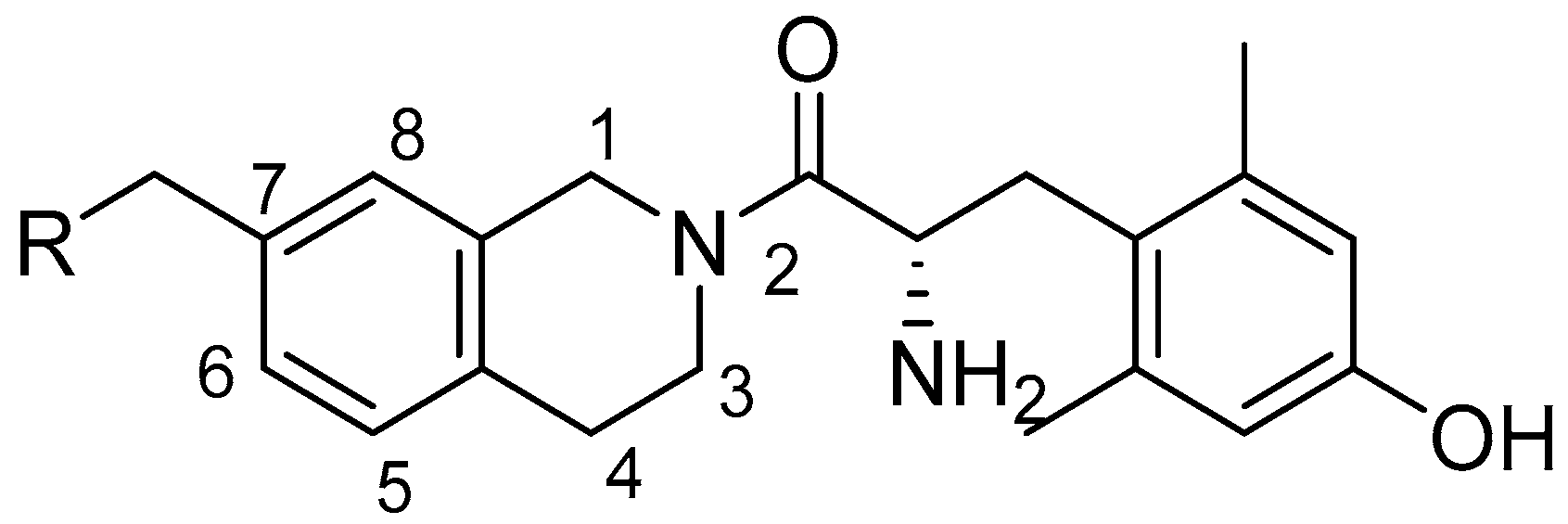
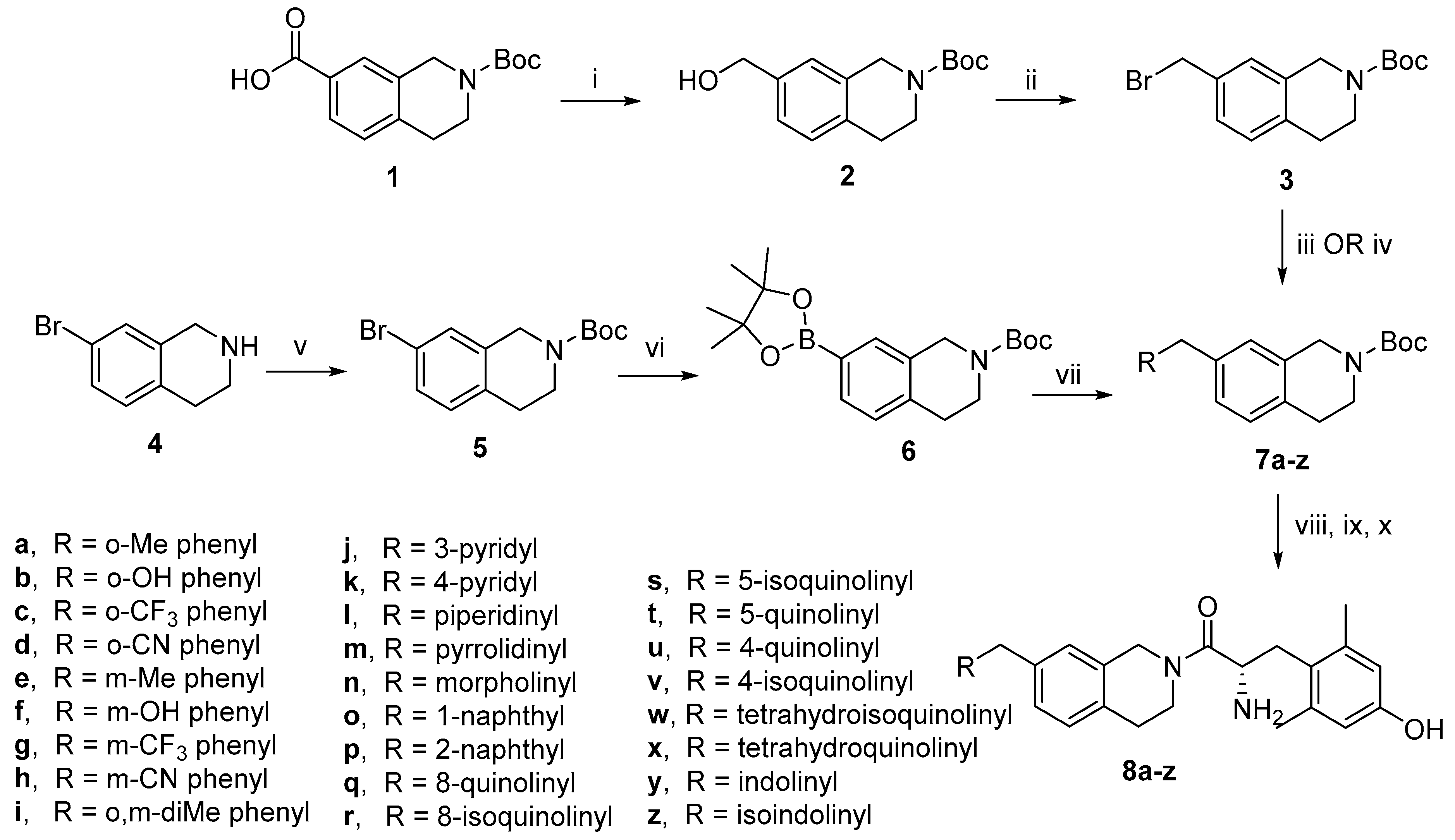
4.1.6. General Procedure F for TFA Boc Deprotection, Peptide Coupling, and TFA Boc Deprotection
The appropriate Boc-protected amine intermediate was dissolved in DCM (1–3 mL). An equal volume of TFA was added, and the reaction mixture stirred at room temperature for 1–1.5 h. The solvent was removed under vacuum to yield the deprotected amine. The amine intermediate (1.0 eq), diBoc-DMT (1.05 eq), and PyBOP (1.0 eq) were combined, and the reaction flask was flushed with argon. Dry DMF (3–12 mL) and DIEA (10 eq) were added. The reaction mixture stirred at room temperature for 6–24 h. The solvent was removed under vacuum, and the coupled product was purified via silica gel chromatography in ethyl acetate/hexanes. The Boc-protected compound was dissolved in DCM (2–2.5 mL). An equal volume of TFA was added, and the reaction mixture stirred at room temperature for 1–1.5 h. The solvent was removed under vacuum, and the product was purified by semi-preparative HPLC and lyophilized.
Tert-butyl 7-(hydroxymethyl)-3,4-dihydroisoquinoline-2(1H)-carboxylate (2). To a solution of compound 1 in dry THF (15 mL), a 2.0 M solution of borane dimethyl sulfide in THF (2.7 mL, 5.41 mmol, 3.0 eq) was added dropwise over 15 min under inert atmosphere. The reaction mixture stirred at room temperature overnight. The reaction was quenched by the addition of methanol (20 mL). The solvent was removed under vacuum. The crude product was dissolved in ethyl acetate and washed with saturated aqueous NaHCO3 and brine. The combined aqueous layers were extracted with ethyl acetate. The combined organic layers were dried over MgSO4, filtered, and concentrated under vacuum to yield the product as a colorless oil (475 mg, 100%). 1H-NMR (CDCl3, 500 MHz) δ 7.16 (d, J = 7.9 Hz, 1H), 7.13 (d, J = 5.8 Hz, 1H), 7.12 (s, 1H), 4.65 (s, 2H), 4.57 (s, 2H), 3.63 (t, J = 5.9 Hz, 2H)), 2.82 (t, J = 5.8 Hz, 2H), 1.49 (s, 9H).
Tert-butyl 7-(bromomethyl)-3,4-dihydroisoquinoline-2(1H)-carboxylate (3). To a solution of compound 2 (950 mg, 3.61 mmol, 1.0 eq) in DCM (40 mL), CBr4 (1.32 g, 3.97 mmol, 1.1 eq) and a solution of PPh3 (1.14 g, 4.33 mmol, 1.2 eq) in DCM (5 mL) were added. The reaction stirred at room temperature for 2 h. The product was purified via silica gel chromatography in ethyl acetate/hexanes to yield a white solid (1.08 g, 92%). 1H-NMR (CDCl3, 500 MHz) δ 7.19 (d, J = 8.3 Hz, 1H), 7.14 (s, 1H), 7.11 (d, J = 7.8 Hz, 1H), 4.56 (s, 2H), 4.47 (s, 2H), 3.64 (t, J = 6.0 Hz, 2H), 2.82 (t, J = 5.9 Hz, 2H), 1.49 (s, 9H).
Tert-butyl 7-bromo-3,4-dihydroisoquinoline-2(1H)-carboxylate (5). 7-bromo-1,2,3,4-tetrahydroisoquinoline 4 (75 µL, 0.50 mmol, 1.0 eq) and di-tert-butyl dicarbonate (120 mg, 0.55 mmol, 1.1 eq) were combined in a microwave vessel equipped with a teflon stirbar. The system was flushed with argon, and the reaction was heated in a microwave to 100 °C for 15 min. The reaction mixture was diluted with DCM and washed with saturated aqueous NaHCO3 and brine. The organic layer was dried over MgSO4, filtered, and concentrated under vacuum to obtain the product as a pale orange oil (145 mg, 99%). 1H-NMR (CDCl3, 500 MHz) δ 7.25 (m, 2H), 6.98 (d, J = 8.0 Hz, 1H), 4.52 (s, 2H), 3.61 (t, J = 5.7 Hz, 2H), 2.75 (t, J = 6.0 Hz, 2H), 1.48 (s, 9H).
Tert-butyl 7-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-3,4-dihydroisoquinoline-2(1H)-carboxylate (6). Intermediate 5 (945 mg, 3.03 mmol, 1.0 eq), bis(pinacolato)diboron (1.54 g, 6.06 mmol, 2.0 eq), Pd(dppf)Cl2 (222 mg, 0.303 mmol, 0.1 eq), and potassium acetate (892 mg, 9.09 mmol, 3.0 eq) were combined in DMSO (20 mL), and the system was flushed with argon. The reaction was heated to 90 °C overnight. The reaction mixture was concentrated under vacuum to remove most DMSO. The remaining mixture was diluted with water and extracted with three portions of DCM. The combined organic layers were washed with water and brine, dried over MgSO4, filtered, and concentrated under vacuum. The product was purified via silica gel chromatography in ethyl acetate/hexanes to yield a pale yellow oil (1.05 g, 96%). 1H-NMR (CDCl3, 500 MHz) δ 7.59 (d, J = 5.7 Hz, 1H), 7.14 (d, J = 5.5 Hz, 1H), 4.58 (s, 2H), 3.63 (br s, 2H), 2.84 (br s, 2H), 1.48 (s, 9H), 1.34 (s, 12H).
Tert-butyl 7-(2-methoxybenzyl)-3,4-dihydroisoquinoline-2(1H)-carboxylate (7a). Compound 7a was synthesized following General Procedure A from compound 3 (60 mg, 0.18 mmol, 1.0 eq), (2-methoxyphenyl)boronic acid (42 mg, 0.28 mmol, 1.5 eq), Pd(dppf)Cl2 (13 mg, 0.02 mmol, 0.1 eq), and K2CO3 (76 mg, 0.55 mmol, 3.0 eq) to yield the product as a colorless oil (37 mg, 57%). 1H-NMR (CDCl3, 500 MHz) δ 7.21 (t, J = 7.6 Hz, 1H), 7.07 (d, J = 7.2 Hz, 1H), 7.03 (s, 2H), 6.95 (s, 1H), 6.88 (t, J = 8.2 Hz, 2H), 4.52 (s, 2H), 3.93 (s, 2H), 3.83 (s, 3H), 3.63 (s, 2H), 2.79 (t, J = 5.8 Hz, 2H), 1.49 (s, 9H).
