
Co-Adsorption of H2O, OH, and Cl on Aluminum and Intermetallic Surfaces and Its Effects on the Work Function Studied by DFT Calculations


Abstract
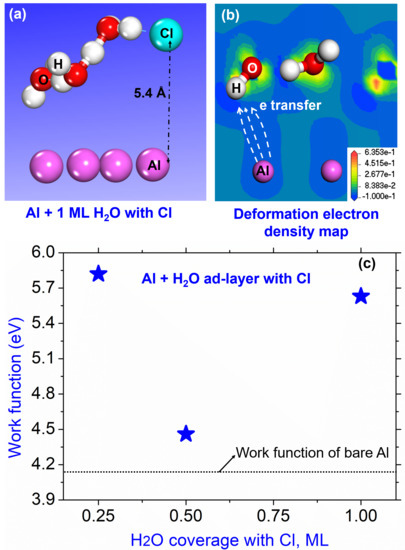
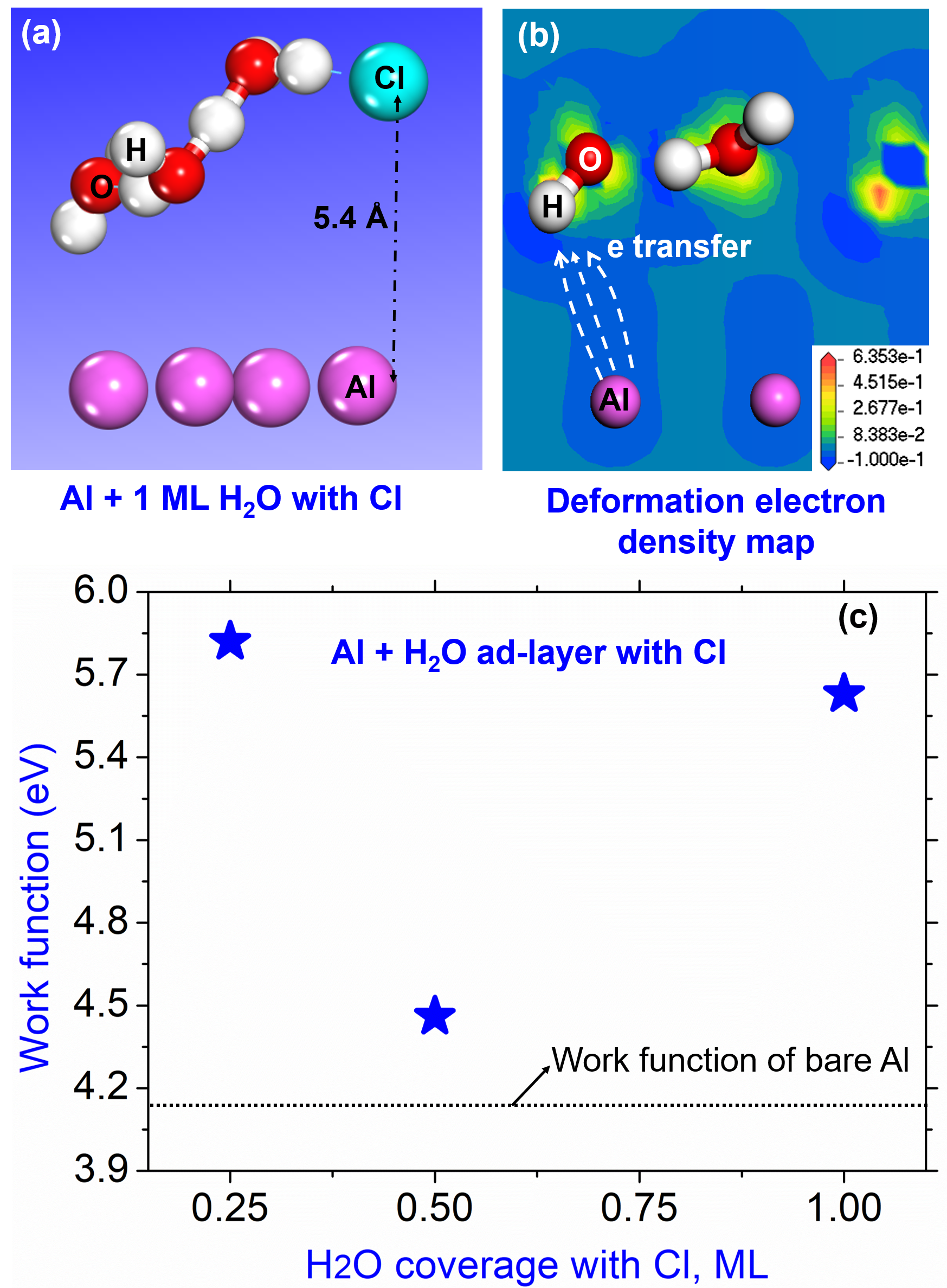{kind=link}
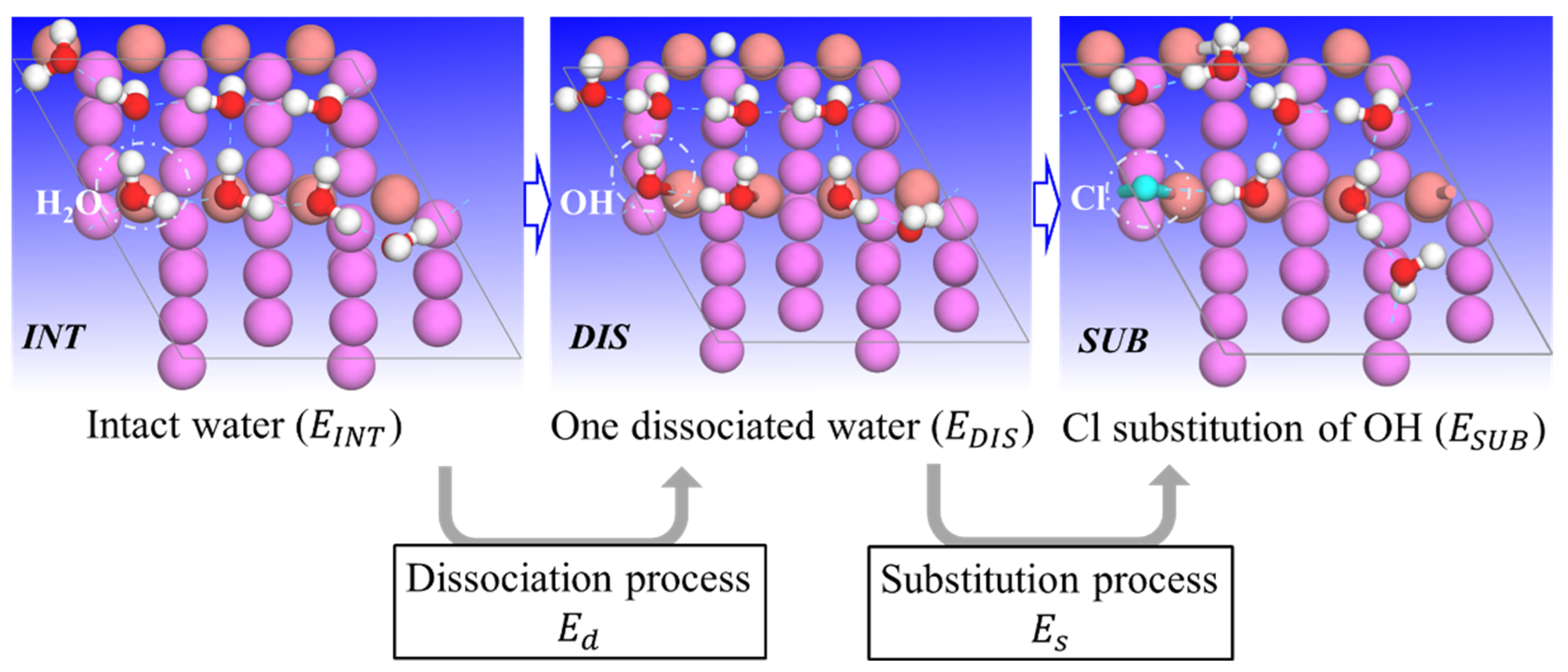{kind=link}
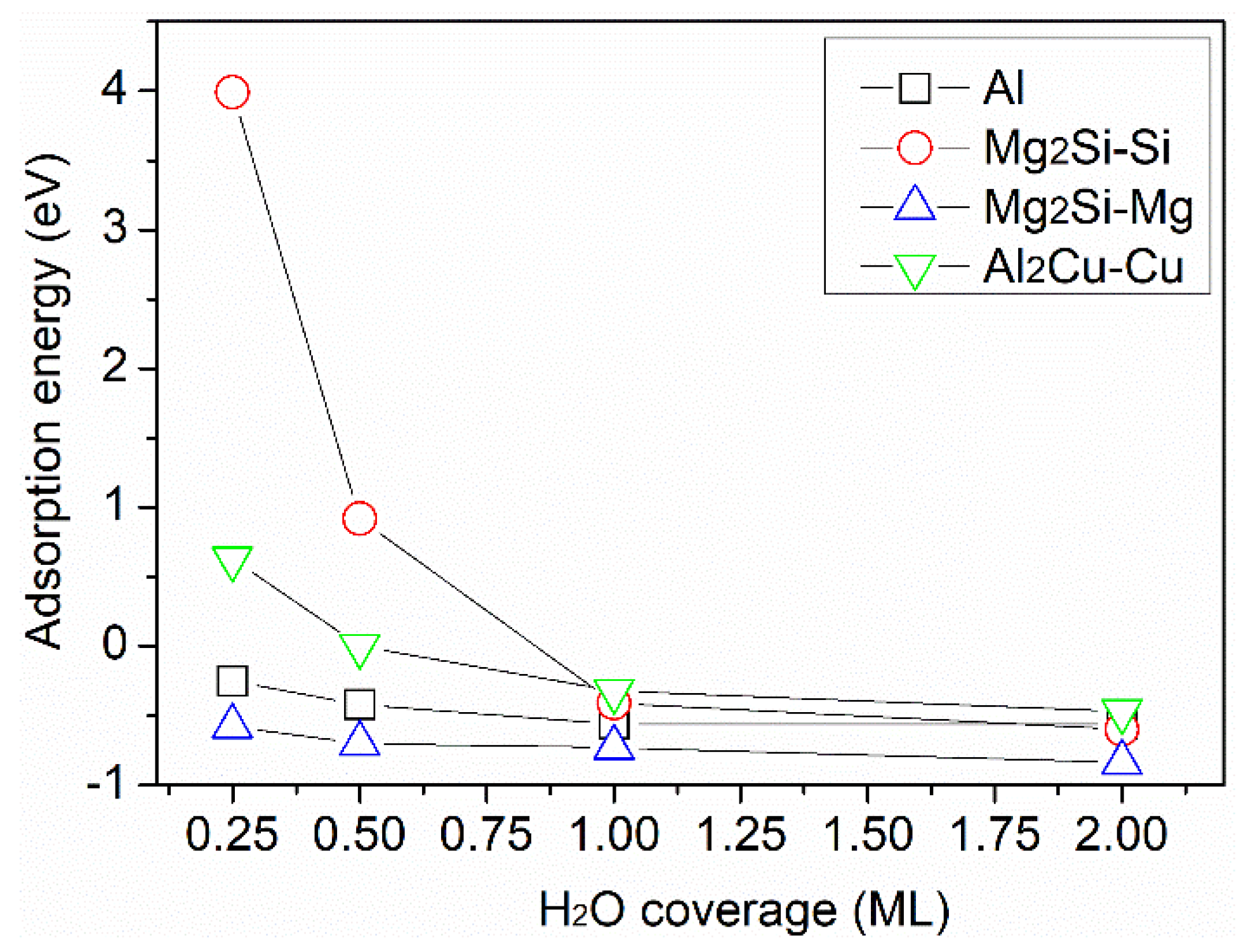{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Computational Details
2.1. Model Construction
2.2. Description of Energetics
3. Results and Discussion
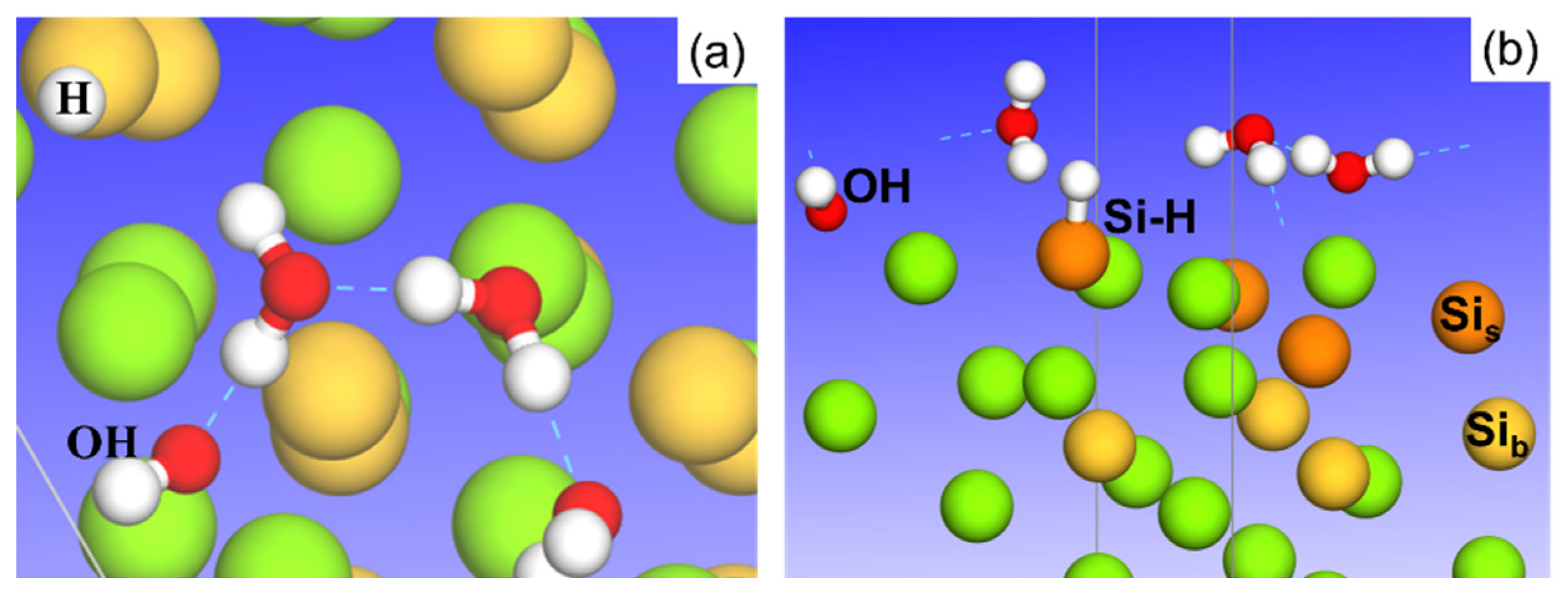
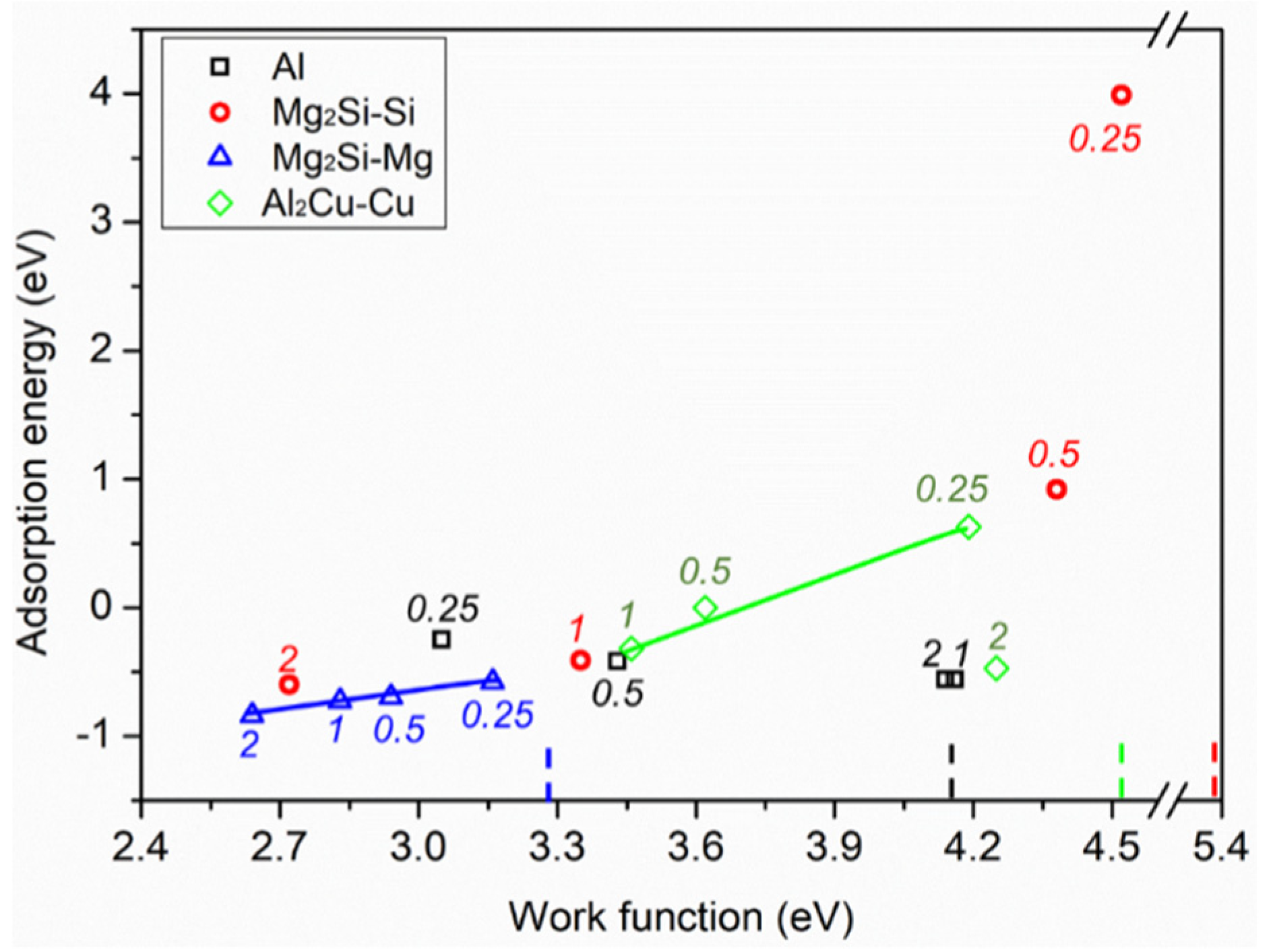
3.1. Adsorption of Pure H2O Ad-Layers
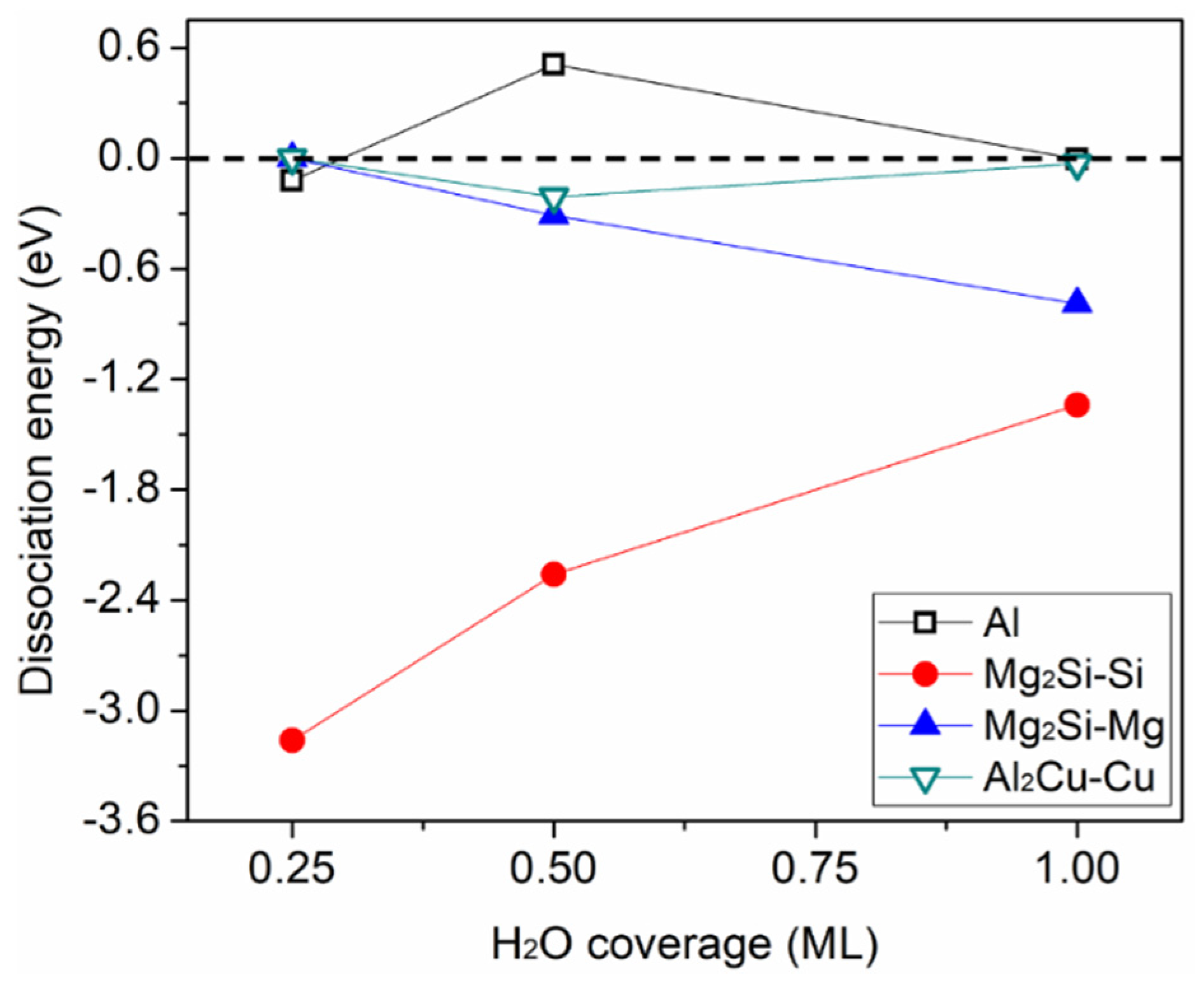
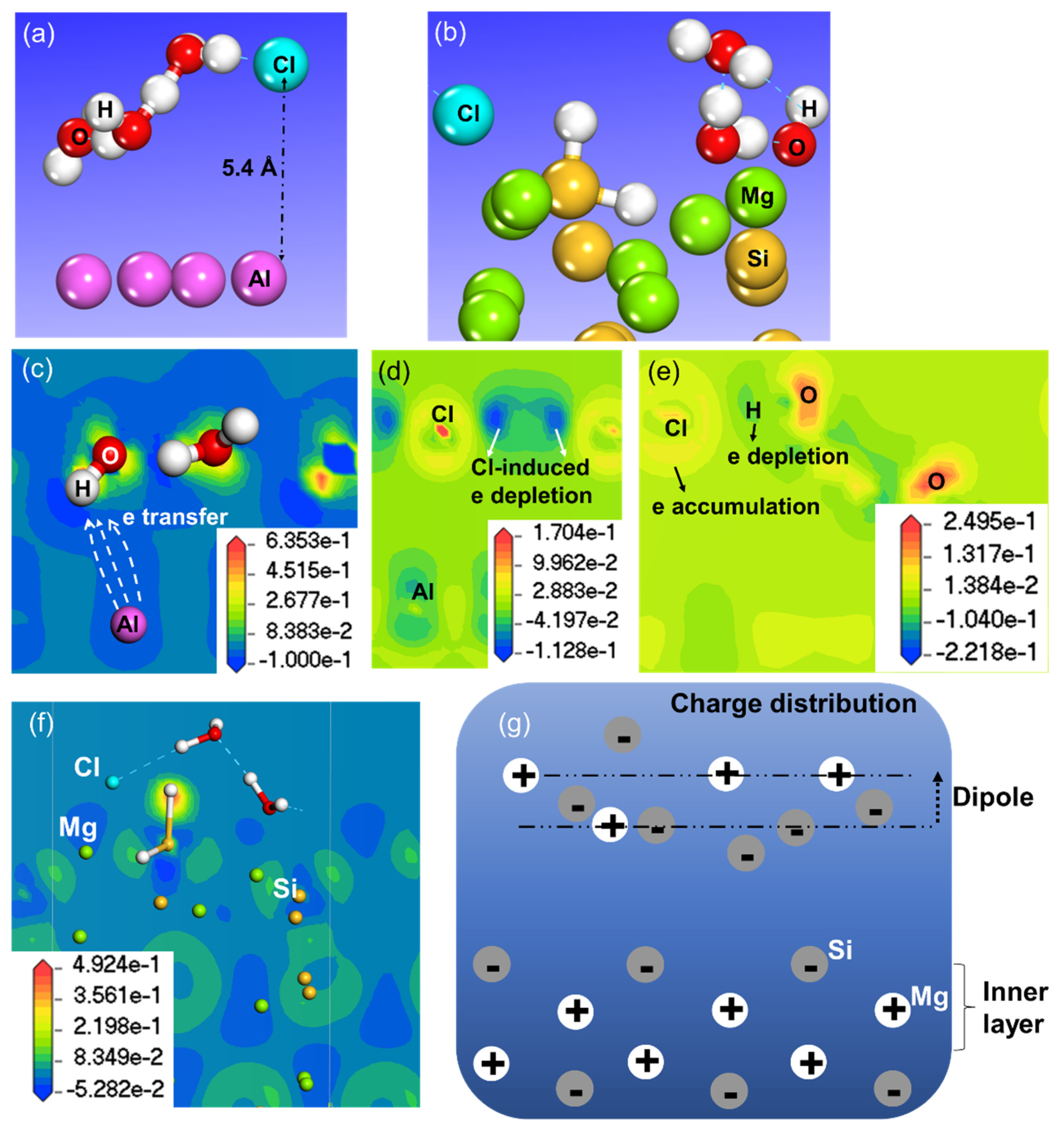
3.2. H2O Dissociation and Cl Substitution of OH
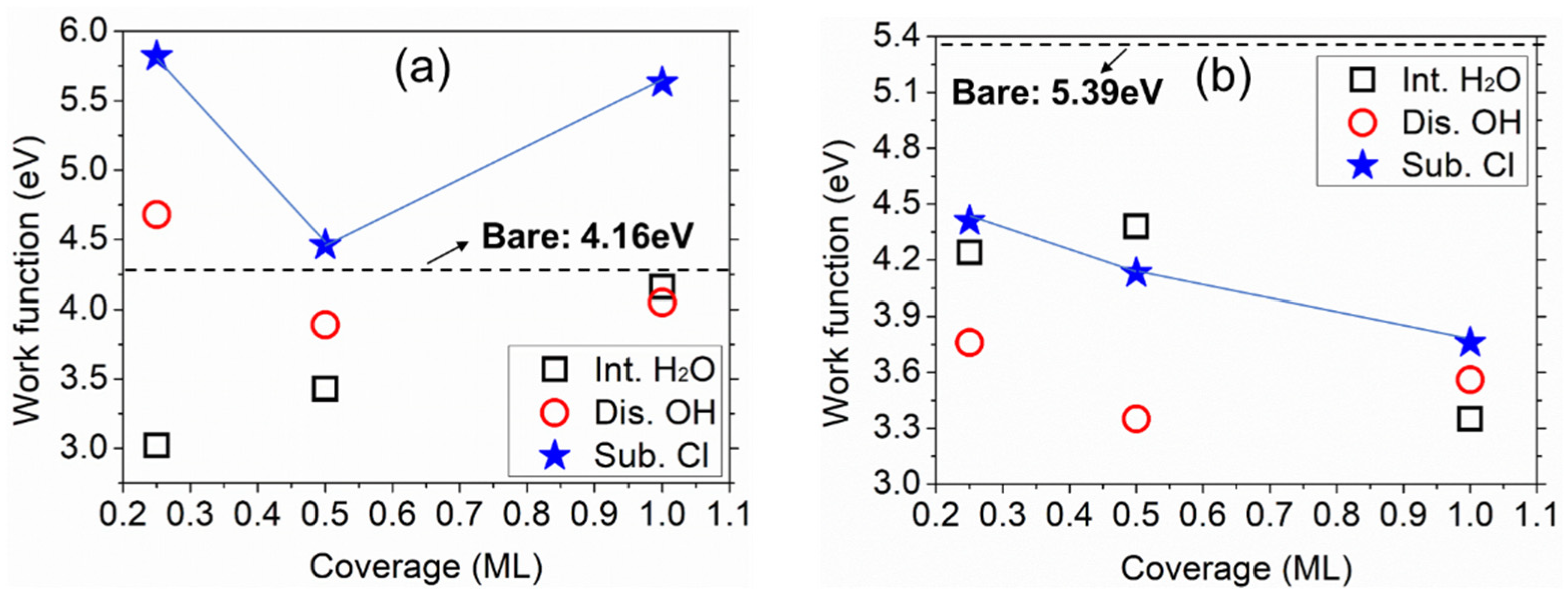
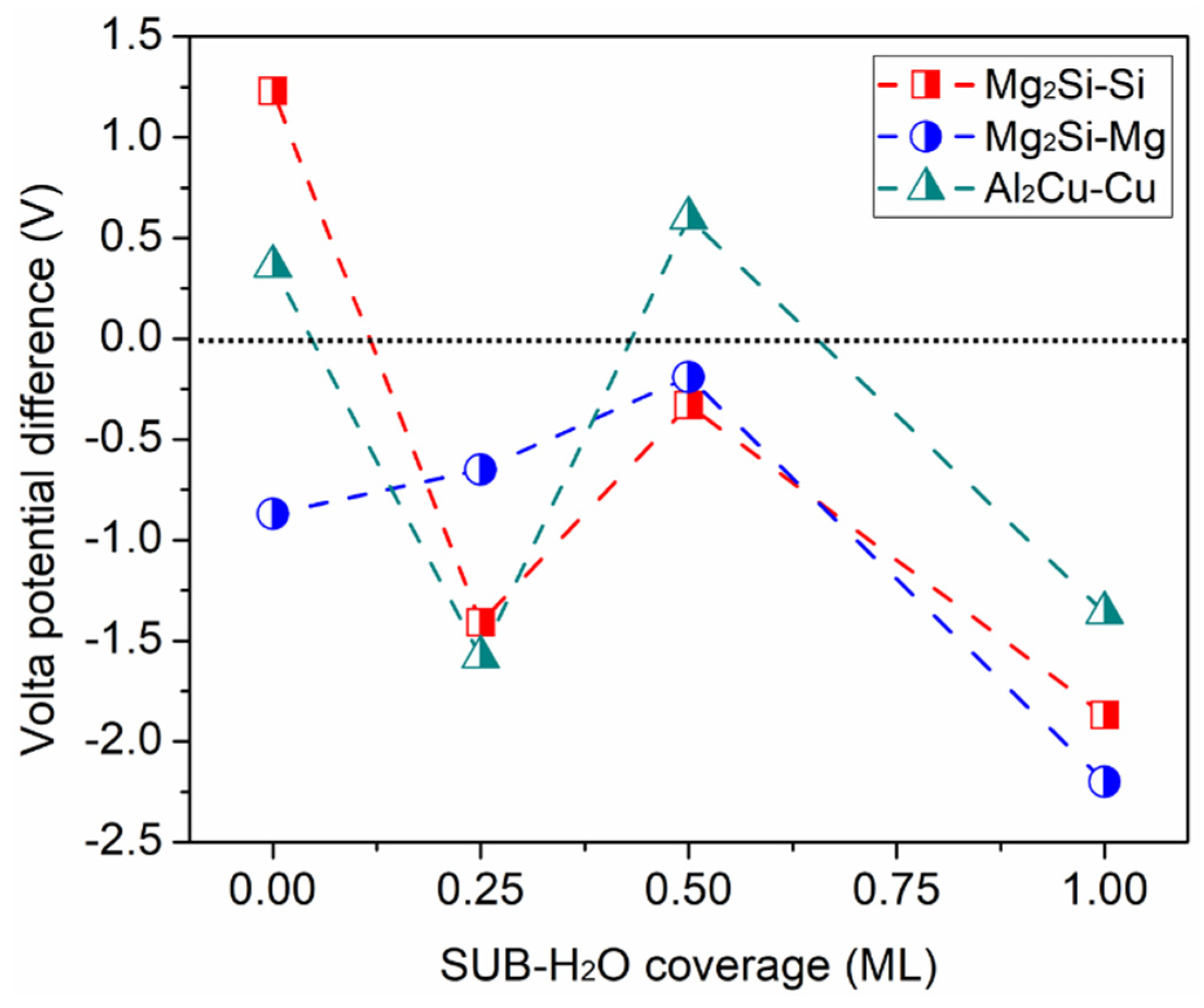
3.3. Effect on Work Function of Cl in the Aqueous Ad-Layer
3.4. Short Summary and Implications
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References and Note
- Birbilis, N.; Buchheit, R.G. Electrochemical characteristics of intermetallic phases in aluminum alloys-an experimental survey and discussion. J. Electrochem. Soc. 2005, 152, B140–B151. [Google Scholar] [CrossRef]
- Lebouil, S.; Tardelli, J.; Rocca, E.; Volovitch, P.; Ogle, K. Dealloying of Al2Cu, Al7Cu2Fe, and Al2CuMg intermetallic phases to form nanoparticulate copper films. Mater. Corros. 2014, 65, 416–424. [Google Scholar] [CrossRef]
- Marcus, P.; Maurice, V.; Strehblow, H.H. Localized corrosion (pitting): A model of passivity breakdown including the role of the oxide layer nanostructure. Corros. Sci. 2008, 50, 2698–2704. [Google Scholar] [CrossRef]
