
Lipase-Catalyzed Chemoselective Ester Hydrolysis of Biomimetically Coupled Aryls for the Synthesis of Unsymmetric Biphenyl Esters


Abstract
1. Introduction
2. Results
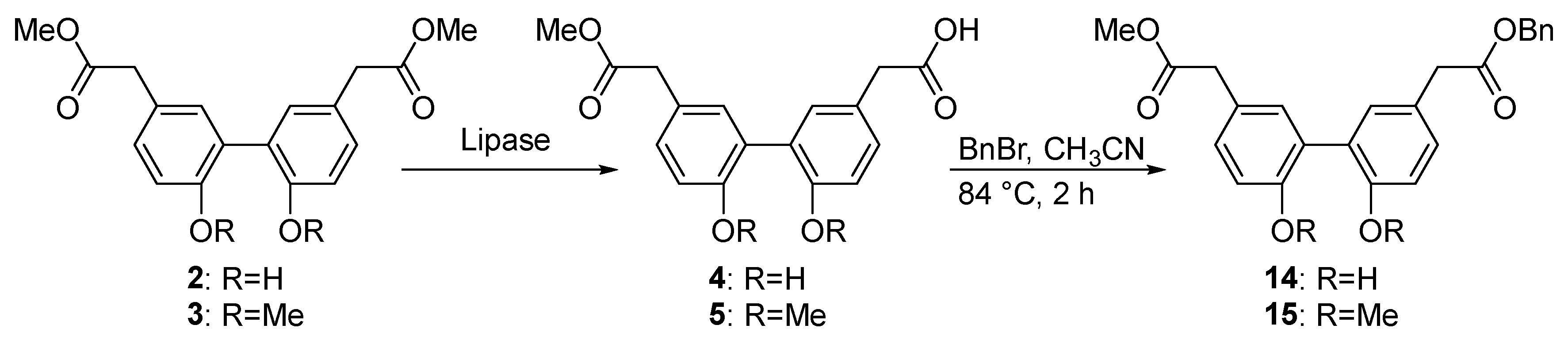
2.1. Biomimetic Synthesis of Biphenyl Ester Building Blocks
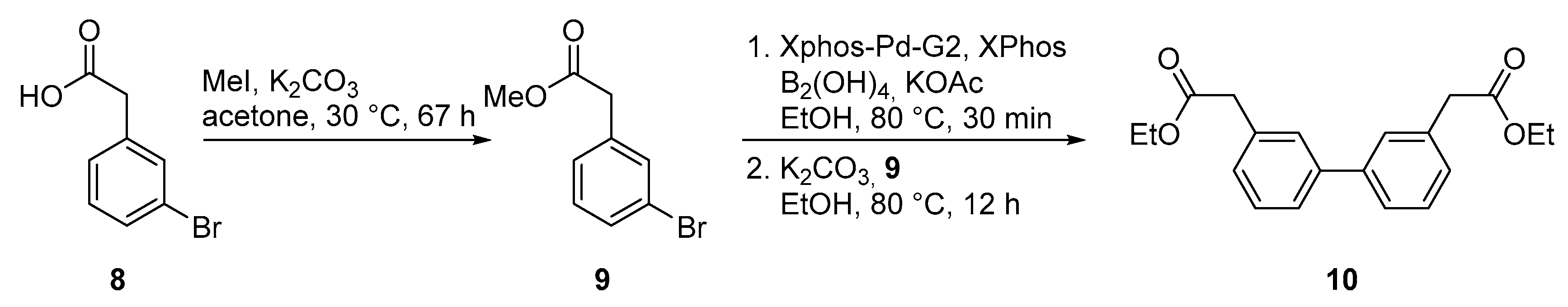
2.2. Chemical Synthesis of Biphenyl Ester Building Blocks without Ortho Substituents
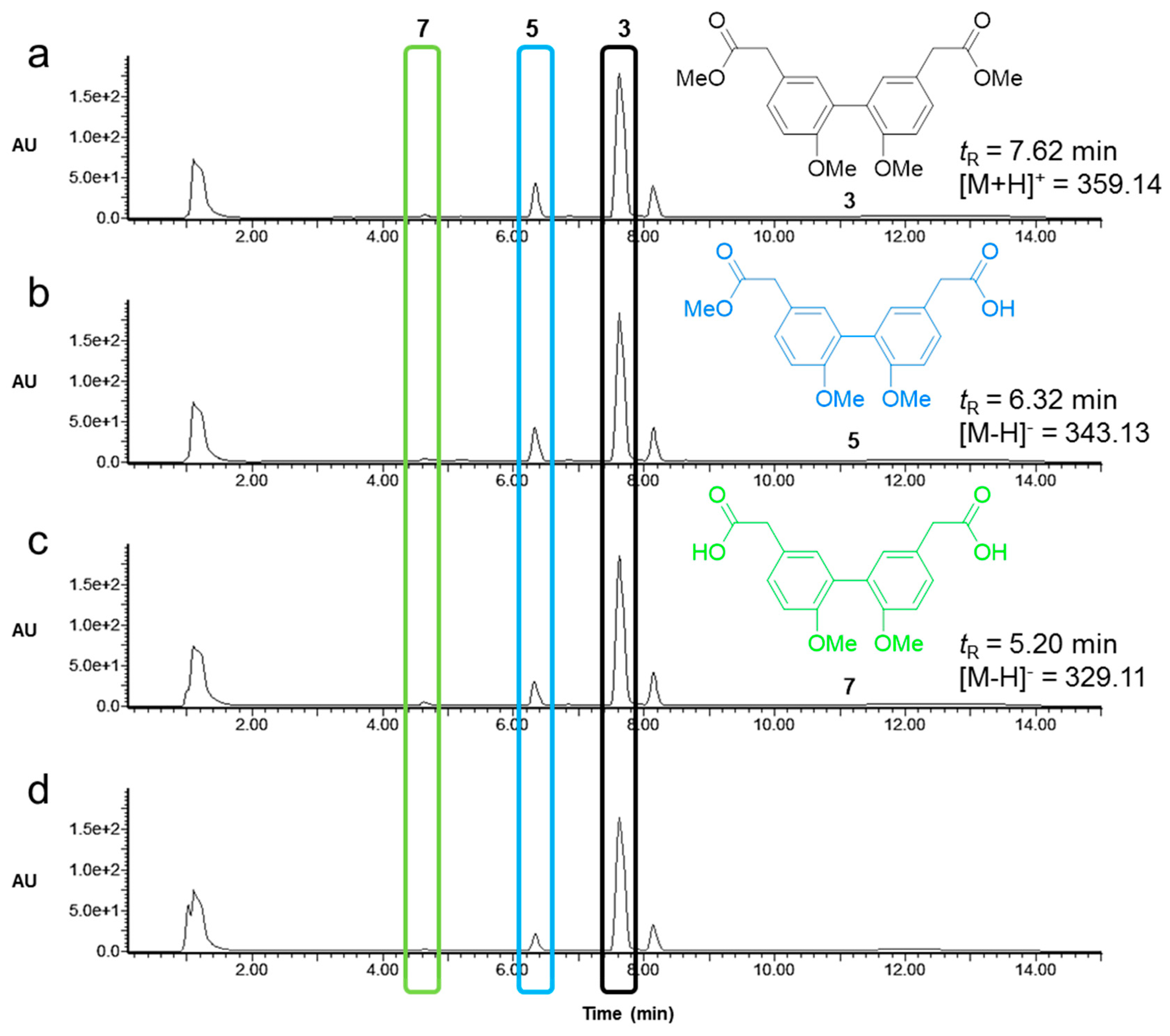
2.3. Activity and Chemoselectivity of Lipases on Biphenyl Ester Hydrolysis
2.4. Molecular Docking Experiments
2.5. Lipase-Catalyzed Biphenyl Ester Conversion in Preparative Scale
3. Discussion
4. Materials and Methods
4.1. Chemical Methods—General Information
4.2. Analytical LC-MS
4.3. Preparative LC-MS
4.4. Dimethyl-2,2‘-(6,6‘-dihydroxy-[1,1‘-biphenyl]-3,3‘-diyl)diacetate (2)
4.5. Dimethyl-2,2′-(6,6′-dimethoxy-[1,1′-biphenyl]-3,3′-diyl)-diacetate (3)
4.6. Methyl-2-(3-bromophenyl)acetate (9)
4.7. Diethyl 2,2′-([1,1′-biphenyl]-3,3′-diyl)diacetate (10)
4.8. Colorimetric Lipase Activity Screening
4.9. Analysis of Lipase Activity and Selectivity by Analytical LC-MS
4.10. Benzyl 2-(2′,6-dihydroxy-5′-(2-methoxy-2-oxoethyl)-[1,1′-biphenyl]-3-yl)acetate (14) and 2-(2′,6-Bis(benzyloxy)-5′-(2-methoxy-2-oxoethyl)-[1,1′-biphenyl]-3-yl)acetic acid (16)
4.11. 2-(2′,6-Dimethoxy-5′-(2-methoxy-2-oxoethyl)-[1,1′-biphenyl]-3-yl)acetic acid (5)
4.12. Benzyl 2-(2′,6-dimethoxy-5′-(2-methoxy-2-oxoethyl)-[1,1′-biphenyl]-3-yl)acetate (15)
4.13. Molecular Docking
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Niwayama, S. Highly Efficient Selective Monohydrolysis of Symmetric Diesters. J. Org. Chem. 2000, 65, 5834–5836. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Estrada, A.A.; Zak, M.; Lee, S.H.; Safina, B.S. A Mild and Selective Method for the Hydrolysis of Esters with Trimethyltin Hydroxide. Angew. Chem. Int. Ed. 2005, 44, 1378–1382. [Google Scholar] [CrossRef]
- Niwayama, S.; Cho, H.; Lin, C. Highly efficient selective monohydrolysis of dialkyl malonates and their derivatives. Tetrahedron Lett. 2008, 49, 4434–4436. [Google Scholar] [CrossRef]