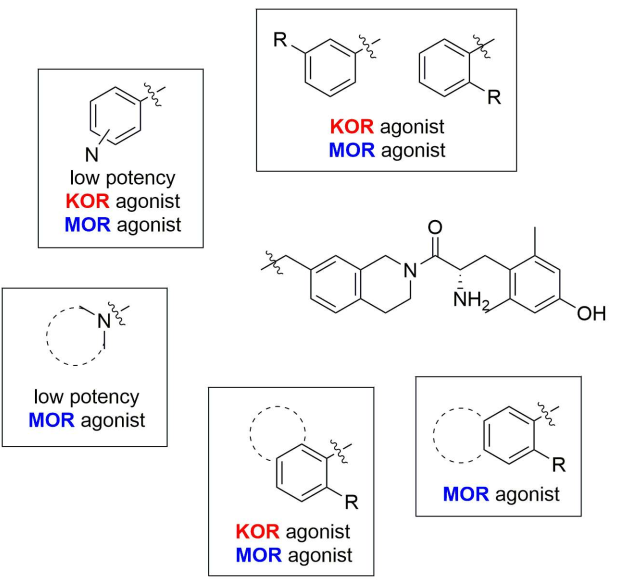
(S)-2-amino-3-(4-hydroxy-2,6-dimethylphenyl)-1-(7-(2-methoxybenzyl)-3,4-dihydroisoquinolin-2(1H)-yl)propan-1-one (8a). Following General Procedure B, intermediate 7a (37 mg, 0.10 mmol) was deprotected to yield the amine intermediate as a colorless oil. This intermediate was coupled to diBoc-DMT (45 mg, 0.10 mmol, 1.05 eq) in the presence of PyBOP (54 mg, 0.10 mmol, 1.0 eq), and DIEA (142 µL, 1.04 mmol, 10 eq) to yield the product as a brown oil. No 6Cl-HOBt was used. TFA deprotection yielded the product as a white solid. 1H-NMR (CD3OD, 500 MHz, rotamers) δ 7.17 (t, J = 7.9 Hz, 2H), 7.05 (t, J = 7.4 Hz, 2H), 6.97 (d, J = 7.7 Hz, 1H), 6.94–6.90 (m, 4H), 6.89–6.82 (m, 4H), 6.50 (d, J = 3.9 Hz, 1H), 6.39 (s, 2H), 6.33 (s, 2H), 4.59–4.45 (m, 4H), 4.15 (d, J = 15.7 Hz, 1H), 3.86 (d, J = 3.7 Hz, 2H), 3.85 (d, J = 2.8 Hz, 2H), 3.78 (d, J = 1.6 Hz, 3H), 3.78 (d, J = 1.2 Hz, 3H), 3.71–3.65 (m, 1H), 3.65–3.58 (m, 1H), 3.33 (d, J = 15.7 Hz, 1H), 3.25–3.17 (m, 2H), 3.09 (d, J = 11.4 Hz, 1H), 2.72–2.65 (m, 1H), 2.65–2.58 (m, 1H), 2.55–2.45 (m, 1H), 2.24 (s, 6H), 2.18 (s, 6H), 1.98–1.87 (m, 1H). HPLC retention time: 39.1 min. HREIMS m/z 445.2494 (calcd. for C28H32N2O3, 445.2486).
Tert-butyl 7-(2-hydroxybenzyl)-3,4-dihydroisoquinoline-2(1H)-carboxylate (7b). Compound 7b was synthesized following General Procedure A from compound 3 (60 mg, 0.18 mmol, 1.0 eq), (2-hydroxyphenyl)boronic acid (38 mg, 0.28 mmol, 1.5 eq), Pd(dppf)Cl2 (13 mg, 0.02 mmol, 0.1 eq), and K2CO3 (76 mg, 0.55 mmol, 3.0 eq) to yield the product as a yellow oil (33 mg, 53%). 1H-NMR (CDCl3, 500 MHz) δ 7.16–7.10 (m, 2H), 7.08–7.01 (m, 2H), 6.96 (s, 1H), 6.89 (t, J = 7.5 Hz, 1H), 6.79 (d, J = 7.9 Hz, 1H), 4.51 (s, 2H), 3.95 (s, 2H), 3.62 (s, 2H), 2.78 (t, J = 5.9 Hz, 2H), 1.48 (s, 9H).
(S)-2-amino-3-(4-hydroxy-2,6-dimethylphenyl)-1-(7-(2-hydroxybenzyl)-3,4-dihydroisoquinolin-2(1H)-yl)propan-1-one (8b). Following General Procedure B, intermediate 7b (33 mg, 0.10 mmol) was deprotected to yield the amine intermediate as an off-white solid. This intermediate was coupled to diBoc-DMT (42 mg, 0.10 mmol, 1.05 eq) in the presence of PyBOP (50 mg, 0.10 mmol, 1.0 eq), and DIEA (132 µL, 0.97 mmol, 10 eq) to yield the product as a brown oil. No 6Cl-HOBt was used. TFA deprotection yielded the product as a white solid. 1H-NMR (CD3OD, 500 MHz, rotamers) δ 7.01 (ddt, J = 10.9, 5.1, 1.8 Hz, 4H), 6.97 (d, J = 7.6 Hz, 3H), 6.92 (d, J = 7.9 Hz, 1H), 6.88 (d, J = 7.9 Hz, 1H), 6.78–6.70 (m, 4H), 6.55 (s, 1H), 6.40 (s, 2H), 6.33 (s, 2H), 4.61 (d, J = 16.9 Hz, 1H), 4.58–4.49 (m, 2H), 4.47 (d, J = 16.5 Hz, 1H), 4.16 (d, J = 15.6 Hz, 1H), 3.87 (s, 2H), 3.86 (s, 2H), 3.75–3.68 (m, 1H), 3.66–3.59 (m, 1H), 3.34 (d, J = 15.8 Hz, 1H), 3.26–3.17 (m, 3H), 3.10–3.05 (m, 2H), 2.72–2.65 (m, 2H), 2.65–2.59 (m, 2H), 2.55–2.45 (m, 1H), 2.24 (s, 6H), 2.19 (s, 6H), 2.03–1.95 (m, 2H). HPLC retention time: 32.8 min. HREIMS m/z 431.2337 (calcd. for C27H30N2O3, 431.2329).
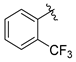
tert-Butyl 7-(2-(trifluoromethyl)benzyl)-3,4-dihydroisoquinoline-2(1H)-carboxylate (7c). Compound 7c was synthesized following General Procedure C from intermediate 6 (100 mg, 0.278 mmol, 1.0 eq), 1-(bromomethyl)-2-(trifluoromethyl)benzene (133 mg, 0.556 mmol, 2.0 eq), Pd(dppf)Cl2 (20 mg, 0.028 mmol, 0.1 eq), and K2CO3 (115 mg, 0.834 mmol, 3.0 eq) to yield a colorless oil (27 mg 25%). 1H-NMR (CDCl3, 400 MHz) δ 7.67 (d, J = 7.9 Hz, 1H), 7.43 (t, J = 7.5 Hz, 1H), 7.31 (t, J = 7.7 Hz, 1H), 7.18 (d, J = 7.9 Hz, 1H), 7.06 (d, J = 7.8 Hz, 1H), 6.95 (d, J = 7.0 Hz, 1H), 6.89 (s, 1H), 4.52 (s, 2H), 4.14 (s, 2H), 3.63 (t, J = 5.4 Hz, 2H), 2.80 (t, J = 5.9 Hz, 2H), 1.48 (s, 9H).
(S)-2-amino-3-(4-hydroxy-2,6-dimethylphenyl)-1-(7-(2-(trifluoromethyl)benzyl)-3,4-dihydroisoquinolin-2(1H)-yl)propan-1-one (8c). Following General Procedure B, intermediate 7c (27 mg, 0.069 mmol, 1.0 eq) was deprotected to yield the amine intermediate as a white solid. This intermediate was coupled to diBoc-DMT (30 mg, 0.074 mmol, 1.05 eq) in the presence of PyBOP (36 mg, 0.070 mmol, 1.0 eq) and DIEA (98 μL, 0.700 mmol, 10 eq) to yield the product as a brown oil. No 6Cl-HOBt was used. TFA deprotection yielded the product as an off-white solid (24 mg, 57%, 3 steps). 1H-NMR (CD3OD, 500 MHz, rotamers) δ 7.70–7.66 (m, 2H), 7.54–7.48 (m, 2H), 7.40–7.35 (m, 2H), 7.28–7.22 (m, 2H), 6.98 (d, J = 7.8 Hz, 1H), 6.95–6.91 (m, 2H), 6.89 (s, 1H), 6.86 (d, 1H), 6.47 (s, 1H), 6.41 (s, 2H), 6.31 (s, 2H), 4.61 (d, J = 17.0 Hz, 1H), 4.58–4.51 (m, 2H), 4.49 (d, J = 17.0 Hz, 1H), 4.18 (d, J = 15.8 Hz, 1H), 4.12 (s, 2H), 4.11 (s, 2H), 3.86–3.80 (m, 1H), 3.59–3.52 (m, 1H), 3.39 (d, J = 15.8 Hz, 1H), 3.26–3.18 (m, 3H), 3.11–3.06 (m, 2H), 2.76–2.69 (m, 2H), 2.68–2.63 (m, 1H), 2.56–2.49 (m, 1H), 2.24 (s, 6H), 2.20 (s, 6H), 2.01–1.92 (m, 1H). 19F-NMR (CD3OD, 470 MHz, rotamers) δ −60.76, −77.10. HPLC retention time: 42.2 min. HREIMS m/z 483.2262 (calcd. for C28H29F3N2O2, 483.2254).
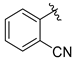
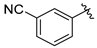
tert-Butyl 7-(2-cyanobenzyl)-3,4-dihydroisoquinoline-2(1H)-carboxylate (7d). Compound 7d was synthesized following General Procedure C from intermediate 6 (75 mg, 0.209 mmol, 1.0 eq), 3-(bromomethyl)phenol (78 mg, 0.418 mmol, 2.0 eq), Pd(dppf)Cl2 (15 mg, 0.021 mmol, 0.1 eq), and K2CO3 (87 mg, 0.627 mmol, 3.0 eq) to yield the product as a colorless oil (26 mg, 37%). 1H-NMR (CDCl3, 500 MHz) δ 7.15 (t, J = 7.8 Hz, 1H), 7.04 (d, J = 7.8 Hz, 1H), 6.99 (d, J = 7.3 Hz, 1H), 6.92 (s, 1H), 6.79–6.74 (m, 1H), 6.69 (d, J = 8.1 Hz, 1H), 6.65 (s, 1H), 4.52 (s, 2H), 3.87 (s, 2H), 3.62 (t, J = 5.9 Hz, 2H), 2.78 (t, J = 5.9 Hz, 2H), 1.49 (s, 9H).
(S)-2-((2-(2-amino-3-(4-hydroxy-2,6-dimethylphenyl)propanoyl)-1,2,3,4-tetrahydroisoquinolin-7-yl)methyl)benzonitrile (8d). Following General Procedure B, intermediate 7d (20 mg, 0.057 mmol, 1.0 eq) was deprotected to yield the amine intermediate as a colorless oil. This intermediate was coupled to diBoc-DMT (24 mg, 0.059 mmol, 1.05 eq) in the presence of PyBOP (29 mg, 0.056 mmol, 1.0 eq) and DIEA (80 μL, 0.560 mmol, 10 eq) to yield the product as a brown oil. No 6Cl-HOBt was used. TFA deprotection yielded the product as a white solid (3 mg, 10%, 3 steps). 1H-NMR (CD3OD, 500 MHz, rotamers) δ 7.70 (s, 1H), 7.68 (s, 1H), 7.62–7.57 (m, 2H), 7.45–7.35 (m, 4H), 7.03 (d, J = 8.1 Hz, 1H), 7.00 (s, 1H), 6.99–6.97 (m, 2H), 6.95 (d, J = 7.8 Hz, 1H), 6.56 (s, 1H), 6.39 (s, 2H), 6.29 (s, 2H), 4.62 (d, J = 16.9 Hz, 1H), 4.57–4.49 (m, 3H), 4.18 (d, J = 15.9 Hz, 1H), 4.15 (s, 2H), 4.13 (d, J = 3.1 Hz, 2H), 3.58–3.51 (m, 1H), 3.43 (d, J = 16.2 Hz, 0H), 3.26–3.18 (m, 4H), 3.06 (dd, J = 13.8, 4.2 Hz, 2H), 2.74–2.69 (m, 2H), 2.69–2.62 (m, 1H), 2.56–2.49 (m, 1H), 2.24 (s, 6H), 2.19 (s, 6H), 2.01–1.95 (m, 1H). HPLC retention time: 34.4 min. HREIMS m/z 440.2336 (calcd. for C28H29N3O2, 440.2333).