- Gründer, Y.; Drünkler, A.; Golks, F.; Wijts, G.; Stettner, J.; Zegenhagen, J.; Magnussen, O.M. Cu (111) in chloride containing acidic electrolytes: Coadsorption of an oxygenated species. J. Electroanal. Chem. 2014, 712, 74–81. [Google Scholar] [CrossRef]
- Liu, M.; Jin, Y.; Zhang, C.H.; Leygraf, C.; Wen, L. Density-functional theory investigation of Al pitting corrosion in electrolyte containing chloride ions. Appl. Surf. Sci. 2015, 357, 2028–2038. [Google Scholar] [CrossRef]
- Bouzoubaa, A.; Diawara, B.; Maurice, M.; Minot, C.; Marcus, P. Ab initio study of the interaction of chlorides with defect-free hydroxylated NiO surfaces. Corros. Sci. 2009, 51, 941–948. [Google Scholar] [CrossRef]
- da Silva, T.H.; Nelson, E.B.; Williamson, I.; Efaw, C.M.; Sapper, E.; Hurley, M.F.; Li, L. First-principles surface interaction studies of aluminum-copper and aluminum-copper-magnesium secondary phases in aluminum alloys. Appl. Surf. Sci. 2018, 439, 910–918. [Google Scholar] [CrossRef]
- Guo, L.Q.; Zhao, X.M.; Bai, Y.; Qiao, L.J. Water adsorption behavior on metal surfaces and its influence on surface potential studied by in situ SPM. Appl. Surf. Sci. 2012, 258, 9087–9091. [Google Scholar] [CrossRef]
- Gossenberger, F.; Roman, T.; Forster-Tonigold, K.; Groß, A. Change of the work function of platinum electrodes induced by halide adsorption. Beilstein J. Nanotech. 2014, 5, 152–161. [Google Scholar] [CrossRef]
- Örnek, C.; Liu, M.; Pan, J.; Jin, Y.; Leygraf, C. Volta potential evolution of intermetallics in aluminum alloy microstructure under thin aqueous adlayers: A combined DFT and experimental study. Top. Catal. 2018, 61, 1169–1182. [Google Scholar] [CrossRef]
- Sarvghad-Moghaddam, M.; Parvizi, R.; Davoodi, A.; Haddad-Sabzevar, M.; Imani, A. Establishing a correlation between interfacial microstructures and corrosion initiation sites in Al/Cu joints by SEM–EDS and AFM–SKPFM. Corros. Sci. 2014, 79, 148–158. [Google Scholar] [CrossRef]