- Preller, M.; Furch, M.; Díaz-Gómez, N.; Kalesse, M.; Manstein, D.J. Biphenyl Compounds for Use in Treating Malaria and Other Parasitic Disorders. U.S. Patent 9,499,471, 22 November 2016. [Google Scholar]
- Sharma, P.; McClees, S.F.; Afaq, F. Pomegranate for prevention and treatment of cancer: An update. Molecules 2017, 22, 177. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Yoshimura, M.; Amakura, Y. Chemical and biological significance of oenothein B and related ellagitannin oligomers with macrocyclic structure. Molecules 2018, 23, 552. [Google Scholar] [CrossRef] [PubMed]
- Yamada, H.; Wakamori, S.; Hirokane, T.; Ikeuchi, K.; Matsumoto, S. Structural Revisions in Natural Ellagitannins. Molecules 2018, 23, 1901. [Google Scholar] [CrossRef]
- Krimmer, S.G.; Klebe, G. Thermodynamics of protein-ligand interactions as a reference for computational analysis: How to assess accuracy, reliability and relevance of experimental data. J. Comput. Aided Mol. Des. 2015, 29, 867–883. [Google Scholar] [CrossRef]
- Li, X.; Deng, Y.; Zheng, Z.; Huang, W.; Chen, L.; Tong, Q.; Ming, Y. Corilagin, a promising medicinal herbal agent. Biomed. Pharmacother. 2018, 99, 43–50. [Google Scholar] [CrossRef]
- Kwong, H.C.; Chidan Kumar, C.S.; Mah, S.H.; Chia, T.S.; Quah, C.K.; Loh, Z.H.; Chandraju, S.; Lim, G.K. Novel biphenyl ester derivatives as tyrosinase inhibitors: Synthesis, crystallographic, spectral analysis and molecular docking studies. PLoS ONE 2017, 12, e0170117. [Google Scholar] [CrossRef]
- Baheti, A.; Tyagi, P.; Thomas, K.R.J.; Hsu, Y.C.; Lin, J.T.s. Simple triarylamine-based dyes containing fluorene and biphenyl linkers for efficient dye-sensitized solar cells. J. Phys. Chem. C 2009, 113, 8541–8547. [Google Scholar] [CrossRef]
- Li, Y.; Xu, Z.; Zhao, S.; Song, D.; Qiao, B.; Zhu, Y.; Meng, J. Benefits of the hydrophobic surface for CH3NH3PbI3 crystalline growth towards highly efficient inverted perovskite solar cells. Molecules 2019, 24, 2027. [Google Scholar] [CrossRef]
- Lestari, W.W.; Lönnecke, P.; Sárosi, M.B.; Streit, H.C.; Adlung, M.; Wickleder, C.; Handke, M.; Einicke, W.D.; Gläser, R.; Hey-Hawkins, E. Syntheses, structures and luminescence properties of novel metal-organic frameworks based on zinc(II), cadmium(II) or lead(II) and a 2,2′-dimethoxy-functionalised biphenyl linker. CrystEngComm 2013, 15, 3874–3884. [Google Scholar] [CrossRef]