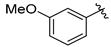
Tert-butyl 7-(3-methoxybenzyl)-3,4-dihydroisoquinoline-2(1H)-carboxylate (7e). Compound 7e was synthesized following General Procedure D from 3-methoxybenzyl bromide (42 μL, 0.30 mmol, 1.0 eq), intermediate 6 (161 mg, 0.45 mmol, 1.5 eq), Pd(dppf)Cl2 (22 mg, 0.03 mmol, 0.1 eq), and K2CO3 (124 mg, 0.90 mmol, 3.0 eq) to yield the product as a colorless oil (42 mg, 40%). 1H-NMR (CDCl3, 500 MHz) δ 7.22 (t, J = 7.8 Hz, 1H), 7.06 (d, J = 7.8 Hz, 1H), 7.01 (d, J = 7.9 Hz, 1H), 6.94 (s, 1H), 6.79 (d, J = 7.5 Hz, 1H), 6.77–6.73 (m, 2H), 4.53 (s, 2H), 3.91 (s, 2H), 3.79 (s, 3H), 3.63 (br s, 2H), 2.80 (t, J = 5.9 Hz, 2H), 1.50 (s, 9H).
(S)-2-amino-3-(4-hydroxy-2,6-dimethylphenyl)-1-(7-(3-methoxybenzyl)-3,4-dihydroisoquinolin-2(1H)-yl)propan-1-one (8e). Following General Procedure B, intermediate 7e (42 mg, 0.119 mmol, 1.0 eq) was deprotected to yield the amine intermediate as a white solid. This intermediate was coupled to diBoc-DMT (50 mg, 0.123 mmol, 1.05 eq) in the presence of PyBOP (61 mg, 0.117 mmol, 1.0 eq), 6Cl-HOBt (20 mg, 0.117 mmol, 1.0 eq), and DIEA (164 µL, 1.17 mmol, 10 eq). Silica gel chromatography yielded the coupled product as a colorless oil (66 mg, 88%). TFA deprotection yielded the product as a white solid. 1H-NMR (CD3OD, 500 MHz, rotamers) δ 7.16 (td, J = 7.8, 3.5 Hz, 2H), 6.99 (dd, J = 7.7, 1.7 Hz, 1H), 6.97–6.93 (m, 3H), 6.91 (d, J = 7.8 Hz, 1H), 6.76–6.70 (m, 6H), 6.49 (s, 1H), 6.40 (s, 2H), 6.32 (s, 2H), 4.61 (d, J = 16.9 Hz, 1H), 4.58–4.53 (m, 2H), 4.50 (d, J = 16.9 Hz, 1H), 4.17 (d, J = 15.7 Hz, 1H), 3.87 (s, 2H), 3.86 (s, 2H), 3.78–3.76 (m, 1H), 3.75 (s, 3H), 3.74 (s, 3H), 3.66–3.53 (m, 1H), 3.37 (d, J = 15.8 Hz, 1H), 3.26–3.17 (m, 3H), 3.11–3.05 (m, 2H), 2.70 (q, J = 7.3, 6.7 Hz, 2H), 2.67–2.62 (m, 1H), 2.52 (dt, J = 16.2, 6.2 Hz, 1H), 2.24 (s, 6H), 2.19 (s, 6H), 2.01–1.94 (m, 1H). HPLC retention time: 37.9 min. EIMS m/z 445.3 (calcd. for C28H32N2O3, 445.24).
tert-Butyl 7-(3-hydroxybenzyl)-3,4-dihydroisoquinoline-2(1H)-carboxylate (7f). Compound 7f was synthesized following General Procedure C from intermediate 6 (75 mg, 0.209 mmol, 1.0 eq), 3-(bromomethyl)phenol (78 mg, 0.418 mmol, 2.0 eq), Pd(dppf)Cl2 (15 mg, 0.021 mmol, 0.1 eq), and K2CO3 (87 mg, 0.627 mmol, 3.0 eq) to yield the product as a colorless oil (26 mg, 37%). 1H-NMR (CDCl3, 500 MHz) δ 7.15 (t, J = 7.8 Hz, 1H), 7.04 (d, J = 7.8 Hz, 1H), 6.99 (d, J = 7.3 Hz, 1H), 6.92 (s, 1H), 6.79–6.74 (m, 1H), 6.69 (d, J = 8.1 Hz, 1H), 6.65 (s, 1H), 4.52 (s, 2H), 3.87 (s, 2H), 3.62 (t, J = 5.9 Hz, 2H), 2.78 (t, J = 5.9 Hz, 2H), 1.49 (s, 9H).
(S)-2-amino-3-(4-hydroxy-2,6-dimethylphenyl)-1-(7-(3-hydroxybenzyl)-3,4-dihydroisoquinolin-2(1H)-yl)propan-1-one (8f). Following General Procedure B, intermediate 7f (26 mg, 0.077 mmol, 1.0 eq) was deprotected to yield the amine intermediate as a colorless oil. This intermediate was coupled to diBoc-DMT (33 mg, 0.080 mmol, 1.05 eq) in the presence of PyBOP (40 mg, 0.076 mmol, 1.0 eq) and DIEA (110 μL, 0.760 mmol, 10 eq) to yield the crude product as a brown oil. No 6Cl-HOBt was used. The crude product was purified via silica gel chromatography in ethyl acetate/hexanes. Subsequent TFA deprotection yielded the product as a white solid (8 mg, 40%, 3 steps). 1H-NMR (CD3OD, 500 MHz, rotamers) δ 7.09–7.03 (m, 2H), 6.99 (d, J = 8.5 Hz, 1H), 6.96–6.93 (m, 3H), 6.91 (d, J = 7.8 Hz, 1H), 6.65 (s, 1H), 6.64 (s, 1H), 6.61–6.57 (m, 3H), 6.49 (s, 1H), 6.40 (s, 2H), 6.32 (s, 2H), 4.62 (d, J = 16.9 Hz, 1H), 4.58–4.52 (m, 2H), 4.49 (d, J = 16.7 Hz, 1H), 4.17 (d, J = 15.7 Hz, 1H), 3.82 (s, 2H), 3.81 (s, 2H), 3.79–3.72 (m, 1H), 3.62–3.56 (m, 1H), 3.37 (d, J = 15.8 Hz, 1H), 3.26–3.17 (m, 3H), 3.07 (dd, J = 13.7, 4.2 Hz, 2H), 2.75–2.67 (m, 2H), 2.67–2.60 (m, 1H), 2.55–2.48 (m, 1H), 2.24 (s, 6H), 2.20 (s, 6H), 2.02–1.95 (m, 1H). HPLC retention time: 30.9 min. HREIMS m/z 431.2331 (calcd. for C27H30N2O3, 431.2329).
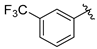
Tert-butyl 7-(3-(trifluoromethyl)benzyl)-3,4-dihydroisoquinoline-2(1H)-carboxylate (7g). Compound 7g was synthesized following General Procedure D from 3-(trifluromethyl)benzyl bromide (33 μL, 0.21 mmol, 1.0 eq), intermediate 6 (114 mg, 0.32 mmol, 1.5 eq), Pd(dppf)Cl2 (15 mg, 0.02 mmol, 0.1 eq), and K2CO3 (88 mg, 0.64 mmol, 3.0 eq) to yield the product as a colorless oil (28 mg, 34%). EIMS m/z 414.2 (calcd. for C22H24F3NO2 + Na, 414.17).
(S)-2-amino-3-(4-hydroxy-2,6-dimethylphenyl)-1-(7-(3-(trifluoromethyl)benzyl)-3,4-dihydroisoquinolin-2(1H)-yl)propan-1-one (8g). Following General Procedure B, intermediate 7g (28 mg, 0.072 mmol, 1.0 eq) was deprotected to yield the amine intermediate as a yellow oil. The crude product was rinsed with two 2 mL portions of diethyl ether to yield a white solid. This intermediate was coupled to diBoc-DMT (56 mg, 0.138 mmol, 1.92 eq) in the presence of PyBOP (68 mg, 0.131 mmol, 1.82 eq), 6Cl-HOBt (22 mg, 0.131 mmol, 1.82 eq), and DIEA (184 µL, 1.31 mmol, 18 eq). Silica gel chromatography yielded the coupled product as a colorless oil (64 mg, 72%). TFA deprotection yielded the product as a white solid. 1H-NMR (CD3OD, 500 MHz, rotamers) δ 7.50–7.43 (m, 8H), 7.02–6.97 (m, 3H), 6.96–6.93 (m, 2H), 6.48 (s, 1H), 6.39 (s, 2H), 6.30 (s, 2H), 4.62 (d, J = 16.9 Hz, 1H), 4.59–4.51 (m, 2H), 4.52 (d, J = 17.3 Hz, 1H), 4.18 (d, J = 15.8 Hz, 1H), 4.00 (s, 2H), 3.99 (s, 2H), 3.81 (dt, J = 12.9, 5.7 Hz, 1H), 3.59–3.52 (m, 1H), 3.40 (d, J = 15.8 Hz, 1H), 3.26–3.17 (m, 3H), 3.08 (dt, J = 13.8, 4.1 Hz, 2H), 2.71 (q, J = 5.4 Hz, 2H), 2.68–2.64 (m, 1H), 2.56–2.49 (m, 1H), 2.23 (s, 6H), 2.19 (s, 6H), 2.00–1.93 (m, 1H). 19F NMR (CD3OD, 470 MHz, rotamers) δ −64.03 (d, J = 28.1 Hz), −77.17. HPLC retention time: 43.3 min. EIMS m/z 483.3 (calcd. for C28H29F3N2O2, 483.22).
Tert-butyl 7-(3-cyanobenzyl)-3,4-dihydroisoquinoline-2(1H)-carboxylate (7h). Compound 7h was synthesized following General Procedure D from 3-(bromomethyl)benzonitrile (40 mg, 0.20 mmol, 1.0 eq), intermediate 6 (109 mg, 0.30 mmol, 1.5 eq), Pd(dppf)Cl2 (15 mg, 0.02 mmol, 0.1 eq), and K2CO3 (84 mg, 0.61 mmol, 3.0 eq) to yield the product as a colorless oil (30 mg, 43%). 1H-NMR (CDCl3, 500 MHz) δ 7.50 (dt, J = 7.4, 1.5 Hz, 1H), 7.45 (s, 1H), 7.43 (t, J = 8.0 Hz, 1H), 7.39 (t, J = 7.6 Hz, 2H), 7.08 (d, J = 7.8 Hz, 1H), 6.96 (d, J = 7.7 Hz, 1H), 6.90 (s, 1H), 4.54 (s, 2H), 3.96 (s, 2H), 3.64 (t, J = 5.9 Hz, 2H), 2.81 (t, J = 5.9 Hz, 2H), 1.49 (s, 9H).
(S)-2-amino-3-(4-hydroxy-2,6-dimethylphenyl)-1-(7-(3-isocyanobenzyl)-3,4-dihydroisoquinolin-2(1H)-yl)propan-1-one (8h). Following General Procedure B, intermediate 7h (30 mg, 0.086 mmol, 1.0 eq) was deprotected to yield the amine intermediate as an off-white solid (21 mg, 84%). The crude product was rinsed with three small portions of diethyl ether. This intermediate was coupled to diBoc-DMT (32 mg, 0.077 mmol, 1.05 eq) in the presence of PyBOP (39 mg, 0.074 mmol, 1.0 eq), 6Cl-HOBt (13 mg, 0.074 mmol, 1.0 eq), and DIEA (104 µL, 0.74 mmol, 10 eq). Silica gel chromatography yielded the coupled product as a white solid (27 mg, 57%). TFA deprotection yielded the product as a white solid. 1H-NMR (CD3OD, 500 MHz, rotamers) δ 7.57–7.51 (m, 6H), 7.48–7.43 (m, 2H), 7.03–6.99 (m, 3H), 6.97–6.94 (m, 2H), 6.48 (s, 1H), 6.39 (s, 2H), 6.29 (s, 2H), 4.63 (d, J = 17.0 Hz, 1H), 4.59–4.49 (m, 2H), 4.53 (d, J = 17.6 Hz, 2H), 4.18 (d, J = 15.8 Hz, 1H), 3.98 (s, 2H), 3.97 (s, 2H), 3.83 (dt, J = 12.3, 5.6 Hz, 1H), 3.59–3.51 (m, 1H), 3.41 (d, J = 15.7 Hz, 1H), 3.27–3.18 (m, 3H), 3.07 (dt, J = 13.8, 4.0 Hz, 2H), 2.74–2.70 (m, 2H), 2.69–2.63 (m, 1H), 2.53 (dt, J = 16.2, 5.9 Hz, 1H), 2.24 (s, 6H), 2.20 (s, 6H), 1.98 (dt, J = 16.0, 5.8 Hz, 1H). HPLC retention time: 35.2 min. EIMS m/z 440.2 (calcd. for C28H29N3O2, 440.23).