- Schmutz, P.; Frankel, G.S. Characterization of AA2024-T3 by scanning Kelvin probe force microscopy. J. Electrochem. Soc. 1998, 145, 2285–2295. [Google Scholar] [CrossRef]
- Rohwerder, M.; Turcu, F. High-resolution Kelvin probe microscopy in corrosion science: Scanning Kelvin probe force microscopy (SKPFM) versus classical scanning Kelvin probe (SKP). Electrochim. Acta 2007, 53, 290–299. [Google Scholar] [CrossRef]
- Migani, A.; Sousa, C.; Illas, F. Chemisorption of atomic chlorine on metal surfaces and the interpretation of the induced work function changes. Surf. Sci. 2005, 574, 297–305. [Google Scholar] [CrossRef]
- Roman, T.; Gross, A. Periodic density-functional calculations on work-function change induced by adsorption of halogens on Cu (111). Phys. Rev. Lett. 2013, 110, 156804. [Google Scholar] [CrossRef]
- Zhu, Q.; Wang, S.Q. Trends and regularities for halogen adsorption on various metal surfaces. J. Electrochem. Soc. 2016, 163, H796–H808. [Google Scholar] [CrossRef]
- Zhou, W.L.; Liu, T.; Li, M.C.; Zhao, T.; Duan, Y.H. Adsorption of bromine on Mg (0001) surface from first-principles calculations. Comput. Mater. Sci. 2016, 111, 47–53. [Google Scholar] [CrossRef]
- Marks, L. Competitive Chloride chemisorption disrupts hydrogen bonding networks: DFT, crystallography, thermodynamics, and morphological consequences. Corros. 2017, 74, 295–311. [Google Scholar] [CrossRef]
- Duan, S.; Xu, X.; Tian, Z.Q.; Luo, Y. Hybrid molecular dynamics and first-principles study on the work function of a Pt (111) electrode immersed in aqueous solution at room temperature. Phys. Rev. B 2012, 86, 045450. [Google Scholar] [CrossRef]
