai, W. Inhibition of androgen receptor activity by histone deacetylase 4 through receptor SUMOylation. Oncogene 2011, 30, 2207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sang, W.; Zhong, Z.; Linghu, K.; Xiong, W.; Tse, A.K.W.; San Cheang, W.; Yu, H.; Wang, Y. Siegesbeckia pubescens Makino inhibits Pam 3 CSK 4-induced inflammation in RAW 264.7 macrophages through suppressing TLR1/TLR2-mediated NF-κB activation. Chin. Med. 2018, 13, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, B.C.Y.; Yu, H.; Su, T.; Fu, X.Q.; Guo, H.; Li, T.; Cao, H.-H.; Tse, A.K.-W.; Kwan, H.-Y.; Yu, Z.-L. A herbal formula comprising Rosae Multiflorae Fructus and Lonicerae Japonicae Flos inhibits the production of inflammatory mediators and the IRAK-1/TAK1 and TBK1/IRF3 pathways in RAW 264.7 and THP-1 cells. J. Ethnopharmacol. 2015, 174, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Zhang, E.; Song, X.; Yin, S.; Fan, L.; Ye, M.; Hu, H. Glycycoumarin prevents hepatic steatosis through activation of adenosine 5,-monophosphate (AMP)-activated protein kinase signaling pathway and up-regulation of BTG1/Tob-1. J. Funct. Foods 2017, 34, 277–286. [Google Scholar] [CrossRef]
- Liu, L.-J.; Wang, W.; Huang, S.-Y.; Hong, Y.; Li, G.; Lin, S.; Tian, J.; Cai, Z.; Wang, H.-M.D.; Ma, D.-L. Inhibition of the Ras/Raf interaction and repression of renal cancer xenografts in vivo by an enantiomeric iridium (III) metal-based compound. Chem. Sci. 2017, 8, 4756–4763. [Google Scholar] [CrossRef] [Green Version]
- Zhong, H.-J.; Lu, L.; Leung, K.-H.; Wong, C.C.; Peng, C.; Yan, S.-C.; Ma, D.-L.; Cai, Z.; Wang, H.-M.D.; Leung, C.-H. An iridium (III)-based irreversible protein–protein interaction inhibitor of BRD4 as a potent anticancer agent. Chem. Sci. 2015, 6, 5400–5408. [Google Scholar] [CrossRef] [Green Version]
- Verma, J.; Khedkar, V.M.; Coutinho, E.C. 3D-QSAR in drug design-a review. Curr. Top. Med. Chem. 2010, 10, 95–115. [Google Scholar] [CrossRef]
- Stumpfe, D.; Ripphausen, P.; Bajorath, J. Virtual compound screening in drug discovery. Future Med. Chem. 2012, 4, 593–602. [Google Scholar] [CrossRef]
- Drwal, M.N.; Griffith, R. Combination of ligand-and structure-based methods in virtual screening. Drug Discov. Today Technol. 2013, 10, e395–e401. [Google Scholar] [CrossRef]
- Leach, A.R.; Gillet, V.J.; Lewis, R.A.; Taylor, R. Three-dimensional pharmacophore methods in drug discovery. J. Med. Chem. 2009, 53, 539–558. [Google Scholar] [CrossRef] [PubMed]
- Andricopulo, A.D.; Guido, R.V.; Oliva, G. Virtual screening and its integration with modern drug design technologies. Curr. Med. Chem. 2008, 15, 37–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, S.; Nie, A.; An, J.; Huang, Z. Structure-based virtual screening of chemical libraries for drug discovery. Curr. Opin. Chem. Biol. 2006, 10, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Konc, J. Binding site comparisons for target-centered drug discovery. Expert Opin. Drug Discov. 2019, 14, 445–454. [Google Scholar] [CrossRef]
- Yasuo, N.; Sekijima, M. Improved Method of Structure-Based Virtual Screening via Interaction-Energy-Based Learning. J. Chem. Inf. Model. 2019, 59, 1050–1061. [Google Scholar] [CrossRef] [Green Version]