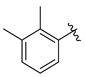
Tert-butyl 7-(2,3-dimethylbenzyl)-3,4-dihydroisoquinoline-2(1H)-carboxylate (7i). Compound 7i was synthesized following General Procedure C from intermediate 6 (100 mg, 0.278 mmol, 1.0 eq), 1-(bromomethyl)-2,3-dimethylbenzene (83 mg, 0.417 mmol, 1.5 eq), Pd(dppf)Cl2 (20 mg, 0.028 mmol, 0.1 eq), and K2CO3 (115 mg, 0.834 mmol, 3.0 eq) to yield the product as a colorless oil (51 mg, 52%). 1H-NMR (CDCl3, 500 MHz) δ 7.09–7.06 (m, 2H), 7.04 (d, J = 7.6 Hz, 1H), 6.99 (d, J = 6.1 Hz, 1H), 6.93 (d, J = 7.9 Hz, 1H), 6.86 (s, 1H), 4.51 (s, 2H), 3.98 (s, 2H), 3.63 (t, J = 6.8 Hz, 2H), 2.79 (t, J = 6.0 Hz, 2H), 2.30 (s, 3H), 2.15 (s, 3H), 1.50 (s, 9H).
(S)-2-amino-1-(7-(2,3-dimethylbenzyl)-3,4-dihydroisoquinolin-2(1H)-yl)-3-(4-hydroxy-2,6-dimethylphenyl)propan-1-one (8i). Following General Procedure B, intermediate 7i (51 mg, 0.145 mmol) was deprotected to yield the amine intermediate. This intermediate was coupled to diBoc-DMT (63 mg, 0.153 mmol, 1.05 eq) in the presence of PyBOP (76 mg, 0.146 mmol, 1.0 eq), and DIEA (199 µL, 1.46 mmol, 10 eq) to yield the product as a brown oil. No 6Cl-HOBt was used. TFA deprotection yielded the product as a white solid. 1H-NMR (CD3OD, 500 MHz, rotamers) δ 7.04–6.97 (m, 5H), 6.97–6.92 (m, 2H), 6.90–6.88 (m, 2H), 6.86–6.82 (m, 2H), 6.41 (s, 1H), 6.40 (s, 2H), 6.30 (s, 2H), 4.58 (d, J = 17.0 Hz, 1H), 4.62–4.48 (m, 2H), 4.46 (d, J = 16.9 Hz, 1H), 4.14 (d, J = 15.7 Hz, 1H), 3.95 (d, J = 2.8 Hz, 2H), 3.93 (s, 2H), 3.83–3.74 (m, 1H), 3.62–3.49 (m, 1H), 3.36 (d, J = 14.8 Hz, 1H), 3.26–3.17 (m, 3H), 3.08 (t, J = 4.9 Hz, 1H), 3.05 (t, J = 4.9 Hz, 1H), 2.72–2.61 (m, 3H), 2.54–2.47 (m, 1H), 2.25 (s, 3H), 2.25 (s, 3H), 2.23 (s, 6H), 2.19 (s, 6H), 2.09 (d, J = 2.2 Hz, 3H), 2.07 (d, J = 1.6 Hz, 3H), 2.00–1.93 (m, 1H). HPLC retention time: 43.2 min. HREIMS m/z 443.2699 (calcd. for C29H34N2O2, 443.2693).
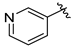
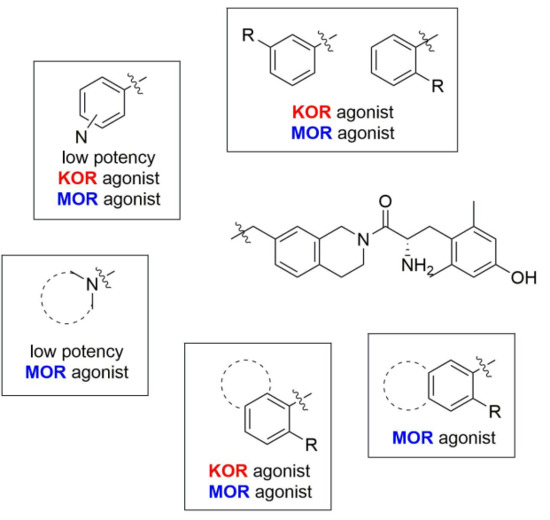
Tert-butyl 7-(pyridin-3-ylmethyl)-3,4-dihydroisoquinoline-2(1H)-carboxylate (7j). Compound 7j was synthesized following General Procedure C from intermediate 6 (104 mg, 0.289 mmol, 1.0 eq), 3-(bromomethyl)pyridine hydrobromide (110 mg, 0.433 mmol, 1.5 eq), Pd(dppf)Cl2 (21 mg, 0.029 mmol, 0.1 eq), and K2CO3 (120 mg, 0.867 mmol, 3.0 eq) to yield the product as a colorless oil (20 mg, 21%).1H-NMR (CDCl3, 500 MHz) δ 8.49 (s, 1H), 8.46 (d, J = 4.1 Hz, 1H), 7.46 (d, J = 7.7 Hz, 1H), 7.20 (dd, J = 7.8, 4.8 Hz, 1H), 7.07 (d, J = 7.9 Hz, 1H), 6.97 (d, J = 7.9 Hz, 1H), 6.91 (s, 1H), 4.52 (s, 2H), 3.93 (s, 2H), 3.62 (br s, 2H), 2.79 (t, J = 6.0 Hz, 2H), 1.48 (s, 9H).
(S)-2-amino-3-(4-hydroxy-2,6-dimethylphenyl)-1-(7-(pyridin-3-ylmethyl)-3,4-dihydroisoquinolin-2(1H)-yl)propan-1-one (8j). Following General Procedure B, intermediate 7j (20 mg, 0.062 mmol, 1.0 eq) was deprotected to yield the amine intermediate as a colorless oil. The crude product was rinsed with several small portions of diethyl ether. This intermediate was coupled to diBoc-DMT (26 mg, 0.064 mmol, 1.05 eq) in the presence of PyBOP (32 mg, 0.061 mmol, 1.0 eq), 6Cl-HOBt (10 mg, 0.061 mmol, 1.0 eq), and DIEA (86 µL, 0.61 mmol, 10 eq). Silica gel chromatography yielded the coupled product (8 mg, 21%, 2 steps). TFA deprotection yielded the product as a white solid. 1H-NMR (CD3OD, 500 MHz, rotamers) δ 8.67–8.61 (m, 4H), 8.29 (d, J = 8.1 Hz, 1H), 8.26 (d, J = 8.0 Hz, 1H), 7.87 (dd, J = 8.1, 5.5 Hz, 1H), 7.83 (dd, J = 8.0, 5.5 Hz, 1H), 7.09–6.98 (m, 5H), 6.56 (s, 1H), 6.39 (s, 2H), 6.23 (s, 2H), 4.64 (d, J = 17.1 Hz, 1H), 4.59–4.52 (m, 2H), 4.53 (d, J = 16.6 Hz, 1H), 4.21 (d, J = 15.8 Hz, 1H), 4.14 (s, 2H), 4.12 (d, J = 3.2 Hz, 2H), 3.97 (dt, J = 13.2, 5.3 Hz, 1H), 3.48 (d, J = 16.1 Hz, 1H), 3.43 (dd, J = 13.0, 6.5 Hz, 1H), 3.26–3.19 (m, 3H), 3.08 (ddd, J = 13.7, 6.4, 4.1 Hz, 2H), 2.72 (t, J = 6.0 Hz, 2H), 2.69–2.62 (m, 1H), 2.57–2.49 (m, 1H), 2.23 (s, 6H), 2.20 (s, 6H), 1.99 (dt, J = 16.1, 6.1 Hz, 1H). HPLC retention time: 16.9 min. EIMS m/z 416.2 (calcd. for C26H29N3O2, 416.23).
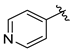
Tert-butyl 7-(pyridin-4-ylmethyl)-3,4-dihydroisoquinoline-2(1H)-carboxylate (7k). Compound 7k was synthesized following General Procedure C from intermediate 6 (112 mg, 0.312 mmol, 1.0 eq), 4-(bromomethyl)pyridine hydrobromide (118 mg, 0.468 mmol, 1.5 eq), Pd(dppf)Cl2 (23 mg, 0.031 mmol, 0.1 eq), and K2CO3 (129 mg, 0.936 mmol, 3.0 eq) to yield the product as a colorless oil (20 mg, 20%). 1H-NMR (CDCl3, 500 MHz) δ 8.49 (d, J = 5.4 Hz, 2H), 7.13–7.05 (m, 3H), 6.97 (d, J = 7.8 Hz, 1H), 6.91 (s, 1H), 4.53 (s, 2H), 3.91 (s, 2H), 3.63 (s, 2H), 2.80 (t, J = 5.0 Hz, 2H), 1.48 (s, 9H).
(S)-2-amino-3-(4-hydroxy-2,6-dimethylphenyl)-1-(7-(pyridin-4-ylmethyl)-3,4-dihydroisoquinolin-2(1H)-yl)propan-1-one (8k). Following General Procedure B, intermediate 7k (20 mg, 0.062 mmol, 1.0 eq) was deprotected to yield the amine intermediate as a cloudy, yellow oil. The crude product was rinsed with several small portions of diethyl ether. This intermediate was coupled to diBoc-DMT (26 mg, 0.064 mmol, 1.05 eq) in the presence of PyBOP (32 mg, 0.061 mmol, 1.0 eq), 6Cl-HOBt (10 mg, 0.061 mmol, 1.0 eq), and DIEA (86 µL, 0.61 mmol, 10 eq). Silica gel chromatography yielded the coupled product (24 mg, 63%, 2 steps). TFA deprotection yielded the product as a white solid. 1H-NMR (CD3OD, 500 MHz, rotamers) δ 8.69–8.65 (m, 4H), 7.81–7.77 (m, 4H), 7.10–7.07 (m, 2H), 7.06 (d, J = 7.8 Hz, 1H), 7.01 (d, 2H), 6.58 (s, 1H), 6.40 (s, 2H), 6.25 (s, 2H), 4.65 (d, J = 17.1 Hz, 1H), 4.58–4.55 (m, 2H), 4.53 (d, J = 16.8 Hz, 1H), 4.25–4.22 (m, 3H), 4.20 (d, J = 3.0 Hz, 2H), 3.96 (dt, J = 12.9, 5.3 Hz, 1H), 3.48 (d, J = 15.0 Hz, 1H), 3.46–3.42 (m, 1H), 3.27–3.19 (m, 3H), 3.08 (ddd, J = 13.7, 6.2, 4.2 Hz, 2H), 2.74 (t, J = 6.1 Hz, 2H), 2.70–2.62 (m, 1H), 2.58–2.51 (m, 1H), 2.24 (s, 6H), 2.21 (s, 6H), 2.00 (dt, J = 16.2, 5.8 Hz, 1H). HPLC retention time: 17.0 min. EIMS m/z 416.3 (calcd. for C26H29N3O2, 416.23).
Tert-butyl 7-(piperidin-1-ylmethyl)-3,4-dihydroisoquinoline-2(1H)-carboxylate (7l). Compound 7l was synthesized following General Procedure E from compound 3 (35 mg, 0.107 mmol, 1.0 eq), piperidine (13 μL, 0.129 mmol, 1.2 eq), and K2CO3 (18 mg, 0.129 mmol, 1.2 eq) to yield the product as an orange oil (27 mg, 77%). 1H-NMR (CDCl3, 500 MHz) δ 7.10 (d, J = 8.0 Hz, 1H), 7.09–7.03 (m, 2H), 4.56 (s, 2H), 3.63 (s, 2H), 3.42 (s, 2H), 2.80 (t, J = 5.7 Hz, 2H), 2.36 (s, 4H), 1.57 (p, J = 5.5 Hz, 4H), 1.48 (s, 9H), 1.47–1.39 (m, 2H).
