Design, Synthesis, and In Vitro Activity of Pyrazine Compounds


Abstract
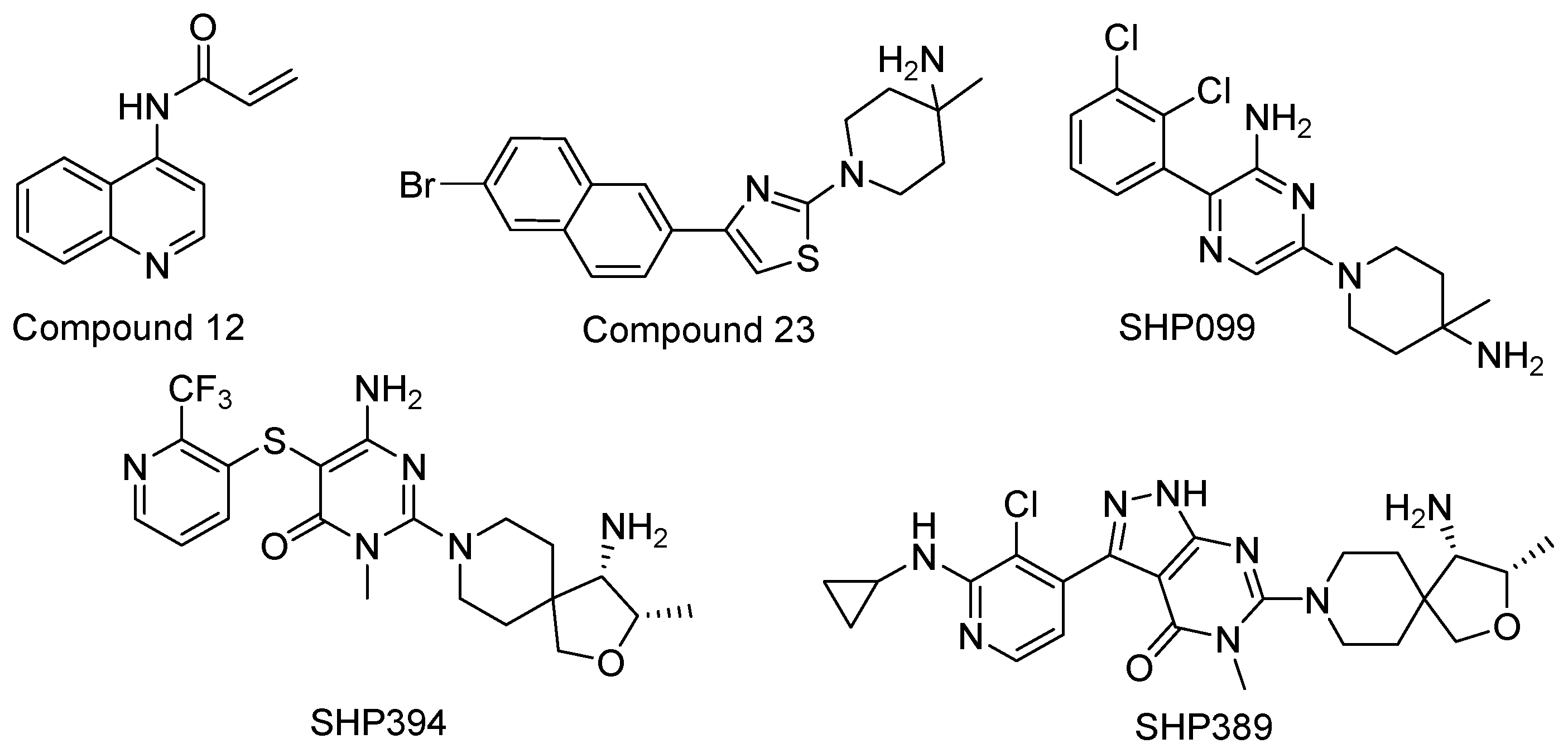
:1. Introduction
2. Results
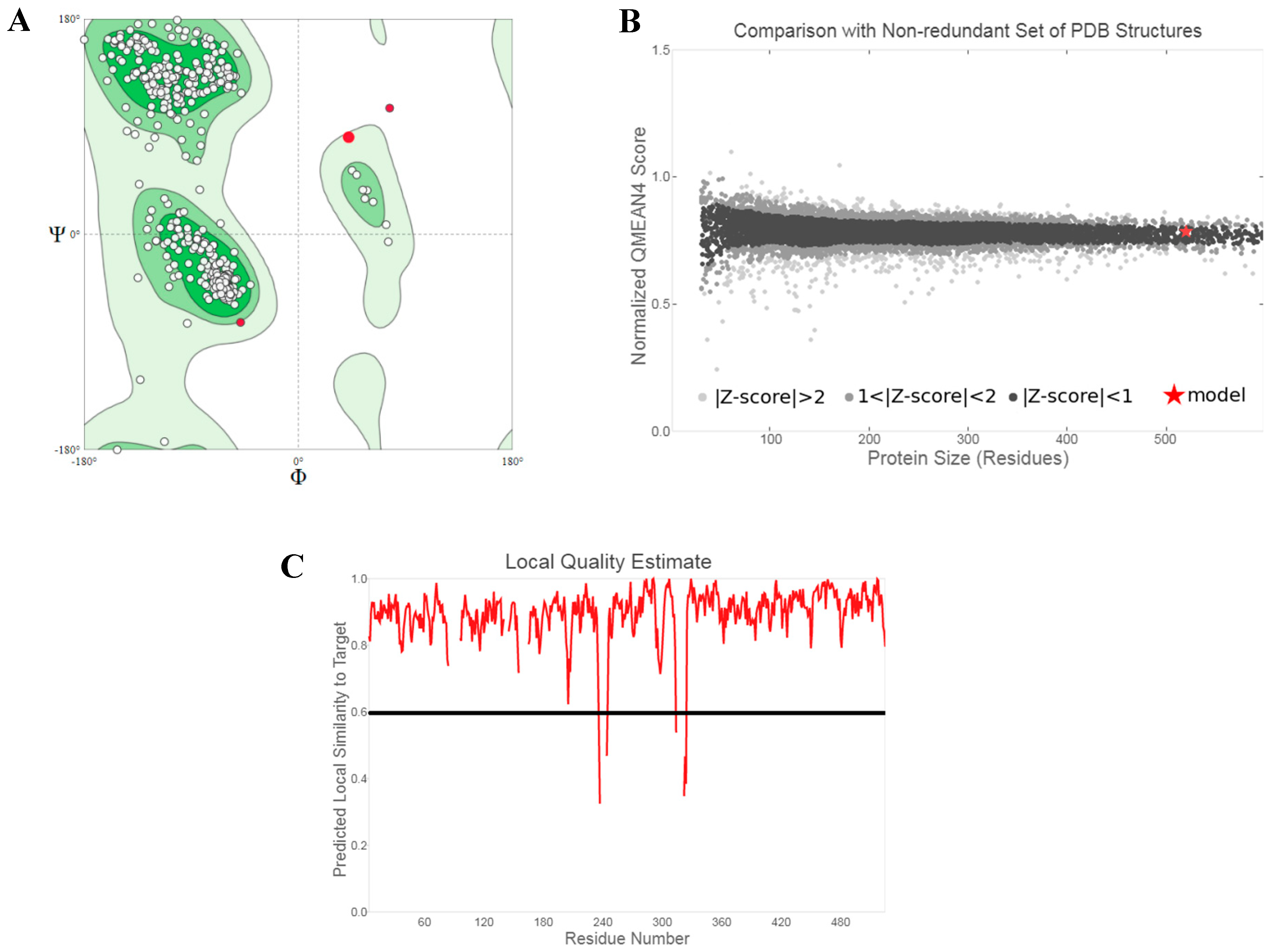