- Bulman Page, P.C.; Kinsey, F.S.; Chan, Y.; Strutt, I.R.; Slawin, A.M.Z.; Jones, G.A. Novel binaphthyl and biphenyl α- and β-amino acids and esters: Organocatalysis of asymmetric Diels-Alder reactions. A combined synthetic and computational study. Org. Biomol. Chem. 2018, 16, 7400–7416. [Google Scholar] [CrossRef]
- Weinberger, M.; Su, P.H.; Peterlik, H.; Lindén, M.; Wohlfahrt-Mehrens, M. Biphenyl-bridged organosilica as a precursor for mesoporous silicon oxycarbide and its application in lithium and sodium ion batteries. Nanomaterials 2019, 9, 754. [Google Scholar] [CrossRef]
- Parashar, S.; Srivastava, P.; Pattanaik, M. Electrode materials for biphenyl-based rectification devices. J. Mol. Model. 2013, 19, 4467–4475. [Google Scholar] [CrossRef]
- Duan, L.; Xie, J.; Zhang, D.; Wang, L.; Dong, G.; Qiao, J.; Qiu, Y. Nanocomposite Thin Film Based on Ytterbium Fluoride and N,N′-Bis(1-naphthyl)-N,N -diphenyl-1,1′-biphenyl-4,4′-diamine and Its Application in Organic Light Emitting Diodes as Hole Transport Layer. J. Phys. Chem. C 2008, 112, 11985–11990. [Google Scholar] [CrossRef]
- Ferro, M.D.; Santos, S.A.O.; Silvestre, A.J.D.; Duarte, M.F. Chromatographic separation of phenolic compounds from extra virgin olive oil: Development and validation of a new method based on a biphenyl HPLC column. Int. J. Mol. Sci. 2019, 20, 201. [Google Scholar] [CrossRef]
- Adlercreutz, P. Immobilisation and application of lipases in organic media. Chem. Soc. Rev. 2013, 42, 6406–6436. [Google Scholar] [CrossRef]
- Casas-Godoy, L.; Duquesne, S.; Bordes, F.; Sandoval, G.; Marty, A. Lipases: An Overview. In Lipases and Phospholipases: Methods and Protocols; Humana Press: Totowa, NJ, USA, 2012; Volume 861, pp. 471–483. ISBN 978-1-61779-599-2. [Google Scholar]
- Reetz, M.T. Lipases as practical biocatalysts. Curr. Opin. Chem. Biol. 2002, 6, 145–150. [Google Scholar] [CrossRef]
- Whitesides, G.M.; Wong, C.H. Enzymes as Catalysts in Synthetic Organic Chemistry [New Synthetic Methods (53)]. Angew. Chem. Int. Ed. 1985, 24, 617–638. [Google Scholar] [CrossRef]