(S)-2-amino-3-(4-hydroxy-2,6-dimethylphenyl)-1-(7-(piperidin-1-ylmethyl)-3,4-dihydroisoquinolin-2(1H)-yl)propan-1-one (8l). Following General Procedure B, intermediate 7l (27 mg, 0.082 mmol, 1.0 eq) was deprotected to yield the amine intermediate as an orange oil. The crude product was rinsed with several small portions of diethyl ether. This intermediate was coupled to diBoc-DMT (35 mg, 0.087 mmol, 1.05 eq) in the presence of PyBOP (43 mg, 0.082 mmol, 1.0 eq), 6Cl-HOBt (14 mg, 0.082 mmol, 1.0 eq), and DIEA (115 µL, 0.82 mmol, 10 eq). TFA deprotection yielded the product as a white solid. 1H-NMR (CD3OD, 500 MHz, rotamers) δ 6.48–6.45 (m, 2H), 6.41 (d, J = 7.8 Hz, 1H), 6.35 (d, J = 7.8 Hz, 1H), 6.32 (d, J = 8.3 Hz, 1H), 5.97 (s, 1H), 5.59 (s, 2H), 5.41 (s, 2H), 3.89 (d, J = 17.3 Hz, 1H), 3.82–3.76 (m, 3H), 3.46 (d, J = 16.0 Hz, 1H), 3.43–3.32 (m, 4H), 3.24 (dt, J = 13.0, 5.2 Hz, 1H), 2.73 (d, J = 16.1 Hz, 2H), 2.67–2.56 (m, 5H), 2.46–2.37 (m, 3H), 2.32–2.25 (m, 2H), 2.10 (q, J = 10.9 Hz, 4H), 1.97 (t, J = 5.7 Hz, 2H), 1.93–1.85 (m, 1H), 1.79 (dt, J = 16.4, 6.5 Hz, 1H), 1.43 (s, 6H), 1.40 (s, 6H), 1.21 (dt, J = 16.4, 5.4 Hz, 1H), 1.13 (s, 2H), 1.10 (s, 3H), 1.01 (d, J = 14.4 Hz, 2H), 0.92 (q, J = 13.2 Hz, 4H), 0.74–0.66 (m, 2H). HPLC retention time: 16.7 min. EIMS m/z 422.3 (calcd. for C26H35N3O2, 422.27).
Tert-butyl 7-(pyrrolidin-1-ylmethyl)-3,4-dihydroisoquinoline-2(1H)-carboxylate (7m). Compound 7m was synthesized following General Procedure E from compound 3 (29 mg, 0.089 mmol, 1.0 eq), pyrrolidine (9 μL, 0.107 mmol, 1.2 eq), and K2CO3 (15 mg, 0.107 mmol, 1.2 eq) to yield the product as a dark yellow-orange oil (28 mg, 100%). 1H-NMR (CDCl3, 500 MHz) δ 7.13–7.10 (m, 1H), 7.10–7.06 (m, 2H), 4.56 (s, 2H), 3.67–3.61 (m, 2H), 3.58 (s, 2H), 2.80 (d, J = 6.1 Hz, 2H), 2.55–2.47 (m, 4H), 1.79 (p, J = 3.0 Hz, 4H), 1.49 (s, 9H).
(S)-2-amino-3-(4-hydroxy-2,6-dimethylphenyl)-1-(7-(pyrrolidin-1-ylmethyl)-3,4-dihydroisoquinolin-2(1H)-yl)propan-1-one (8m). Following General Procedure B, intermediate 7m (28 mg, 0.088 mmol, 1.0 eq) was deprotected to yield the amine intermediate. The crude product was rinsed with several small portions of diethyl ether. This intermediate was coupled to diBoc-DMT (37 mg, 0.091 mmol, 1.05 eq) in the presence of PyBOP (45 mg, 0.087 mmol, 1.0 eq), 6Cl-HOBt (15 mg, 0.087 mmol, 1.0 eq), and DIEA (122 µL, 0.87 mmol, 10 eq). TFA deprotection yielded the product as a white solid. 1H-NMR (CD3OD, 500 MHz, rotamers) δ 7.31–7.26 (m, 2H), 7.24 (d, J = 7.6 Hz, 2H), 7.17 (d, J = 7.9 Hz, 1H), 7.13 (d, J = 7.8 Hz, 1H), 6.79 (s, 1H), 6.41 (s, 2H), 6.23 (s, 2H), 4.70 (d, J = 17.2 Hz, 1H), 4.61 (d, J = 17.0 Hz, 1H), 4.62–4.51 (m, 2H), 4.31 (s, 2H), 4.31–4.22 (m, 3H), 4.03 (dt, J = 11.7, 5.2 Hz, 1H), 3.53 (d, J = 16.0 Hz, 1H), 3.51–3.41 (m, 5H), 3.29–3.25 (m, 1H), 3.26–3.21 (m, 2H), 3.20–3.13 (m, 4H), 3.12–3.06 (m, 2H), 2.79 (t, J = 6.0 Hz, 2H), 2.76–2.67 (m, 1H), 2.59 (dt, J = 16.4, 6.0 Hz, 1H), 2.24 (s, 6H), 2.22 (s, 6H), 2.19–2.16 (m, 4H), 2.05–1.97 (m, 5H). HPLC retention time: 15.3 min. HREIMS m/z 408.2649 (calcd. for C25H33N3O2, 408.2646).
tert-butyl 7-(morpholinomethyl)-3,4-dihydroisoquinoline-2(1H)-carboxylate (7n). Compound 7n was synthesized following General Procedure E from compound 3 (31 mg, 0.095 mmol, 1.0 eq), morpholine (10 μL, 0.114 mmol, 1.2 eq), and K2CO3 (16 mg, 0.114 mmol, 1.2 eq) to yield the product as a pale yellow oil (32 mg, 100%). 1H-NMR (CDCl3, 500 MHz) δ 7.11 (d, J = 7.8 Hz, 1H), 7.08 (s, 2H), 4.56 (s, 2H), 3.70 (t, J = 4.7 Hz, 4H), 3.64 (t, J = 6.5 Hz, 2H), 3.45 (s, 2H), 2.81 (t, J = 5.9 Hz, 2H), 2.43 (t, J = 4.6 Hz, 4H), 1.49 (s, 9H).
(S)-2-amino-3-(4-hydroxy-2,6-dimethylphenyl)-1-(7-(morpholinomethyl)-3,4-dihydroisoquinolin-2(1H)-yl)propan-1-one (8n). Following General Procedure B, intermediate 7n (28 mg, 0.088 mmol, 1.0 eq) was deprotected to yield the amine intermediate. The crude product was rinsed with several small portions of diethyl ether. This intermediate was coupled to diBoc-DMT (37 mg, 0.091 mmol, 1.05 eq) in the presence of PyBOP (45 mg, 0.087 mmol, 1.0 eq), 6Cl-HOBt (15 mg, 0.087 mmol, 1.0 eq), and DIEA (122 µL, 0.87 mmol, 10 eq). TFA deprotection yielded the product as a white solid. 1H-NMR (CD3OD, 500 MHz, rotamers) δ 7.30–7.28 (m, 2H), 7.24 (d, J = 7.8 Hz, 1H), 7.17 (d, J = 7.6 Hz, 1H), 7.14 (d, J = 7.8 Hz, 1H), 6.81 (s, 1H), 6.40 (s, 2H), 6.22 (s, 2H), 4.70 (d, J = 17.3 Hz, 1H), 4.63–4.55 (m, 3H), 4.31 (s, 2H), 4.29–4.21 (m, 3H), 4.13–4.05 (m, 1H), 4.08–3.99 (m, 4H), 3.74 (q, J = 12.7 Hz, 4H), 3.55 (d, J = 16.0 Hz, 1H), 3.42–3.32 (m, 5H), 3.28–3.25 (m, 1H), 3.25–3.20 (m, 2H), 3.21–3.12 (m, 4H), 3.13–3.06 (m, 2H), 2.79 (t, J = 6.1 Hz, 2H), 2.75–2.66 (m, 1H), 2.60 (dt, J = 16.5, 5.9 Hz, 1H), 2.24 (s, 6H), 2.22 (s, 6H), 2.03 (dt, J = 11.1, 5.7 Hz, 1H). HPLC retention time: 14.0 min. HREIMS m/z 424.2597 (calcd. for C25H33N3O3, 424.2595).
Tert-butyl 7-(naphthalen-1-ylmethyl)-3,4-dihydroisoquinoline-2(1H)-carboxylate (7o). Compound 7o was synthesized following General Procedure C from intermediate 6 (100 mg, 0.278 mmol, 1.0 eq), 1-(bromomethyl)naphthalene (123 mg, 0.556 mmol, 2.0 eq), Pd(dppf)Cl2 (20 mg, 0.028 mmol, 0.1 eq), and K2CO3 (115 mg, 0.834 mmol, 3.0 eq) to yield the product as a yellow oil (41 mg, 39%). 1H-NMR (CDCl3, 500 MHz) δ 8.03–7.98 (m, 1H), 7.89–7.86 (m, 1H), 7.78 (d, J = 8.2 Hz, 1H), 7.49–7.45 (m, 2H), 7.43 (d, J = 7.7 Hz, 1H), 7.31 (d, J = 7.1 Hz, 1H), 7.03 (s, 2H), 6.95 (s, 1H), 4.50 (s, 2H), 4.41 (s, 2H), 3.63 (s, 2H), 2.80 (t, J = 5.9 Hz, 2H), 1.49 (s, 9H).
(S)-2-amino-3-(4-hydroxy-2,6-dimethylphenyl)-1-(7-(naphthalen-1-ylmethyl)-3,4-dihydroisoquinolin-2(1H)-yl)propan-1-one (8o). Following General Procedure B, intermediate 7o (41 mg, 0.110 mmol, 1.0 eq) was deprotected to yield the amine intermediate. This intermediate was coupled to diBoc-DMT (47 mg, 0.115 mmol, 1.05 eq) in the presence of PyBOP (57 mg, 0.110 mmol, 1.0 eq), 6Cl-HOBt (57 mg, 0.330 mmol, 3.0 eq), and DIEA (154 µL, 1.1 mmol, 10 eq). Silica gel chromatography yielded the coupled product (57 mg, 78%, 2 steps). TFA deprotection yielded the product as a white solid. 1H-NMR (CD3OD, 500 MHz, rotamers) δ 7.98–7.94 (m, 2H), 7.86–7.83 (m, 2H), 7.76 (d, J = 8.2 Hz, 2H), 7.46–7.39 (m, 6H), 7.35 (d, J = 7.0 Hz, 1H), 7.32 (d, J = 6.9 Hz, 1H), 7.00 (d, J = 7.8 Hz, 1H), 6.97–6.94 (m, 2H), 6.92 (s, 1H), 6.88 (d, J = 7.8 Hz, 1H), 6.47 (s, 1H), 6.38 (s, 2H), 6.32 (s, 2H), 4.55 (d, J = 17.1 Hz, 1H), 4.52–4.47 (m, 2H), 4.44 (d, J = 16.9 Hz, 1H), 4.38 (s, 4H), 4.10 (d, J = 15.8 Hz, 1H), 3.79 (dt, J = 11.9, 5.6 Hz, 1H), 3.56–3.49 (m, 1H), 3.36 (d, J = 15.8 Hz, 1H), 3.24–3.15 (m, 3H), 3.05 (dt, J = 13.7, 5.1 Hz, 2H), 2.68 (t, J = 6.2 Hz, 2H), 2.66–2.60 (m, 1H), 2.49 (dt, J = 16.0, 6.2 Hz, 1H), 2.21 (s, 6H), 2.16 (s, 6H), 1.94 (dt, J = 16.1, 6.0 Hz, 1H). HPLC retention time: 43.3 min. EIMS m/z 465.2 (calcd. for C31H32N2O2, 465.25).