2.1. Structure Retrieval and Validation
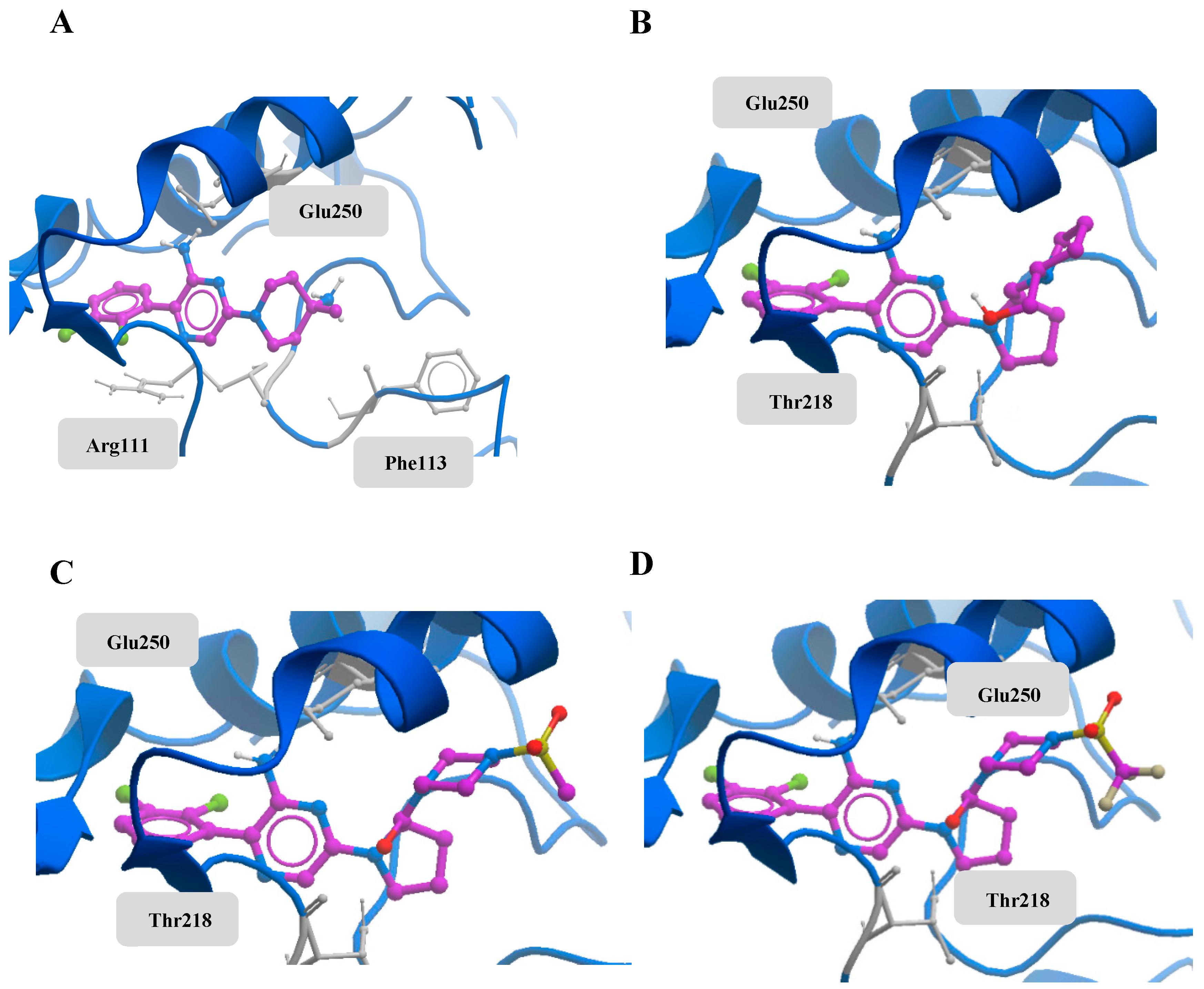
2.2. Small Molecule Docking
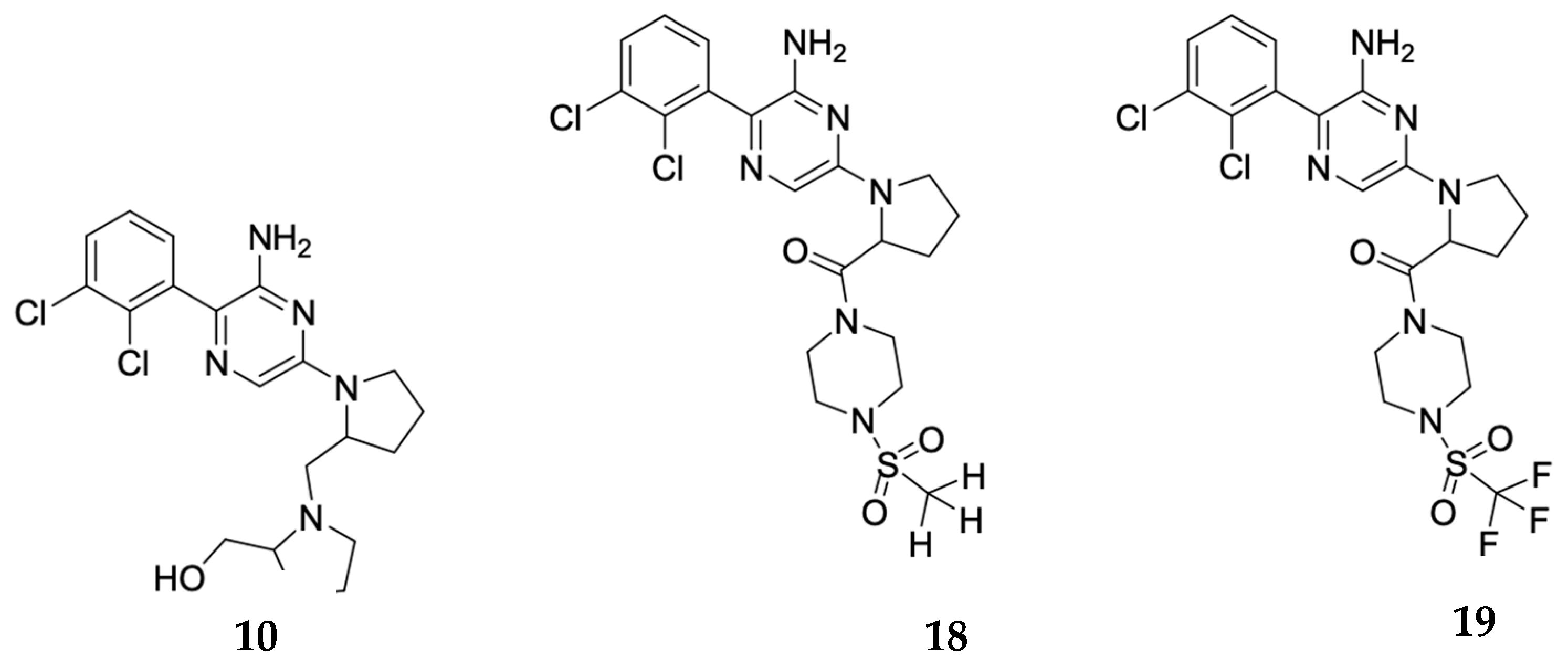
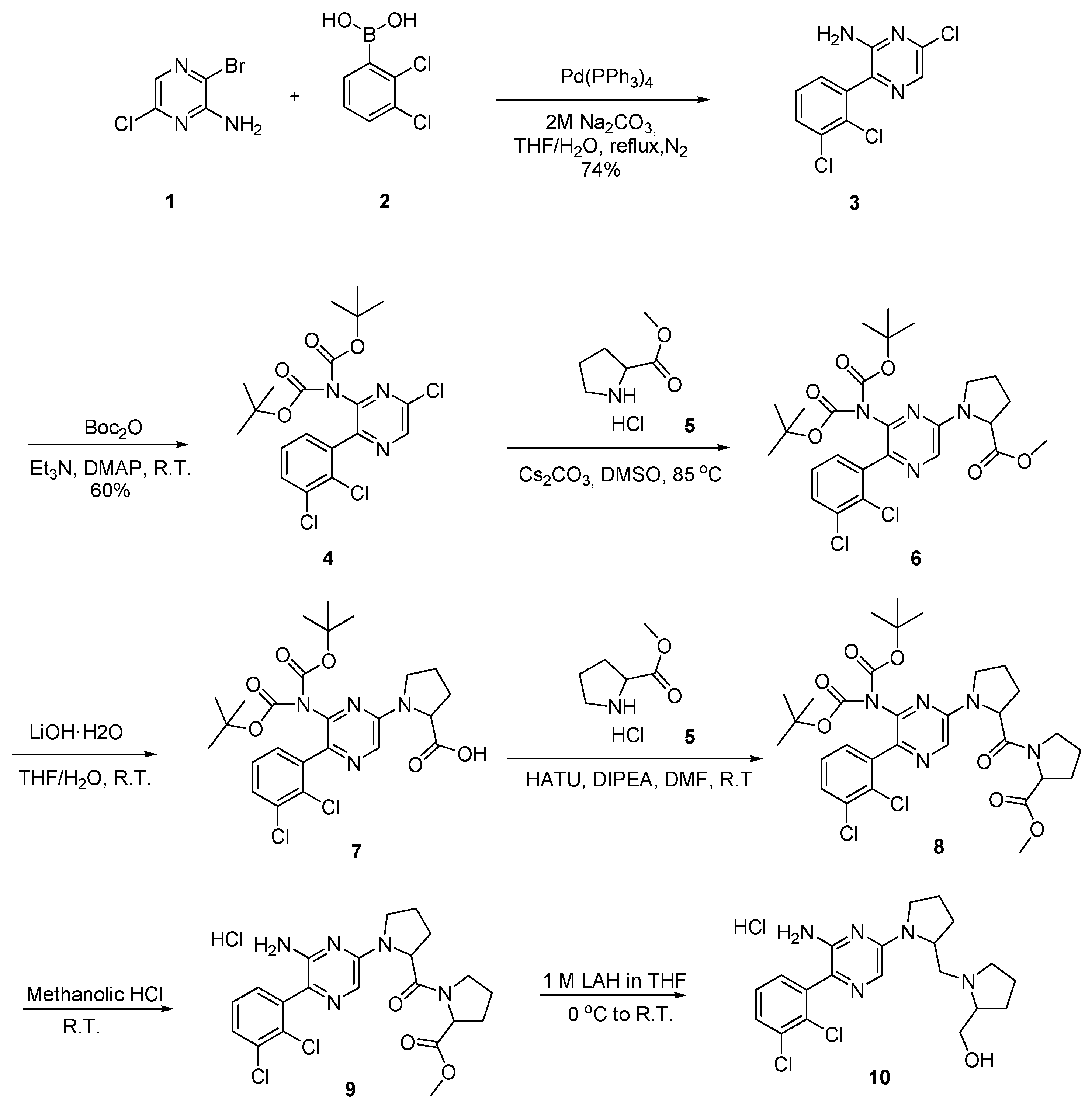
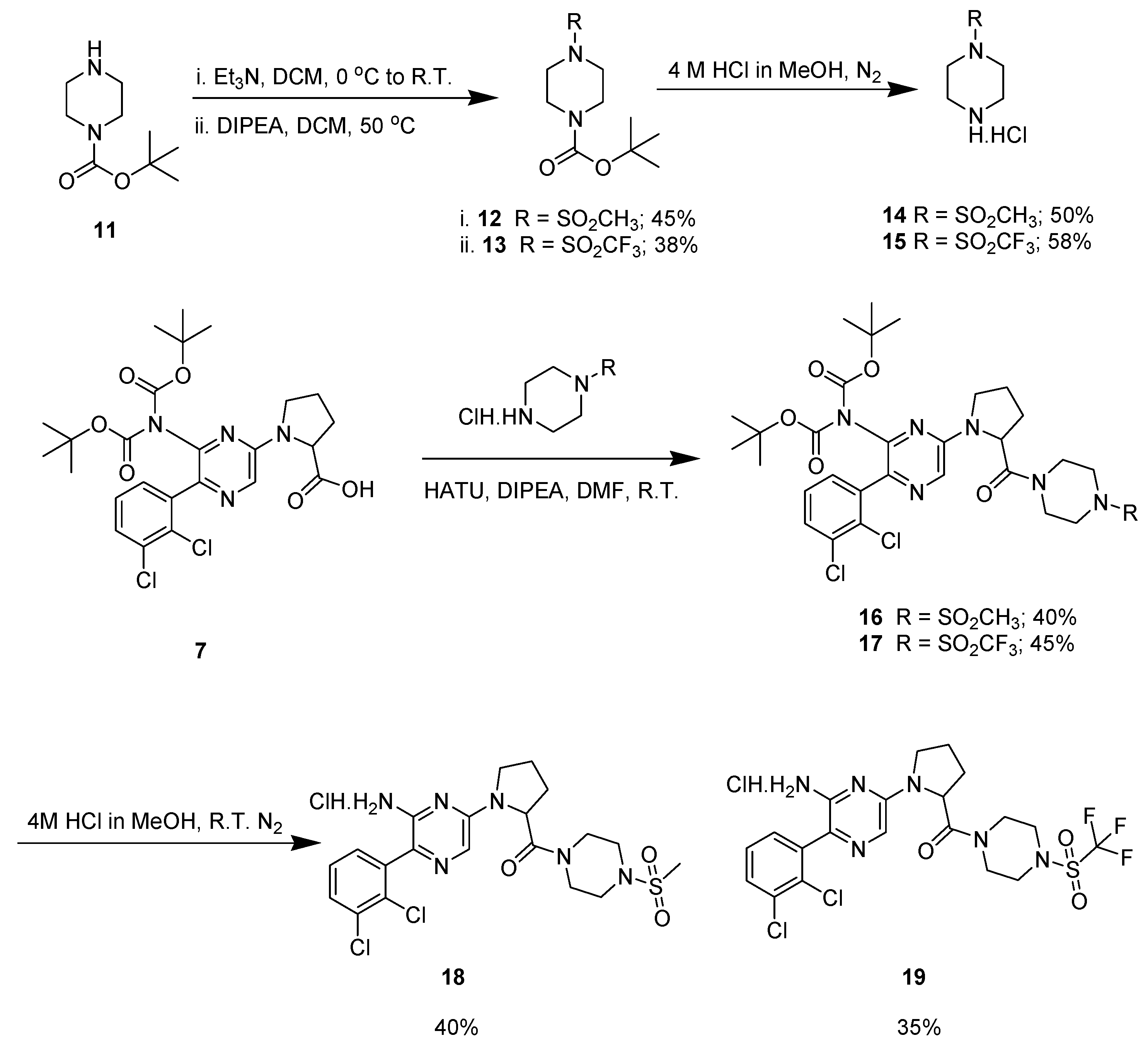
2.3. Synthetic Strategy
2.4. Biological Effect of Intermediates (21, 22) in Human Cancer Cell Lines
2.5. Biological Effect of Intermediate 9 and final 10 in Human Cancer Cell Lines
3. Discussion
4. Materials and Methods
4.1. Small Molecule Docking
4.2. Chemistry
4.2.1. General Information
4.2.2. Experimental Procedures and Analytical Data
4.3. Cell Culture
4.4. Cell Viability Analysis
4.5. Apoptosis Assessment
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- World Health Organization. Available online: https://www.who.int/health-topics/cancer#tab=tab_1 (accessed on 10 November 2019).