- Fischer, M.; Coleman, R.G.; Fraser, J.S.; Shoichet, B.K. Incorporation of protein flexibility and conformational energy penalties in docking screens to improve ligand discovery. Nat. Chem. 2014, 6, 575. [Google Scholar] [CrossRef]
- Hospital, A.; Goñi, J.R.; Orozco, M.; Gelpí, J.L. Molecular dynamics simulations: Advances and applications. Adv. Appl. Bioinform. Chem AABC 2015, 8, 37. [Google Scholar]
- De Vivo, M.; Masetti, M.; Bottegoni, G.; Cavalli, A. Role of molecular dynamics and related methods in drug discovery. J. Med. Chem. 2016, 59, 4035–4061. [Google Scholar] [CrossRef]
- Perricone, U.; Gulotta, M.R.; Lombino, J.; Parrino, B.; Cascioferro, S.; Diana, P.; Cirrincione, G.; Padova, A. An overview of recent molecular dynamics applications as medicinal chemistry tools for the undruggable site challenge. MedChemComm 2018, 9, 920–936. [Google Scholar] [CrossRef] [Green Version]
- Saez, N.J.; Deplazes, E.; Cristofori-Armstrong, B.; Chassagnon, I.R.; Lin, X.; Mobli, M.; Mark, A.E.; Rash, L.D.; King, G.F. Molecular dynamics and functional studies define a hot spot of crystal contacts essential for PcTx1 inhibition of acid-sensing ion channel 1a. Br. J. Pharm. 2015, 172, 4985–4995. [Google Scholar] [CrossRef]
- Biswas, R.; Ghosh, S.; Bagchi, A. A structural perspective on the interactions of TRAF6 and B asigin during the onset of melanoma: A molecular dynamics simulation study. J. Mol. Recognit 2017, 30, e2643. [Google Scholar] [CrossRef]
- Rees, D.C.; Congreve, M.; Murray, C.W.; Carr, R. Fragment-based lead discovery. Nat. Rev. Drug Discov. 2004, 3, 660. [Google Scholar] [CrossRef] [PubMed]
- Hughes, J.D.; Blagg, J.; Price, D.A.; Bailey, S.; DeCrescenzo, G.A.; Devraj, R.V.; Ellsworth, E.; Fobian, Y.M.; Gibbs, M.E.; Gilles, R.W. Physiochemical drug properties associated with in vivo toxicological outcomes. Bioorg. Med. Chem. Lett. 2008, 18, 4872–4875. [Google Scholar] [CrossRef] [PubMed]
- Ripphausen, P.; Stumpfe, D.; Bajorath, J. Analysis of structure-based virtual screening studies and characterization of identified active compounds. Future Med. Chem. 2012, 4, 603–613. [Google Scholar] [CrossRef] [PubMed]
- Schierz, A.C. Virtual screening of bioassay data. J. Cheminf. 2009, 1, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorne, N.; Auld, D.S.; Inglese, J. Apparent activity in high-throughput screening: Origins of compound-dependent assay interference. Curr. Opin.Chem. Biol. 2010, 14, 315–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Huang, M.; Zou, X. Docking-based inverse virtual screening: Methods, applications, and challenges. Biophys. Rep. 2018, 4, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, D.-L.; Chan, D.S.-H.; Leung, C.-H. Drug repositioning by structure-based virtual screening. Chem. Soc. Rev. 2013, 42, 2130–2141. [Google Scholar] [CrossRef]
- Spahn, V.; Del Vecchio, G.; Rodriguez-Gaztelumendi, A.; Temp, J.; Labuz, D.; Kloner, M.; Reidelbach, M.; Machelska, H.; Weber, M.; Stein, C. Opioid receptor signaling, analgesic and side effects induced by a computationally designed pH-dependent agonist. Sci. Rep. 2018, 8, 8965. [Google Scholar] [CrossRef] [Green Version]
- Gimeno, A.; Ojeda-Montes, M.J.; Tomás-Hernández, S.; Cereto-Massagué, A.; Beltrán-Debón, R.; Mulero, M.; Pujadas, G.; Garcia-Vallvé, S. The light and dark sides of virtual screening: What is there to know? Int. J. Mol. Sci. 2019, 20, 1375. [Google Scholar] [CrossRef] [Green Version]