Tert-butyl 7-(naphthalen-2-ylmethyl)-3,4-dihydroisoquinoline-2(1H)-carboxylate (7p). Compound 7p was synthesized following General Procedure C from intermediate 6 (100 mg, 0.278 mmol, 1.0 eq), 2-(bromomethyl)naphthalene (123 mg, 0.556 mmol, 2.0 eq), Pd(dppf)Cl2 (20 mg, 0.028 mmol, 0.1 eq), and K2CO3 (115 mg, 0.834 mmol, 3.0 eq) to yield the product (42 mg, 40%). 1H-NMR (CDCl3, 500 MHz) δ 7.79 (q, J = 8.1, 7.7 Hz, 3H), 7.65 (s, 1H), 7.45 (p, J = 7.1, 6.5 Hz, 2H), 7.32 (d, J = 8.5 Hz, 1H), 7.06 (s, 2H), 6.97 (s, 1H), 4.53 (s, 2H), 4.11 (s, 2H), 3.64 (s, 2H), 2.81 (t, J = 6.0 Hz, 2H), 1.49 (s, 9H).
(S)-2-amino-3-(4-hydroxy-2,6-dimethylphenyl)-1-(7-(naphthalen-2-ylmethyl)-3,4-dihydroisoquinolin-2(1H)-yl)propan-1-one (8p). Following General Procedure B, intermediate 7p (42 mg, 0.112 mmol, 1.0 eq) was deprotected to yield the amine intermediate. Half of this intermediate (18 mg, 0.058 mmol, 1.0 eq) was coupled to diBoc-DMT (25 mg, 0.061 mmol, 1.05 eq) in the presence of PyBOP (30 mg, 0.058 mmol, 1.0 eq), 6Cl-HOBt (10 mg, 0.058 mmol, 1.0 eq), and DIEA (81 µL, 0.58 mmol, 10 eq). TFA deprotection yielded the product as a brown solid. 1H-NMR (CD3OD, 500 MHz, rotamers) δ 7.81–7.72 (m, 6H), 7.63 (d, J = 6.7 Hz, 2H), 7.46–7.38 (m, 4H), 7.29 (d, J = 8.3 Hz, 2H), 7.05 (d, J = 8.5 Hz, 1H), 7.02–6.96 (m, 3H), 6.93 (d, J = 7.8 Hz, 1H), 6.53 (s, 1H), 6.40 (s, 2H), 6.32 (s, 2H), 4.61 (d, J = 16.9 Hz, 1H), 4.57–4.48 (m, 2H), 4.50 (d, J = 16.8 Hz, 1H), 4.16 (d, J = 15.8 Hz, 1H), 4.07 (s, 2H), 4.06 (s, 2H), 3.77 (dt, J = 12.1, 5.7 Hz, 1H), 3.61–3.53 (m, 1H), 3.38 (d, J = 15.8 Hz, 1H), 3.24–3.17 (m, 3H), 3.06 (dt, J = 13.8, 5.2 Hz, 2H), 2.71 (q, J = 5.8 Hz, 2H), 2.67–2.61 (m, 1H), 2.52 (dt, J = 15.7, 6.3 Hz, 1H), 2.22 (s, 6H), 2.18 (s, 6H), 1.98 (dt, J = 16.2, 5.9 Hz, 1H). HPLC retention time: 43.5 min. EIMS m/z 465.3 (calcd. for C31H32N2O2, 465.25).
Tert-butyl 7-(quinolin-8-ylmethyl)-3,4-dihydroisoquinoline-2(1H)-carboxylate (7q). Compound 7q was synthesized following General Procedure A from compound 3 (75 mg, 0.23 mmol, 1.0 eq), quinolin-8-ylboronic acid (60 mg, 0.345 mmol, 1.5 eq), Pd(dppf)Cl2 (17 mg, 0.023 mmol, 0.1 eq), and K2CO3 (95 mg, 0.69 mmol, 3.0 eq) to yield the product as a colorless oil (18 mg, 21%). 1H-NMR (CDCl3, 500 MHz) δ 8.97 (dd, J = 4.2, 1.8 Hz, 1H), 8.15 (dd, J = 8.3, 1.8 Hz, 1H), 7.74–7.65 (m, 1H), 7.46 (s, 1H), 7.44 (s, 1H), 7.41 (dd, J = 8.3, 4.2 Hz, 1H), 7.13 (dd, J = 7.6, 1.8 Hz, 1H), 7.07 (s, 1H), 7.04 (d, J = 7.9 Hz, 1H), 4.64 (s, 2H), 4.52 (s, 2H), 3.62 (s, 2H), 2.79 (t, J = 5.3 Hz, 2H), 1.48 (s, 9H).
(S)-2-amino-3-(4-hydroxy-2,6-dimethylphenyl)-1-(7-(quinolin-8-ylmethyl)-3,4-dihydroisoquinolin-2(1H)-yl)propan-1-one (8q). Following General Procedure F, intermediate 7q (18 mg, 0.048 mmol) was deprotected to yield the amine intermediate. This intermediate was coupled to diBoc-DMT (21 mg, 0.05 mmol, 1.05 eq) in the presence of PyBOP (25 mg, 0.048 mmol, 1.0 eq), and DIEA (84 µL, 0.48 mmol, 10 eq) to yield the product. No 6Cl-HOBt was used. TFA deprotection yielded the product as a white solid. 1H-NMR (CD3OD, 500 MHz, rotamers) δ 9.03 (td, J = 4.9, 1.7 Hz, 2H), 8.79 (dd, J = 8.3, 1.7 Hz, 1H), 8.75 (dd, J = 8.3, 1.7 Hz, 1H), 8.04 (dd, J = 8.1, 1.6 Hz, 1H), 8.00 (dd, J = 8.1, 1.4 Hz, 1H), 7.81 (ddd, J = 13.0, 8.3, 4.9 Hz, 2H), 7.75–7.66 (m, 3H), 7.63 (dd, J = 7.3, 1.4 Hz, 1H), 7.04 (dd, J = 7.8, 1.8 Hz, 1H), 7.01 (d, J = 1.7 Hz, 1H), 6.96 (s, 2H), 6.94 (d, J = 7.9 Hz, 1H), 6.58 (s, 1H), 6.38 (s, 2H), 6.24 (s, 2H), 4.61 (d, J = 15.8 Hz, 1H), 4.58 (s, 2H), 4.56 (s, 2H), 4.56–4.52 (m, 2H), 4.47 (d, J = 17.0 Hz, 1H), 4.17 (d, J = 15.9 Hz, 1H), 3.84 (dt, J = 12.9, 5.5 Hz, 1H), 3.54–3.47 (m, 1H), 3.38 (d, J = 15.9 Hz, 1H), 3.25–3.16 (m, 3H), 3.11–3.05 (m, 2H), 2.69 (q, J = 5.5 Hz, 2H), 2.66–2.59 (m, 1H), 2.52 (dt, J = 16.2, 7.4, 4.8 Hz, 1H), 2.21 (s, 6H), 2.18 (s, 6H), 1.98 (ddd, J = 16.2, 6.4, 4.4 Hz, 1H). HPLC retention time: 25.0 min. EIMS m/z 466.3 (calcd. for C30H31N3O2, 466.24).
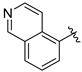
Tert-butyl 7-(isoquinolin-8-ylmethyl)-3,4-dihydroisoquinoline-2(1H)-carboxylate (7r). Compound 7r was synthesized following General Procedure A from compound 3 (77 mg, 0.24 mmol, 1.0 eq), isoquinolin-8-ylboronic acid (49 mg, 0.283 mmol, 1.2 eq), Pd(dppf)Cl2 (18 mg, 0.024 mmol, 0.1 eq), and K2CO3 (98 mg, 0.71 mmol, 3.0 eq) to yield the product as a pale pink oil (55 mg, 62%). 1H-NMR (CDCl3, 500 MHz) δ 9.47 (s, 1H), 8.52 (d, J = 5.6 Hz, 1H), 7.73 (d, J = 8.3 Hz, 1H), 7.68–7.60 (m, 2H), 7.40 (d, J = 7.0 Hz, 1H), 7.02 (q, J = 8.0 Hz, 2H), 6.94 (s, 1H), 4.48 (s, 4H), 3.61 (s, 2H), 2.77 (t, J = 6.1 Hz, 2H), 1.47 (s, 9H).
(S)-2-amino-3-(4-hydroxy-2,6-dimethylphenyl)-1-(7-(isoquinolin-8-ylmethyl)-3,4-dihydroisoquinolin-2(1H)-yl)propan-1-one (8r). Following General Procedure F, intermediate 7r (27 mg, 0.072 mmol) was deprotected to yield the amine intermediate. This intermediate was coupled to diBoc-DMT (31 mg, 0.076 mmol, 1.05 eq) in the presence of PyBOP (37 mg, 0.072 mmol, 1.0 eq), and DIEA (125 µL, 0.72 mmol, 10 eq) to yield the product. No 6Cl-HOBt was used. TFA deprotection yielded the product as a white solid. 1H-NMR (CD3OD, 500 MHz, rotamers) δ 9.73 (s, 1H), 9.72 (s, 1H), 8.54 (t, J = 5.0 Hz, 2H), 8.38 (d, J = 6.3 Hz, 1H), 8.35 (d, J = 6.2 Hz, 1H), 8.17–8.10 (m, 3H), 8.08 (q, J = 7.6 Hz, 1H), 7.82 (d, J = 6.6 Hz, 1H), 7.76 (d, J = 6.9 Hz, 1H), 7.07–7.02 (m, 2H), 7.00–6.94 (m, 3H), 6.53 (s, 1H), 6.36 (s, 2H), 6.17 (s, 2H), 4.61 (s, 2H), 4.60 (s, 2H), 4.57 (d, J = 14.3 Hz, 1H), 4.54 (dd, J = 11.9, 4.1 Hz, 2H), 4.48 (d, J = 17.0 Hz, 1H), 4.16 (d, J = 15.8 Hz, 1H), 3.97 (dt, J = 12.9, 5.3 Hz, 1H), 3.45 (d, J = 15.8 Hz, 1H), 3.38 (dt, J = 13.2, 6.7 Hz, 1H), 3.24–3.15 (m, 3H), 3.07 (ddd, J = 13.6, 11.5, 4.1 Hz, 2H), 2.69 (t, J = 6.0 Hz, 2H), 2.62 (ddd, J = 12.5, 7.5, 4.7 Hz, 1H), 2.52 (ddd, J = 16.2, 7.5, 4.8 Hz, 1H), 2.19 (s, 6H), 2.17 (s, 6H), 1.96 (ddd, J = 16.3, 6.9, 4.7 Hz, 1H). HPLC retention time: 21.4 min. EIMS m/z 466.3 (calcd. for C30H31N3O2, 466.24).
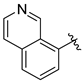
Tert-butyl 7-(isoquinolin-5-ylmethyl)-3,4-dihydroisoquinoline-2(1H)-carboxylate (7s). Compound 7s was synthesized following General Procedure A from compound 3 (73 mg, 0.224 mmol, 1.0 eq), isoquinolin-5-ylboronic acid (46 mg, 0.269 mmol, 1.2 eq), Pd(dppf)Cl2 (16 mg, 0.022 mmol, 0.1 eq), and K2CO3 (93 mg, 0.672 mmol, 3.0 eq) to yield the product as a yellow oil (48 mg, 57%). 1H-NMR (CDCl3, 500 MHz) δ 9.26 (s, 1H), 8.50 (d, J = 6.0 Hz, 1H), 7.88 (d, J = 8.1 Hz, 1H), 7.76 (d, J = 6.0 Hz, 1H), 7.55 (t, J = 7.6 Hz, 1H), 7.50 (d, J = 7.1 Hz, 1H), 7.04 (d, J = 7.8 Hz, 1H), 6.97 (s, 1H), 6.91 (s, 1H), 4.49 (s, 2H), 4.36 (s, 2H), 3.62 (t, J = 5.9 Hz, 2H), 2.78 (t, J = 5.9 Hz, 2H), 1.47 (s, 9H).