- Meng, S.; Wang, E.G.; Gao, S. Water adsorption on metal surfaces: A general picture from density functional theory studies. Phys. Rev. B 2004, 69, 195404. [Google Scholar] [CrossRef]
- Schnur, S.; Groß, A. Properties of metal–water interfaces studied from first principles. New J. Phys. 2009, 11, 125003. [Google Scholar] [CrossRef]
- Pedroza, L.S.; Poissier, A.; Fernández-Serra, M.V. Local order of liquid water at metallic electrode surfaces. J. Chem. Phys. 2015, 142, 034706. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.L.; Chen, Z.X. Density functional slab model studies of water adsorption on flat and stepped Cu surfaces. Surf. Sci. 2007, 601, 954–964. [Google Scholar] [CrossRef]
- Tzvetkov, G.; Zubavichus, Y.; Koller, G.; Schmidt, T.; Heske, C.; Umbach, E.; Grunze, M.; Ramsey, M.G.; Netzer, F.P. Growth of H2O layers on an ultra-thin Al2O3 film: From monomeric species to ice. Surf. Sci. 2003, 543, 131–140. [Google Scholar] [CrossRef]
- Musumeci, F.; Pollack, G.H. Influence of water on the work function of certain metals. Chem. Phys. Lett. 2012, 536, 65–67. [Google Scholar] [CrossRef] [PubMed]
- Langenbach, E.; Spitzer, A.; Lüth, H. The adsorption of water on Pt (111) studied by irreflection and UV-photoemission spectroscopy. Surf. Sci. 1984, 147, 179–190. [Google Scholar] [CrossRef]
- McCrum, I.T.; Akhade, S.A.; Janik, M.J. Electrochemical specific adsorption of halides on Cu 111, 100, and 211: A Density Functional Theory study. Electrochim. Acta 2015, 173, 302–309. [Google Scholar] [CrossRef]
- Gossenberger, F.; Roman, T.; Groß, A. Hydrogen and halide co-adsorption on Pt (111) in an electrochemical environment: A computational perspective. Electrochim. Acta 2016, 216, 152–159. [Google Scholar] [CrossRef]