- Malo, N.; Hanley, J.A.; Cerquozzi, S.; Pelletier, J.; Nadon, R. Statistical practice in high-throughput screening data analysis. Nat. Biotechnol. 2006, 24, 167. [Google Scholar] [CrossRef] [PubMed]
- Farhadi, T.; Hashemian, S.M. Computer-aided design of amino acid-based therapeutics: A review. Dru Des. Devel. Ther. 2018, 12, 1239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eichler, J. Peptides as protein binding site mimetics. Curr. Opin. Chem. Biol. 2008, 12, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Sheng, C.; Dong, G.; Miao, Z.; Zhang, W.; Wang, W. State-of-the-art strategies for targeting protein–protein interactions by small-molecule inhibitors. Chem. Soc. Rev. 2015, 44, 8238–8259. [Google Scholar] [CrossRef]
- Fletcher, S.; Hamilton, A.D. Targeting protein–protein interactions by rational design: Mimicry of protein surfaces. J. R. Soc. Interface 2006, 3, 215–233. [Google Scholar] [CrossRef] [Green Version]
- Toniolo, C.; Bonora, G.M.; Bavoso, A.; Benedetti, E.; di Blasio, B.; Pavone, V.; Pedone, C. Preferred conformations of peptides containing α, α-disubstituted α-amino acids. Biopolym. Orig. Res. Biomol. 1983, 22, 205–215. [Google Scholar]
- Ernst, J.T.; Becerril, J.; Park, H.S.; Yin, H.; Hamilton, A.D. Design and application of an α-helix-mimetic scaffold based on an oligoamide-foldamer strategy: Antagonism of the bak BH3/Bcl-xL complex. Angew Chem. Int. 2003, 42, 535–539. [Google Scholar] [CrossRef]
- Venkatachalam, C. Stereochemical criteria for polypeptides and proteins. V. Conformation of a system of three linked peptide units. Biopolym. Orig. Res. Biomol. 1968, 6, 1425–1436. [Google Scholar] [CrossRef] [Green Version]
- Chou, K.-C. Prediction of tight turns and their types in proteins. Anal. Biochem. 2000, 286, 1–16. [Google Scholar] [CrossRef]
- Bartfai, T.; Behrens, M.M.; Gaidarova, S.; Pemberton, J.; Shivanyuk, A.; Rebek, J. A low molecular weight mimic of the Toll/IL-1 receptor/resistance domain inhibits IL-1 receptor-mediated responses. Proc. Natl. Acad. Sci. 2003, 100, 7971–7976. [Google Scholar] [CrossRef] [Green Version]
- Azzarito, V.; Long, K.; Murphy, N.S.; Wilson, A.J. Inhibition of α-helix-mediated protein–protein interactions using designed molecules. Nat. Chem. 2013, 5, 161. [Google Scholar] [CrossRef] [PubMed]
- Ripka, A.S.; Rich, D.H. Peptidomimetic design. Curr. Opin. Chem. Biol. 1998, 2, 441–452. [Google Scholar] [CrossRef]
- Floris, M.; Moro, S. Mimicking peptides… in silico. Mol. Inf. 2012, 31, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Jansen, J.M.; Martin, E.J. Target-biased scoring approaches and expert systems in structure-based virtual screening. Curr. Opin. Chem. Biol. 2004, 8, 359–364. [Google Scholar] [CrossRef]
- Leung, K.-H.; Liu, L.-J.; Lin, S.; Lu, L.; Zhong, H.-J.; Susanti, D.; Rao, W.; Wang, M.; Che, W.I.; Chan, D.S.-H. Discovery of a small-molecule inhibitor of STAT3 by ligand-based pharmacophore screening. Methods 2015, 71, 38–43. [Google Scholar] [CrossRef]
- Zhong, H.-J.; Liu, L.-J.; Chan, D.S.-H.; Wang, H.-M.; Chan, P.W.H.; Ma, D.-L.; Leung, C.-H. Structure-based repurposing of FDA-approved drugs as inhibitors of NEDD8-activating enzyme. Biochimie 2014, 102, 211–215. [Google Scholar] [CrossRef]