(S)-2-amino-3-(4-hydroxy-2,6-dimethylphenyl)-1-(7-(isoquinolin-5-ylmethyl)-3,4-dihydroisoquinolin-2(1H)-yl)propan-1-one (8s). Following General Procedure F, intermediate 7s (24 mg, 0.064 mmol) was deprotected to yield the amine intermediate. This intermediate was coupled to diBoc-DMT (28 mg, 0.067 mmol, 1.05 eq) in the presence of PyBOP (33 mg, 0.064 mmol, 1.0 eq), and DIEA (111 µL, 0.64 mmol, 10 eq) to yield the product. No 6Cl-HOBt was used. TFA deprotection yielded the product as a white solid. 1H-NMR (CD3OD, 500 MHz, rotamers) δ 9.67 (d, J = 7.1 Hz, 2H), 8.52 (d, J = 7.0 Hz, 2H), 8.44 (t, J = 7.6 Hz, 2H), 8.35 (t, J = 8.9 Hz, 2H), 8.04–7.91 (m, 4H), 7.04–6.92 (m, 5H), 6.51 (s, 1H), 6.37 (s, 2H), 6.23 (s, 2H), 4.58 (d, J = 17.4 Hz, 2H), 4.56–4.50 (m, 2H), 4.53 (s, 2H), 4.52 (s, 2H), 4.47 (d, J = 17.1 Hz, 1H), 4.15 (d, J = 15.8 Hz, 1H), 3.90 (dt, J = 12.9, 5.4 Hz, 1H), 3.48–3.39 (m, 1H), 3.41 (d, J = 15.8 Hz, 1H), 3.24–3.16 (m, 3H), 3.07 (ddd, J = 13.3, 8.5, 4.1 Hz, 2H), 2.69 (t, 2H), 2.62 (ddd, J = 12.6, 7.5, 4.7 Hz, 1H), 2.51 (ddd, J = 16.2, 7.3, 4.8 Hz, 1H), 2.21 (s, 6H), 2.17 (s, 6H), 1.96 (ddd, J = 16.2, 6.9, 4.9 Hz, 1H). HPLC retention time: 21.5 min. EIMS m/z 466.3 (calcd. for C30H31N3O2, 466.24).
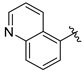
Tert-butyl 7-(quinolin-5-ylmethyl)-3,4-dihydroisoquinoline-2(1H)-carboxylate (7t). Compound 7t was synthesized following General Procedure A from compound 3 (50 mg, 0.153 mmol, 1.0 eq), quinolin-5-ylboronic acid (32 mg, 0.184 mmol, 1.2 eq), Pd(dppf)Cl2 (11 mg, 0.015 mmol, 0.1 eq), and K2CO3 (63 mg, 0.459 mmol, 3.0 eq) to yield the product as a yellow oil (36 mg, 63%). 1H-NMR (CDCl3, 500 MHz) δ 8.90 (dd, J = 4.2, 1.7 Hz, 1H), 8.30 (ddd, J = 8.6, 1.7, 0.9 Hz, 1H), 8.04 (d, J = 8.5 Hz, 1H), 7.66 (t, J = 7.8 Hz, 1H), 7.39–7.33 (m, 2H), 7.03 (d, J = 7.6 Hz, 1H), 6.99–6.85 (m, 2H), 4.48 (s, 2H), 4.39 (s, 2H), 3.60 (t, J = 6.1 Hz, 2H), 2.77 (t, J = 6.0 Hz, 2H), 1.47 (s, 9H).
(S)-2-amino-3-(4-hydroxy-2,6-dimethylphenyl)-1-(7-(quinolin-5-ylmethyl)-3,4-dihydroisoquinolin-2(1H)-yl)propan-1-one (8t). Following General Procedure F, intermediate 7t (36 mg, 0.096 mmol) was deprotected to yield the amine intermediate. This intermediate was coupled to diBoc-DMT (41 mg, 0.100 mmol, 1.05 eq) in the presence of PyBOP (49 mg, 0.095 mmol, 1.0 eq), and DIEA (123 µL, 0.95 mmol, 10 eq) to yield the product. No 6Cl-HOBt was used. TFA deprotection yielded the product as a white solid. 1H-NMR (CD3OD, 500 MHz, rotamers) δ 9.15 (dd, J = 8.5, 5.5 Hz, 2H), 9.10 (td, J = 5.1, 1.5 Hz, 2H), 8.13 (t, J = 9.1 Hz, 2H), 8.05 (ddd, J = 18.3, 8.7, 7.1 Hz, 2H), 7.93 (ddd, J = 8.7, 7.4, 5.2 Hz, 2H), 7.78 (d, J = 7.1 Hz, 1H), 7.74 (d, J = 7.1 Hz, 1H), 7.04–6.92 (m, 5H), 6.50 (s, 1H), 6.36 (s, 2H), 6.23 (s, 2H), 4.59 (d, J = 16.4 Hz, 1H), 4.55 (s, 2H), 4.54 (s, 2H), 4.54–4.52 (m, 2H), 4.47 (d, J = 17.1 Hz, 1H), 4.15 (d, J = 15.8 Hz, 1H), 3.90 (dt, J = 12.9, 5.4 Hz, 1H), 3.43 (dt, J = 13.1, 6.6 Hz, 1H), 3.41 (d, J = 15.9 Hz, 1H), 3.24–3.16 (m, 3H), 3.07 (ddd, J = 13.3, 8.5, 4.1 Hz, 2H), 2.69 (t, J = 6.0 Hz, 2H), 2.63 (ddd, J = 12.5, 7.4, 4.7 Hz, 1H), 2.50 (ddd, J = 16.2, 7.4, 4.8 Hz, 1H), 2.20 (s, 6H), 2.17 (s, 6H), 1.94 (ddd, J = 16.1, 6.8, 4.6 Hz, 1H). HPLC retention time: 21.6 min. EIMS m/z 466.3 (calcd. for C30H31N3O2, 466.24).
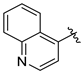
Tert-butyl 7-(quinolin-4-ylmethyl)-3,4-dihydroisoquinoline-2(1H)-carboxylate (7u). Compound 7u was synthesized following General Procedure A from compound 3 (50 mg, 0.153 mmol, 1.0 eq), quinolin-4-ylboronic acid (32 mg, 0.184 mmol, 1.2 eq), Pd(dppf)Cl2 (11 mg, 0.015 mmol, 0.1 eq), and K2CO3 (63 mg, 0.459 mmol, 3.0 eq) to yield the product as a colorless oil (32 mg, 56%). 1H-NMR (CDCl3, 500 MHz) δ 8.83 (d, J = 4.4 Hz, 1H), 8.13 (d, J = 8.4 Hz, 1H), 8.03 (dd, J = 8.5, 1.3 Hz, 1H), 7.70 (t, J = 7.7 Hz, 1H), 7.54 (t, J = 7.7 Hz, 1H), 7.15 (d, J = 4.2 Hz, 1H), 7.07 (d, J = 7.6 Hz, 1H), 7.03–6.88 (m, 2H), 4.50 (s, 2H), 4.40 (s, 2H), 3.63 (t, J = 5.4 Hz, 2H), 2.80 (t, J = 5.8 Hz, 2H), 1.47 (s, 9H).
(S)-2-amino-3-(4-hydroxy-2,6-dimethylphenyl)-1-(7-(quinolin-4-ylmethyl)-3,4-dihydroisoquinolin-2(1H)-yl)propan-1-one (8u). Following General Procedure F, intermediate 7u (32 mg, 0.085 mmol) was deprotected to yield the amine intermediate. This intermediate was coupled to diBoc-DMT (37 mg, 0.089 mmol, 1.05 eq) in the presence of PyBOP (44 mg, 0.085 mmol, 1.0 eq), and DIEA (148 µL, 0.85 mmol, 10 eq) to yield the product. No 6Cl-HOBt was used. TFA deprotection yielded the product as a white solid. 1H-NMR (CD3OD, 500 MHz, rotamers) δ 9.07 (dd, J = 9.2, 5.6 Hz, 2H), 8.55 (t, J = 9.6 Hz, 2H), 8.26 (t, J = 8.8 Hz, 2H), 8.18–8.11 (m, 2H), 8.00–7.93 (m, 2H), 7.78 (d, J = 5.6 Hz, 2H), 7.12 (dd, J = 7.7, 1.8 Hz, 1H), 7.10 (s, 1H), 7.05 (s, 2H), 7.01 (d, J = 7.8 Hz, 1H), 6.60 (s, 1H), 6.37 (s, 2H), 6.20 (s, 2H), 4.74 (s, 2H), 4.71 (s, 2H), 4.61 (d, J = 17.2 Hz, 1H), 4.55 (dd, J = 12.0, 4.1 Hz, 2H), 4.51 (d, J = 17.1 Hz, 1H), 4.20 (d, J = 15.8 Hz, 1H), 3.99 (dt, J = 12.9, 5.3 Hz, 1H), 3.48 (d, J = 15.8 Hz, 1H), 3.40 (dt, J = 13.2, 6.7 Hz, 1H), 3.25–3.17 (m, 3H), 3.08 (ddd, J = 13.9, 9.9, 4.1 Hz, 2H), 2.73 (t, J = 6.1 Hz, 2H), 2.65 (ddd, J = 12.5, 7.4, 4.8 Hz, 1H), 2.54 (ddd, J = 16.4, 7.4, 4.9 Hz, 1H), 2.21 (s, 6H), 2.18 (s, 6H), 1.98 (ddd, J = 16.3, 7.0, 4.7 Hz, 1H). HPLC retention time: 21.2 min. EIMS m/z 466.3 (calcd. for C30H31N3O2, 466.24).
Tert-butyl 7-(isoquinolin-4-ylmethyl)-3,4-dihydroisoquinoline-2(1H)-carboxylate (7v). Compound 7v was synthesized following General Procedure A from compound 3 (50 mg, 0.153 mmol, 1.0 eq), isoquinolin-4-ylboronic acid (32 mg, 0.184 mmol, 1.2 eq), Pd(dppf)Cl2 (11 mg, 0.015 mmol, 0.1 eq), and K2CO3 (63 mg, 0.459 mmol, 3.0 eq) to yield the crude product. Silica gel chromatography yielded a mixture of products. This mixture was used directly in the next step.
(S)-2-amino-3-(4-hydroxy-2,6-dimethylphenyl)-1-(7-(isoquinolin-4-ylmethyl)-3,4-dihydroisoquinolin-2(1H)-yl)propan-1-one (8v). Following General Procedure F, intermediate 7v (19 mg, 0.051 mmol) was deprotected. The crude product was purified by semi-preparative HPLC to yield the product as a white solid (21 mg, 100%). EIMS calcd. for [C19H18N2 + H]+: 275.15, found: 275.2. The amine was coupled to diBoc-DMT (23 mg, 0.057 mmol, 1.05 eq) in the presence of PyBOP (28 mg, 0.054 mmol, 1.0 eq), and DIEA (94 µL, 0.54 mmol, 10 eq) to yield the product. No 6Cl-HOBt was used. TFA deprotection yielded the product as a white solid. 1H-NMR (CD3OD, 500 MHz, rotamers) δ 9.65 (d, J = 6.0 Hz, 2H), 8.50 (t, J = 8.3 Hz, 2H), 8.42–8.35 (m, 4H), 8.22–8.14 (m, 2H), 8.04–7.97 (m, 2H), 7.10 (dd, J = 7.8, 1.8 Hz, 1H), 7.07 (s, 1H), 7.02 (s, 2H), 6.97 (d, J = 7.9 Hz, 1H), 6.56 (s, 1H), 6.37 (s, 2H), 6.21 (s, 2H), 4.61 (d, J = 16.1 Hz, 1H), 4.57 (s, 2H), 4.56 (s, 2H), 4.55–4.52 (m, 2H), 4.49 (d, J = 17.2 Hz, 1H), 4.17 (d, J = 15.8 Hz, 1H), 3.95 (dt, J = 12.9, 5.3 Hz, 1H), 3.45 (d, J = 15.5 Hz, 1H), 3.43–3.38 (m, 1H), 3.25–3.17 (m, 3H), 3.07 (ddd, J = 13.7, 8.1, 4.1 Hz, 2H), 2.72 (t, J = 6.0 Hz, 2H), 2.65 (ddd, J = 12.7, 7.3, 4.8 Hz, 1H), 2.52 (ddd, J = 16.4, 7.1, 4.8 Hz, 1H), 2.21 (s, 6H), 2.17 (s, 6H), 1.95 (dt, J = 16.4, 6.2 Hz, 1H). HPLC retention time: 21.5 min. EIMS m/z 466.3 (calcd. for C30H31N3O2, 466.24).
Tert-butyl 7-((3,4-dihydroisoquinolin-2(1H)-yl)methyl)-3,4-dihydroisoquinoline-2(1H)-carboxylate (7w). Compound 7w was synthesized following General Procedure E from compound 3 (50 mg, 0.153 mmol, 1.0 eq), 1,2,3,4-tetrahydroisoquinoline (23 μL, 0.184 mmol, 1.2 eq), and K2CO3 (25 mg, 0.184 mmol, 1.2 eq) to yield the product as a colorless oil (24 mg, 41%). 1H-NMR (CDCl3, 500 MHz) δ 7.19 (d, J = 7.5 Hz, 1H), 7.15 (s, 1H), 7.12–7.08 (m, 4H), 6.99 (d, J = 7.0 Hz, 1H), 4.57 (s, 2H), 3.66 (br s, 2H), 3.64 (s, 2H), 3.63 (s, 2H), 2.90 (t, J = 6.0 Hz, 2H), 2.83 (t, J = 5.9 Hz, 2H), 2.75 (t, J = 5.9 Hz, 2H), 1.49 (s, 9H).