- Bange, K.; Grider, D.; Sass, J.K. Coadsorption of water and ions on Cu (110): Models for the double layer. Surf. Sci. 1983, 126, 437–443. [Google Scholar] [CrossRef]
- Wasileski, S.A.; Janik, M.J. A first-principles study of molecular oxygen dissociation at an electrode surface: A comparison of potential variation and coadsorption effects. Phys. Chem. Chem. Phys. 2008, 10, 3613–3627. [Google Scholar] [CrossRef]
- Dieter, G.E. Mechanical Metallurgy; McGraw-Hill Press: New York, NY, USA, 1986. [Google Scholar]
- Villars, P.; Calvert, L.D. Pearson’s Handbook of Crystallographic Data for Intermetallic Phases; American Society of Metals: Cleveland, OH, USA, 1985. [Google Scholar]
- Jin, Y.; Liu, M.; Zhang, C.H.; Leygraf, C.; Wen, L.; Pan, J. First-principle calculation of Volta potential of intermetallic particles in aluminum alloys and practical implications. J. Electrochem. Soc. 2017, 164, C465–C473. [Google Scholar] [CrossRef]
- Delley, B. An all-electron numerical method for solving the local density functional for polyatomic molecules. J. Chem. Phys. 1990, 92, 508–517. [Google Scholar] [CrossRef]
- Delley, B. From molecules to solids with the DMol3 approach. J. Chem. Phys. 2000, 113, 7756–7764. [Google Scholar] [CrossRef]
- Pineau, N.; Minot, C.; Maurice, V.; Marcus, P. Density functional theory study of the interaction of Cl− with passivated nickel surfaces. Electrochem. Solid-Sate Lett. 2003, 6, B47–B51. [Google Scholar] [CrossRef]
- Björneholm, O.; Hansen, M.; Hodgson, A.; Liu, L.; Limmer, D.; Michaelides, A.; Pedevilla, P.; Rossmeisl, J.; Shen, H.; Tocci, G.; et al. Water at interfaces. Chem. Rev. 2016, 116, 7698–7726. [Google Scholar]
- Thiel, P.A.; Madey, T.E. The interaction of water with solid surfaces: Fundamental aspects. Surf. Sci. Rep. 1987, 7, 211–385. [Google Scholar] [CrossRef]