- Hof, P.; Pluskey, S.; Dhe-Paganon, S.; Eck, M.J.; Shoelson, S.E. Crystal structure of the tyrosine phosphatase SHP-2. Cell 1998, 92, 441–450. [Google Scholar] [CrossRef]
- Zhang, R.Y.; Yu, Z.H.; Zeng, L.; Zhang, S.; Bai, Y.; Miao, J.; Chen, L.; Xie, J.; Zhang, Z.Y. SHP2 phosphatase, as a novel therapeutic target for melanoma treatment. Oncotarget 2016, 45, 73817–73829. [Google Scholar] [CrossRef] [PubMed]
- Xiao, P.; Guo, Y.; Zhang, H.; Zhang, X.; Cheng, H.; Cao, Q.; Ke, Y. Myeloid-restricted ablation of Shp2 restrains melanoma growth by amplifying the reciprocal promotion of CXCL9 and IFN-γ production in tumor microenvironment. Oncogene 2018, 37, 5088–5100. [Google Scholar] [CrossRef] [PubMed]
- Han, T.; Xiang, D.T.; Sun, W.; Liu, N.; Sun, H.L.; Wen, W.; Shen, W.F.; Wang, R.Y.; Chen, C.; Wang, X.; et al. PTPN11/Shp2 overexpression enhances liver cancer progression and predicts poor prognosis of patients. J. Hepatol. 2015, 63, 651–660. [Google Scholar] [CrossRef]
- Xiang, D.; Cheng, Z.; Liu, H.; Wang, X.; Han, T.; Sun, W.; Li, X.; Yang, W.; Chen, C.; Xia, M.; et al. Shp2 promotes liver cancer stem cell expansion by augmenting β-catenin signaling and predicts chemotherapeutic response of patients. Hepatology 2017, 65, 1566–1580. [Google Scholar] [CrossRef]
- Mainardi, S.; Mulero-Sánchez, A.; Prahallad, A.; Germano, G.; Bosma, A.; Krimpenfort, P.; Lieftink, C.; Steinberg, J.D.; Wit, N.; Gonçalves-Ribeiro, S.; et al. SHP2 is required for growth of KRAS-mutant non-small-cell lung cancer in vivo. Nat. Med. 2018, 24, 961–967. [Google Scholar] [CrossRef]
- Ruess, D.A.; Heynen, G.J.; Ciecielski, K.J.; Ai, J.; Berninger, A.; Kabacaoglu, D.; Görgülü, K.; Dantes, Z.; Wörmann, S.M.; Diakopoulos, K.N.; et al. KRAS-driven cancers depend on PTPN11/SHP2 phosphatase. Nat. Med. 2018, 24, 954–960. [Google Scholar] [CrossRef]
- Lu, H.; Liu, C.; Velazquez, R.; Wang, H.; Dunkl, L.M.; Kazic-Legueux, M.; Haberkorn, A.; Billy, E.; Manchado, E.; Brachmann, S.M.; et al. SHP2 Inhibition Overcomes RTK-Mediated Pathway Reactivation in KRAS-Mutant Tumors Treated with MEK Inhibitors. Mol. Cancer Ther. 2019, 18, 1323–1334. [Google Scholar] [CrossRef]
- Garcia Fortanet, J.; Chen, C.H.-T.; Chen, Y.-N.P.; Chen, Z.; Deng, Z.; Firestone, B.; Fekkes, P.; Fodor, M.; Fortin, P.D.; Fridrich, C.; et al. Allosteric Inhibition of SHP2: Identification of a Potent, Selective, and Orally Efficacious Phosphatase Inhibitor. J. Med. Chem. 2016, 59, 7773–7782. [Google Scholar] [CrossRef]
- Chen, Y.N.P.; LaMarche, M.J.; Chan, H.M.; Fekkes, P.; Garcia-Fortanet, G.; Acker, M.G.; Antonakos, B.; Chen, C.H.T.; Chen, Z.; Cooke, V.G.; et al. Allosteric inhibition of SHP2 phosphatase inhibits cancers driven by receptor tyrosine kinases. Nature 2016, 535, 148–152. [Google Scholar] [CrossRef]
- LaRochelle, J.R.; Fodor, M.; Ellegast, J.M.; Liu, X.; Vemulapalli, V.; Mohseni, M.; Stams, T.; Buhrlage, S.J.; Stegmaier, K.; LaMarche, M.J.; et al. Identification of an allosteric benzothiazolopyrimidone inhibitor of the oncogenic protein tyrosine phosphatase SHP2. Bioorg. Med. Chem. 2017, 25, 6479–6485. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Xu, G.; Li, X.; Xu, H.; Li, Q.; Liu, W.; Kirby, K.A.; LohJun Li, M.L.; Sarafianos, S.G.; Qu, C.K. Small Molecule Inhibitor that Stabilizes the Autoinhibited Conformation of the Oncogenic Tyrosine Phosphatase SHP2. J. Med. Chem. 2019, 62, 1125–1137. [Google Scholar] [CrossRef] [PubMed]