(S)-2-amino-1-(7-((3,4-dihydroisoquinolin-2(1H)-yl)methyl)-3,4-dihydroisoquinolin-2(1H)-yl)-3-(4-hydroxy-2,6-dimethylphenyl)propan-1-one (8w). Following General Procedure F, intermediate 7w (24 mg, 0.063 mmol, 1.0 eq) was deprotected to yield the amine intermediate. This intermediate was coupled to diBoc-DMT (27 mg, 0.067 mmol, 1.05 eq) in the presence of PyBOP (33 mg, 0.064 mmol, 1.0 eq), and DIEA (111 µL, 0.64 mmol, 10 eq) to yield the product. No 6Cl-HOBt was used. TFA deprotection yielded the product as a white solid. 1H-NMR (CD3OD, 500 MHz, rotamers) δ 7.36–7.33 (m, 2H), 7.32–7.23 (m, 7H), 7.21–7.13 (m, 4H), 6.85 (s, 1H), 6.41 (s, 2H), 6.21 (s, 2H), 4.72 (d, J = 17.3 Hz, 1H), 4.62 (d, J = 17.3 Hz, 1H), 4.64–4.57 (m, 2H), 4.44 (s, 2H), 4.42–4.35 (m, 5H), 4.29 (d, J = 16.0 Hz, 1H), 4.10 (dt, J = 13.0, 5.0 Hz, 1H), 3.73 (br s, 1H), 3.57 (d, J = 16.1 Hz, 1H), 3.46–3.35 (m, 2H), 3.28–3.26 (m, 1H), 3.26–3.15 (m, 6H), 3.15–3.07 (m, 2H), 2.80 (t, J = 6.0 Hz, 2H), 2.76–2.67 (m, 1H), 2.62 (dt, J = 16.5, 6.0 Hz, 1H), 2.24 (s, 6H), 2.22 (s, 6H), 2.04 (dt, J = 6.9, 5.0 Hz, 1H). HPLC retention time: 21.3 min. HREIMS m/z 470.2799 (calcd. for C30H35N3O2, 470.2802).
Tert-butyl 7-((3,4-dihydroquinolin-1(2H)-yl)methyl)-3,4-dihydroisoquinoline-2(1H)-carboxylate (7x). Compound 7x was synthesized following General Procedure E from compound 3 (50 mg, 0.153 mmol, 1.0 eq), 1,2,3,4-tetrahydroquinoline (23 μL, 0.184 mmol, 1.2 eq), and K2CO3 (25 mg, 0.184 mmol, 1.2 eq). The reaction mixture was diluted with water. The aqueous layer was extracted with several portions of ethyl acetate. Combined organic layers were dried over MgSO4, filtered, and concentrated under vacuum. Silica gel chromatography yielded a mixture of the desired product and 1,2,3,4-tetrahydroquinoline. The product was partitioned between ethyl acetate and 2 M NaOH. The aqueous layer was extracted with ethyl acetate. Some 1,2,3,4-tetrahydroquinoline remained. The mixture was resubmitted to reaction conditions with additional 3 (25 mg, 0.076, 0.5 eq) and K2CO3 (25 mg, 0.184 mmol, 1.2 eq). Silica gel chromatography yielded the desired product as a colorless oil (24 mg, 28%).1H-NMR (CDCl3, 500 MHz) δ 7.08 (s, 1H), 7.02 (s, 1H), 6.99 (t, J = 6.5 Hz, 2H), 6.59 (t, J = 7.3 Hz, 1H), 6.51 (d, J = 8.5 Hz, 1H), 4.55 (s, 2H), 4.44 (s, 2H), 3.65 (s, 2H), 3.36 (t, J = 5.7 Hz, 2H), 2.92–2.75 (m, 4H), 2.03 (p, J = 6.1 Hz, 2H), 1.50 (s, 9H).
(S)-2-amino-1-(7-((3,4-dihydroquinolin-1(2H)-yl)methyl)-3,4-dihydroisoquinolin-2(1H)-yl)-3-(4-hydroxy-2,6-dimethylphenyl)propan-1-one (8x). Following General Procedure F, intermediate 7x (24 mg, 0.063 mmol, 1.0 eq) was deprotected to yield the amine intermediate. This intermediate was coupled to diBoc-DMT (27 mg, 0.067 mmol, 1.05 eq) in the presence of PyBOP (33 mg, 0.064 mmol, 1.0 eq), and DIEA (111 µL, 0.64 mmol, 10 eq) to yield the product. No 6Cl-HOBt was used. TFA deprotection yielded the product as a white solid. 1H-NMR (CD3OD, 500 MHz, rotamers) δ 7.09 (d, J = 7.9 Hz, 1H), 7.04 (d, J = 8.1 Hz, 2H), 7.01 (d, J = 7.9 Hz, 1H), 6.99–6.89 (m, 5H), 6.64–6.51 (m, 5H), 6.40 (s, 2H), 6.27 (s, 2H), 4.62 (d, J = 17.1 Hz, 1H), 4.58–4.53 (m, 2H), 4.51 (d, J = 17.0 Hz, 2H), 4.45 (s, 2H), 4.44 (s, 2H), 4.19 (d, J = 15.8 Hz, 1H), 3.93 (dt, J = 12.9, 5.4 Hz, 1H), 3.51–3.43 (m, 1H), 3.46 (d, J = 15.7 Hz, 1H), 3.40–3.34 (m, 4H), 3.26–3.19 (m, 3H), 3.11–3.05 (m, 2H), 2.81 (q, J = 5.9 Hz, 4H), 2.73 (t, J = 6.2 Hz, 2H), 2.69–2.62 (m, 1H), 2.54 (dt, J = 16.3, 6.1 Hz, 1H), 2.23 (s, 6H), 2.20 (s, 6H), 2.06–1.95 (m, 5H). HPLC retention time: 35.9 min. HREIMS m/z 470.2800 (calcd. for C30H35N3O2, 470.2802).
Tert-butyl 7-(indolin-1-ylmethyl)-3,4-dihydroisoquinoline-2(1H)-carboxylate (7y). Compound 7y was synthesized following General Procedure E from compound 3 (50 mg, 0.153 mmol, 1.0 eq), indoline (21 μL, 0.184 mmol, 1.2 eq), and K2CO3 (25 mg, 0.184 mmol, 1.2 eq) to yield the product as a brown oil (37 mg, 66%). 1H-NMR (CDCl3, 500 MHz) δ 7.18 (d, J = 7.9 Hz, 1H), 7.15–7.08 (m, 3H), 7.07 (t, J = 7.6 Hz, 1H), 6.69 (t, J = 7.3 Hz, 1H), 6.52 (d, J = 7.8 Hz, 1H), 4.58 (s, 2H), 4.21 (s, 2H), 3.66 (s, 2H), 3.32 (t, J = 8.2 Hz, 2H), 2.99 (t, J = 8.3 Hz, 2H), 2.84 (t, J = 6.0 Hz, 2H), 1.51 (s, 9H).
(S)-2-amino-3-(4-hydroxy-2,6-dimethylphenyl)-1-(7-(indolin-1-ylmethyl)-3,4-dihydroisoquinolin-2(1H)-yl)propan-1-one (8y). Following General Procedure F, intermediate 7y (37 mg, 0.102 mmol, 1.0 eq) was deprotected to yield the amine intermediate. This intermediate was coupled to diBoc-DMT (43 mg, 0.105 mmol, 1.05 eq) in the presence of PyBOP (52 mg, 0.100 mmol, 1.0 eq), and DIEA (174 µL, 1.0 mmol, 10 eq) to yield the product. No 6Cl-HOBt was used. TFA deprotection yielded the product as a white solid. 1H-NMR (CD3OD, 500 MHz, rotamers) δ 7.24–7.18 (m, 3H), 7.18–7.11 (m, 4H), 7.06 (d, J = 7.9 Hz, 1H), 7.03–6.94 (m, 4H), 6.89 (d, J = 8.5 Hz, 1H), 6.67 (s, 1H), 6.41 (s, 2H), 6.27 (s, 2H), 4.63 (d, J = 16.9 Hz, 1H), 4.60–4.56 (m, 2H), 4.53 (d, J = 17.2 Hz, 1H), 4.40–4.32 (m, 4H), 4.21 (d, J = 15.8 Hz, 1H), 3.89 (dt, J = 12.7, 5.5 Hz, 1H), 3.55–3.50 (m, 1H), 3.50–3.46 (m, 4H), 3.44 (d, J = 15.9 Hz, 1H), 3.28–3.17 (m, 3H), 3.13–3.06 (m, 2H), 2.99 (t, J = 7.6 Hz, 4H), 2.74 (t, J = 4.7 Hz, 2H), 2.72–2.65 (m, 1H), 2.56 (dt, J = 16.3, 6.0 Hz, 1H), 2.23 (s, 6H), 2.20 (s, 6H), 1.98 (dt, J = 16.2, 5.9 Hz, 1H). HPLC retention time: 30.1 min. HREIMS m/z 456.2644 (calcd. for C29H33N3O2, 456.2646).
Tert-butyl 7-(isoindolin-2-ylmethyl)-3,4-dihydroisoquinoline-2(1H)-carboxylate (7z). Compound 7z was synthesized following General Procedure E from compound 3 (50 mg, 0.153 mmol, 1.0 eq), isoindoline HCl (29 mg, 0.184 mmol, 1.2 eq), and K2CO3 (25 mg, 0.184 mmol, 1.2 eq) to yield the product as an orange oil (22 mg, 39%). 1H-NMR (CDCl3, 500 MHz) δ 7.29 (s, 1H), 7.21 (d, J = 7.8 Hz, 1H), 7.18 (s, 4H), 7.12 (d, J = 7.6 Hz, 1H), 4.58 (s, 2H), 3.93 (s, 4H), 3.88 (s, 2H), 3.65 (t, J = 8.2 Hz, 2H), 2.84 (t, J = 6.2 Hz, 2H), 1.49 (s, 9H).
(S)-2-amino-3-(4-hydroxy-2,6-dimethylphenyl)-1-(7-(isoindolin-2-ylmethyl)-3,4-dihydroisoquinolin-2(1H)-yl)propan-1-one (8z). Following General Procedure F, intermediate 7z (22 mg, 0.060 mmol, 1.0 eq) was deprotected to yield the amine intermediate. This intermediate was coupled to diBoc-DMT (26 mg, 0.063 mmol, 1.05 eq) in the presence of PyBOP (31 mg, 0.060 mmol, 1.0 eq), and DIEA (105 µL, 0.600 mmol, 10 eq) to yield the product. No 6Cl-HOBt was used. TFA deprotection yielded the product as a white solid. 1H-NMR (CD3OD, 500 MHz, rotamers) δ 7.41–7.38 (m, 8H), 7.37–7.34 (m, 2H), 7.30 (dd, J = 7.9, 1.8 Hz, 1H), 7.19 (dd, J = 7.9, 3.5 Hz, 1H), 7.16 (dd, J = 8.0, 3.7 Hz, 1H), 6.86 (s, 1H), 6.42 (s, 2H), 6.24 (s, 2H), 4.72 (d, J = 17.2 Hz, 1H), 4.70–4.64 (m, 8H), 4.61 (d, J = 15.3 Hz, 1H), 4.65–4.57 (m, 2H), 4.56 (s, 2H), 4.53 (d, J = 10.7 Hz, 2H), 4.29 (d, J = 16.0 Hz, 1H), 4.09 (dt, J = 13.2, 4.9 Hz, 1H), 3.57 (d, J = 15.9 Hz, 1H), 3.44–3.37 (m, 1H), 3.29–3.25 (m, 1H), 3.24 (q, J = 13.0 Hz, 2H), 3.15–3.07 (m, 2H), 2.80 (t, J = 4.3 Hz, 2H), 2.75–2.67 (m, 1H), 2.61 (dt, J = 16.2, 5.6 Hz, 1H), 2.25 (s, 6H), 2.23 (s, 6H), 2.07–2.00 (m, 1H). HPLC retention time: 20.1 min. HREIMS m/z 456.2645 (calcd. for C29H33N3O2, 456.2646).


 ,
, 

{kind=link}
{kind=link}
{kind=link}