- Lyne, P.D. Structure-based virtual screening: An overview. Drug Discov. Today 2002, 7, 1047–1055. [Google Scholar] [CrossRef]
- Smadbeck, J.; Peterson, M.B.; Zee, B.M.; Garapaty, S.; Mago, A.; Lee, C.; Giannis, A.; Trojer, P.; Garcia, B.A.; Floudas, C.A. De novo peptide design and experimental validation of histone methyltransferase inhibitors. PLoS ONE 2014, 9, e90095. [Google Scholar] [CrossRef]
- Woolfson, D.N.; Bartlett, G.J.; Burton, A.J.; Heal, J.W.; Niitsu, A.; Thomson, A.R.; Wood, C.W. De novo protein design: How do we expand into the universe of possible protein structures? Curr. Opin. Struct. Biol. 2015, 33, 16–26. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Quan, L.; Lyu, J.; He, Z.; Wang, X.; Meng, J.; Zhao, Z.; Zhu, L.; Liu, X.; Li, H. Discovery of peptide inhibitors targeting human programmed death 1 (PD-1) receptor. Oncotarget 2016, 7, 64967. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Gómez, G.; Hawkins, J.C.; Philipp, J.; Künze, G.; Wodtke, R.; Löser, R.; Fahmy, K.; Pisabarro, M.T. Rational structure-based rescaffolding approach to De Novo design of interleukin 10 (IL-10) receptor-1 mimetics. PLoS ONE 2016, 11, e0154046. [Google Scholar] [CrossRef] [PubMed]
- Schulz, M.N.; Hubbard, R.E. Recent progress in fragment-based lead discovery. Curr. Opin. Pharm. 2009, 9, 615–621. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.-H.; Kim, N.D.; Seong, B.-L. Pharmacophore-based virtual screening: A review of recent applications. Expert Opin. Drug Discov. 2010, 5, 205–222. [Google Scholar] [CrossRef] [PubMed]
- Sun, H. Pharmacophore-based virtual screening. Curr. Med. Chem. 2008, 15, 1018–1024. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.; Alst, T.; Havelkova, M.; Strøm, M.B. Antimicrobial activity of small β-peptidomimetics based on the pharmacophore model of short cationic antimicrobial peptides. J. Med. Chem. 2009, 53, 595–606. [Google Scholar] [CrossRef]
- Caporuscio, F.; Tafi, A.; González, E.; Manetti, F.; Esté, J.A.; Botta, M. A dynamic target-based pharmacophoric model mapping the CD4 binding site on HIV-1 gp120 to identify new inhibitors of gp120–CD4 protein–protein interactions. Bioorg. Med. Chem. Lett. 2009, 19, 6087–6091. [Google Scholar] [CrossRef]
- Hall, P.R.; Leitão, A.; Ye, C.; Kilpatrick, K.; Hjelle, B.; Oprea, T.I.; Larson, R.S. Small molecule inhibitors of hantavirus infection. Bioorg. Med. Chem. Lett. 2010, 20, 7085–7091. [Google Scholar] [CrossRef] [Green Version]
- Atatreh, N.; Ghattas, M.A.; Bardaweel, S.K.; Al Rawashdeh, S.; Al Sorkhy, M. Identification of new inhibitors of Mdm2–p53 interaction via pharmacophore and structure-based virtual screening. Drug Des. Dev. 2018, 12, 3741. [Google Scholar] [CrossRef] [Green Version]
- Taylor, P.; Blackburn, E.; Sheng, Y.; Harding, S.; Hsin, K.Y.; Kan, D.; Shave, S.; Walkinshaw, M. Ligand discovery and virtual screening using the program LIDAEUS. Br. J. Pharm. 2008, 153 (Suppl. 1), S55–S67. [Google Scholar] [CrossRef] [Green Version]
- Stahura, F.L.; Bajorath, J. New methodologies for ligand-based virtual screening. Curr. Pharm. Des. 2005, 11, 1189–1202. [Google Scholar] [CrossRef]