- Michaelides, A. Simulating ice nucleation, one molecule at a time, with the ‘DFT microscope’. Faraday Discuss. 2007, 136, 287–297. [Google Scholar] [CrossRef]
- Michaelides, A.; Alavi, A.; King, D.A. Insight into H2O-ice adsorption and dissociation on metal surfaces from first-principles simulations. Phys. Rev. B 2004, 69, 113404. [Google Scholar] [CrossRef]
- Guo, F.Y.; Long, C.G.; Zhang, J.; Zhang, Z.; Liu, C.H.; Yu, K. Adsorption and dissociation of H2O on Al (1 1 1) surface by density functional theory calculation. Appl. Surf. Sci. 2015, 324, 584–589. [Google Scholar] [CrossRef]
- Taheri, P.; Pohl, K.; Grundmeier, G.; Flores, J.R.; Hannour, F.; de Wit, J.H.W.; Mol, J.M.C.; Terryn, H. Effects of surface treatment and carboxylic acid and anhydride molecular dipole moments on the Volta potential values of zinc surfaces. J. Phys. Chem. C 2013, 117, 1712–1721. [Google Scholar] [CrossRef]
- Cai, N.; Zhou, G.; Müller, K.; Starr, D.E. Comparative study of the passivation of Al (111) by molecular oxygen and water vapor. J. Phys. Chem. C 2012, 117, 172–178. [Google Scholar] [CrossRef]
- Digne, M.; Raybaud, P.; Sautet, P.; Guillaume, D.; Toulhoat, H. Atomic scale insights on chlorinated γ-alumina surfaces. J. Am. Chem. Soc. 2008, 130, 11030–11039. [Google Scholar] [CrossRef] [PubMed]
- Huheey, J.E.; Ketter, E.A.; Ketter, R.L.; Medhi, O.K. Inorganic Chemistry: Principles of Structure and Reactivity; Pearson Education India: Delhi, India, 1993. [Google Scholar]
- Bouzoubaa, A.; Costa, D.; Diawara, B.; Audiffren, N.; Marcus, P. Insight of DFT and atomistic thermodynamics on the adsorption and insertion of halides onto the hydroxylated NiO (111) surface. Corros. Sci. 2010, 52, 2643–2652. [Google Scholar] [CrossRef]