- Marsh-Armstrong, B.; Fajnzylber, J.M.; Korntner, S.; Plaman, B.A.; Bishop, A.C. The Allosteric Site on SHP2′s Protein Tyrosine Phosphatase Domain is Targetable with Druglike Small Molecules. ACS Omega 2018, 3, 15763–15770. [Google Scholar] [CrossRef] [PubMed]
- Sarver, P.; Acker, M.G.; Bagdanoff, J.; Chen, Z.; Chen, Y.N.P.; Chan, H.; Firestone, B.; Fodor, M.; Fortanet, J.; Hao, H.; et al. 6-Amino-3-Methylpyrimidinones as Potent, Selective, and Orally Efficacious SHP2 Inhibitors. J. Med. Chem. 2019, 62, 1793–1802. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Si, X.; Gu, S.; Wang, M.; Shen, J.; Li, H.; Shen, J.; Li, D.; Fang, Y.; Liu, C.; et al. Allosteric Inhibitors of SHP2 with Therapeutic Potential for Cancer Treatment. J. Med. Chem. 2017, 60, 10205–10219. [Google Scholar] [CrossRef] [PubMed]
- Bagdanoff, J.T.; Chen, J.; Acker, M.; Chen, Y.N.; Chan, H.; Dore, M.; Firestone, B.; Fodor, M.; Fortanet, J.; Hentemann, M.; et al. Optimization of Fused Bicyclic Allosteric SHP2 Inhibitors. J. Med. Chem. 2019, 62, 1781–1792. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; De Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, 296–303. [Google Scholar] [CrossRef]
- Chen, V.B.; Arendall, W.B.; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef]
- Benkert, P.; Biasini, M.; Schwede, T. Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics 2011, 27, 343–350. [Google Scholar] [CrossRef]
- Burger, M.T.; Han, W.; Lan, J.; Nishiguchi, G.; Bellamacina, C.; Lindval, M.; Atallah, G.; Ding, Y.; Mathur, M.; McBride, C.; et al. Structure Guided Optimization, in Vitro Activity, and inVivo Activity of Pan-PIM Kinase Inhibitors. ACS Med. Chem. Lett. 2013, 4, 1193–1197. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, A.L.; Höglund, M.; Lindhagen, E.; Aleskog, A.; Hassan, S.B.; Ekholm, C.; Fhölenhag, K.; Jensen, A.J.; Löthgren, A.; Scobie, M.; et al. Identification of AKN-032, a Novel 2-Aminopyrazine Tyrosine Kinase Inhibitor, with Significant Preclinical Activity in Acute Myeloid Leukemia. Biochem. Pharmacol. 2010, 80, 1507–1516. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Malkani, S.; Lombardo, M.; Yang, Y.; Mills, S.G.; Chapman, K.; Thompson, J.E.; Zhang, W.X.; Wang, R.; Cubbon, R.M.; et al. Design, Synthesis, and Biological Evaluation of Aminopyrazine Derivatives as Inhibitors of Mitogen-Activated Protein Kinase-Activated Protein Kinase 2 (MK-2). Bioorg. Med. Chem. Lett. 2015, 25, 5402–5408. [Google Scholar] [CrossRef] [PubMed]
- Fodor, M.; Price, E.; Wang, P.; Lu, H.; Argintaru, A.; Chen, Z.; Glick, M.; Hao, X.H.; Kato, M.; Koenig, R.; et al. Dual Allosteric Inhibition of SHP2 Phosphatase. ACS Chem. Biol. 2018, 13, 647–656. [Google Scholar] [CrossRef] [PubMed]
- Abagyan, R.; Totrov, M.; Kuznetsov, D. ICM-A new method for protein modeling and design: Applications to docking and structure prediction from the distorted native conformation. J. Comput. Chem. 1994, 15, 488–506. [Google Scholar] [CrossRef]
- Abagyan, R.; Totrov, M. Biased probability Monte Carlo conformational searches and electrostatic calculations for peptides and proteins. J. Mol. Biol. 1994, 235, 983–1002. [Google Scholar] [CrossRef]