- Neves, B.J.; Braga, R.C.; Melo-Filho, C.C.; Moreira Filho, J.T.; Muratov, E.N.; Andrade, C.H. QSAR-based virtual screening: Advances and applications in drug discovery. Front Pharm. 2018, 9, 1275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolber, G.; Seidel, T.; Bendix, F.; Langer, T. Molecule-pharmacophore superpositioning and pattern matching in computational drug design. Drug Discov. Today 2008, 13, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Liao, C.; Liu, Z.; T Hagler, A.; Gu, Q.; Xu, J. Chemical structure similarity search for ligand-based virtual screening: Methods and computational resources. Curr. Drug Targets 2016, 17, 1580–1585. [Google Scholar] [CrossRef] [PubMed]
- Maggiora, G.; Vogt, M.; Stumpfe, D.; Bajorath, J. Molecular similarity in medicinal chemistry: Miniperspective. J. Med. Chem. 2013, 57, 3186–3204. [Google Scholar] [CrossRef]
- Geppert, H.; Vogt, M.; Bajorath, J. Current trends in ligand-based virtual screening: Molecular representations, data mining methods, new application areas, and performance evaluation. J. Chem. Inf. Model. 2010, 50, 205–216. [Google Scholar] [CrossRef]
- Švajger, U.; Brus, B.; Turk, S.; Sova, M.; Hodnik, V.; Anderluh, G.; Gobec, S. Novel toll-like receptor 4 (TLR4) antagonists identified by structure-and ligand-based virtual screening. Eur. J. Med. Chem. 2013, 70, 393–399. [Google Scholar] [CrossRef]
- Varney, K.M.; Bonvin, A.M.; Pazgier, M.; Malin, J.; Yu, W.; Ateh, E.; Oashi, T.; Lu, W.; Huang, J.; Diepeveen-de Buin, M. Turning defense into offense: Defensin mimetics as novel antibiotics targeting lipid II. PLoS Pathog. 2013, 9, e1003732. [Google Scholar] [CrossRef] [Green Version]
- Ambaye, N.D.; Gunzburg, M.J.; Lim, R.C.; Price, J.T.; Wilce, M.C.; Wilce, J.A. The Discovery of Phenylbenzamide Derivatives as Grb7-Based Antitumor Agents. ChemMedChem 2013, 8, 280–288. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Types | Pros | Cons | |
|---|---|---|---|
| SBVS | 1) Pharmacophore-based models | Uses protein structure | Increased screening time |
| 2) Molecular docking | Not biased toward existing ligand structures | Higher false positives | |
| 3) Binding site comparisons | Takes protein flexibility into consideration | Oversimplification of scoring functions | |
| LBVS | 1) Similarity methods | Simple and fast | Requires existing ligands |
| 2) QSAR modeling | Less computationally intensive | Poor accuracy | |
| 3) Pharmacophore-based models | Protein structure information may remain unknown | Lack of consideration of protein structural framework |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, K.-J.; Lei, P.-M.; Liu, H.; Wu, C.; Leung, C.-H.; Ma, D.-L. Mimicking Strategy for Protein–Protein Interaction Inhibitor Discovery by Virtual Screening. Molecules 2019, 24, 4428. https://doi.org/10.3390/molecules24244428
Wu K-J, Lei P-M, Liu H, Wu C, Leung C-H, Ma D-L. Mimicking Strategy for Protein–Protein Interaction Inhibitor Discovery by Virtual Screening. Molecules. 2019; 24(24):4428. https://doi.org/10.3390/molecules24244428
Chicago/Turabian StyleWu, Ke-Jia, Pui-Man Lei, Hao Liu, Chun Wu, Chung-Hang Leung, and Dik-Lung Ma. 2019. "Mimicking Strategy for Protein–Protein Interaction Inhibitor Discovery by Virtual Screening" Molecules 24, no. 24: 4428. https://doi.org/10.3390/molecules24244428