- Ren, J.; Meng, S. First-principles study of water on copper and noble metal (110) surfaces. Phys. Rev. B 2008, 77, 054110. [Google Scholar] [CrossRef]
- Jin, X.; Yang, W.; Qian, Z.; Wang, Y.; Bi, S. DFT study on the interaction between monomeric aluminium and chloride ion in aqueous solution. Dalton Trans. 2011, 40, 5052–5058. [Google Scholar] [CrossRef]
- Leung, T.C.; Kao, C.L.; Su, W.S.; Feng, Y.J.; Chan, C.T. Relationship between surface dipole, work function and charge transfer: Some exceptions to an established rule. Phys. Rev. B 2003, 68, 195408. [Google Scholar] [CrossRef]
- Zhu, Y.; Sun, K.; Frankel, G.S. Intermetallic phases in aluminum alloys and their roles in localized corrosion. J. Electrochem. Soc. 2018, 165, C807–C820. [Google Scholar] [CrossRef]
- Xue, M.; Xie, J.; Li, W.; Yang, C.; Ai, Y.; Wang, F.; Ou, J.; Yao, J. Dependence of electron work function of Al-Mg alloys on surface structures and relative humidity. Physica B 2011, 406, 4240–4244. [Google Scholar] [CrossRef]
- Crispin, X.; Geskin, V.; Crispin, A.; Cornil, J.; Lazzaroni, R.; Salaneck, W.R.; Bredas, J.L. Characterization of the interface dipole at organic/metal interfaces. J. Am. Chem. Soc. 2002, 124, 8131–8141. [Google Scholar] [CrossRef]
- Migani, A.; Illas, F. A systematic study of the structure and bonding of halogens on low-index transition metal surfaces. J. Phys. Chem. B 2006, 110, 11894–11906. [Google Scholar] [CrossRef]
- Andreatta, F.; Terryn, H.; de Wit, J.H.W. Effect of solution heat treatment on galvanic coupling between intermetallics and matrix in AA7075-T6. Corros. Sci. 2003, 45, 1733–1746. [Google Scholar] [CrossRef]
- Rossmeisl, J.; Nørskov, J.K.; Taylor, C.D.; Janik, M.J.; Neurock, M. Calculated phase diagrams for the electrochemical oxidation and reduction of water over Pt (111). J. Phys. Chem. B 2006, 110, 21833–21839. [Google Scholar] [CrossRef] [PubMed]