- Totrov, M.; Abagyan, R. Rapid boundary element solvation electrostatics calculations in folding simulations: Successful folding of a 23-residue peptide. Biopolymers 2001, 60, 124–133. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Intermediate 9 | 24h | 48h | 72h | ||||||
|---|---|---|---|---|---|---|---|---|---|
| MTT | SRB | CVE | MTT | SRB | CVE | MTT | SRB | CVE | |
| MCF7 | - | - | 0.1 µM (17%) | 0.001 µM (27%) | - | 0.001 µM (9%) | 0.001 µM (12%), 0.01 µM (11%), 1 µM (9%) | 0.1 µM (7%), 1 µM (10%) | |
| HCT116 | 0.01 µM (18%), 0.1 µM (16%) | - | - | - | - | - | - | - | |
| MOR | 0.001 µM (20%), 0.1 µM (13%) | - | - | 0.001 µM (19%), 0.01 µM (16%) | 0.01 µM (13%) | - | 0.001 µM (30%), 0.01 µM (31%) | 0.01 µM (25%) | 0.1 µM (9%) |
| Intermediate 10 | 24 h | 48 h | 72 h | ||||||
|---|---|---|---|---|---|---|---|---|---|
| MTT | SRB | CVE | MTT | SRB | CVE | MTT | SRB | CVE | |
| MCF7 | 0.001 µM (13%), 0.1 µM (20%) | - | 1 µM (10%) | - | - | 0.01 µM (6%) | - | - | 0.001 µM (8%), 0.01 µM (11%), 0.1 µM (15%), 1 µM (8%) |
| MDA-MB-231 | 0.01 µM (16%), 0.1 µM (30%) | - | - | - | - | - | 0.1 µM (13%) | - | - |
| HCT116 | 0.001 µM (19%), 0.01 µM (14%), 0.1 µM (32%) | - | - | - | - | - | 0.01 µM (25%), 0.1 µM (22%) | - | - |
| MOR | 0.001 µM (17%), 0.01 µM (28%), 0.1 µM (27%) | - | - | 0.01 µM (31%), 0.1 µM (23%) | 1 µM (24%) | - | 0.001 µM (25%), 0.01 µM (38%), 0.1 µM (33%) | - | - |
| MCF7 | Untreated (48h) | 0.001 µM (48 h) | Untreated (72h) | 1 µM (72 h) |
|---|---|---|---|---|
| Living Cells | 88.48% | 86.53% | 78.21% | 78.21% |
| Early Apoptotic | 0.41% | 0.38% | 1.62% | 1.51% |
| Late Apoptotic | 1.84% | 1.38% | 6.45% | 5.91% |
| Dead Cells | 1.3% | 2.34% | 1.52% | 2.34% |
| MOR | Untreated | 0.01 µM (48 h) | Untreated | 0.01 µM (72 h) | Untreated | 0.1 µM (72 h) |
|---|---|---|---|---|---|---|
| Living Cells | 43.11% | 42.53% | 61.21% | 64.78% | 61.21% | 61.26% |
| Early Apoptotic | 9.99% | 7.75% | 4.56% | 4.59% | 4.56% | 5.63% |
| Late Apoptotic | 33.85% | 29.62% | 20.64% | 12.06% | 20.64% | 15.22% |
| Dead Cells | 6.05% | 13.14% | 9.23% | 13.35% | 9.23% | 12.76% |
| MDA-MB-231 | Untreated | 0.1 µM (24 h) |
|---|---|---|
| Living Cells | 69.6% | 56.79% |
| Early Apoptotic | 3.02% | 3.16% |
| Late Apoptotic | 14.24% | 27.72% |
| Dead Cells | 2.39% | 2.52% |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parsonidis, P.; Shaik, M.; Serafeim, A.P.; Vlachou, I.; Daikopoulou, V.; Papasotiriou, I. Design, Synthesis, and In Vitro Activity of Pyrazine Compounds. Molecules 2019, 24, 4389. https://doi.org/10.3390/molecules24234389
Parsonidis P, Shaik M, Serafeim AP, Vlachou I, Daikopoulou V, Papasotiriou I. Design, Synthesis, and In Vitro Activity of Pyrazine Compounds. Molecules. 2019; 24(23):4389. https://doi.org/10.3390/molecules24234389
Chicago/Turabian StyleParsonidis, Panagiotis, Mahammad Shaik, Athanasia Panagiota Serafeim, Ioanna Vlachou, Vasiliki Daikopoulou, and Ioannis Papasotiriou. 2019. "Design, Synthesis, and In Vitro Activity of Pyrazine Compounds" Molecules 24, no. 23: 4389. https://doi.org/10.3390/molecules24234389
APA StyleParsonidis, P., Shaik, M., Serafeim, A. P., Vlachou, I., Daikopoulou, V., & Papasotiriou, I. (2019). Design, Synthesis, and In Vitro Activity of Pyrazine Compounds. Molecules, 24(23), 4389. https://doi.org/10.3390/molecules24234389