- Liu, M. a,b; Busch, M. c; Grönbeck, H. c; Jin, Y. a; Leygraf, C. b; Pan, J. b Aqueous environment around chloride ion studied by first-principle theory, molecular dynamics and x-ray absorption fine structure. (a: University of Science and Technology Beijing, Beijing, China. b: Biotechnology and Health, KTH Royal Institute of Technology, Stockholm, Sweden. c: Chalmers University of Technology, Göteborg). Unpublished work, 2019.
- Wei, X.; Dong, C.; Chen, Z.; Xiao, K.; Li, X. A DFT study of the adsorption of O2 and H2O on Al (111) surfaces. RSC Adv. 2016, 6, 56303–56312. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, M.; Jin, Y.; Pan, J.; Leygraf, C. Co-Adsorption of H2O, OH, and Cl on Aluminum and Intermetallic Surfaces and Its Effects on the Work Function Studied by DFT Calculations. Molecules 2019, 24, 4284. https://doi.org/10.3390/molecules24234284
Liu M, Jin Y, Pan J, Leygraf C. Co-Adsorption of H2O, OH, and Cl on Aluminum and Intermetallic Surfaces and Its Effects on the Work Function Studied by DFT Calculations. Molecules. 2019; 24(23):4284. https://doi.org/10.3390/molecules24234284
Chicago/Turabian StyleLiu, Min, Ying Jin, Jinshan Pan, and Christofer Leygraf. 2019. "Co-Adsorption of H2O, OH, and Cl on Aluminum and Intermetallic Surfaces and Its Effects on the Work Function Studied by DFT Calculations" Molecules 24, no. 23: 4284. https://doi.org/10.3390/molecules24234284
APA StyleLiu, M., Jin, Y., Pan, J., & Leygraf, C. (2019). Co-Adsorption of H2O, OH, and Cl on Aluminum and Intermetallic Surfaces and Its Effects on the Work Function Studied by DFT Calculations. Molecules, 24(23), 4284. https://doi.org/10.3390/molecules24234284