- Carrea, G.; Riva, S. Properties and Synthetic Applications of Enzymes in Organic Solvents. Angew. Chem. Int. Ed. 2000, 39, 2226–2254. [Google Scholar] [CrossRef]
- Sarmah, N.; Revathi, D.; Sheelu, G.; Yamuna Rani, K.; Sridhar, S.; Mehtab, V.; Sumana, C. Recent advances on sources and industrial applications of lipases. Biotechnol. Prog. 2018, 34, 5–28. [Google Scholar] [CrossRef]
- Carvalho, A.C.L.D.M.; Fonseca, T.D.S.; De Mattos, M.C.; De Oliveira, M.D.C.F.; De Lemos, T.M.L.G.; Molinari, F.; Romano, D.; Serra, I. Recent advances in lipase-mediated preparation of pharmaceuticals and their intermediates. Int. J. Mol. Sci. 2015, 16, 29682–29716. [Google Scholar] [CrossRef] [PubMed]
- Schmid, R.D.; Verger, R. Lipasen: Grenzflächen-Enzyme mit attraktiven Anwendungen. Angew. Chem. 1998, 110, 1694–1720. [Google Scholar] [CrossRef]
- Singh, A.K.; Mukhopadhyay, M. Overview of fungal lipase: A review. Appl. Biochem. Biotechnol. 2012, 166, 486–520. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.J. Enzyme catalysed deracemisation and dynamic kinetic resolution reactions. Curr. Opin. Chem. Biol. 2004, 8, 114–119. [Google Scholar] [CrossRef] [PubMed]
- García-Urdiales, E.; Alfonso, I.; Gotor, V. Enantioselective enzymatic desymmetrizations in organic synthesis. Chem. Rev. 2005, 105, 313–354. [Google Scholar] [CrossRef]
- Hirose, Y.; Kariya, K.; Sasaki, I.; Kurono, Y.; Ebiike, H.; Achiwa, K. Drastic solvent effect on lipase-catalyzed enantioselective hydrolysis of prochiral 1,4-dihydropyridines. Tetrahedron Lett. 1992, 33, 7157–7160. [Google Scholar] [CrossRef]
- Okuyama, K.; Shingubara, K.; Tsujiyama, S.I.; Suzuki, K.; Matsumoto, T. Enantiodivergent synthesis of tetra-ortho-substituted biphenyls by enzymatic desymmetrization. Synlett 2009, 2009, 941–944. [Google Scholar]
- Chong, J.; Poutaraud, A.; Hugueney, P. Metabolism and roles of stilbenes in plants. Plant Sci. 2009, 177, 143–155. [Google Scholar] [CrossRef]
- Wink, M. Evolution of secondary metabolites from an ecological and molecular phylogenetic perspective. Phytochemistry 2003, 64, 3–19. [Google Scholar] [CrossRef]
- Quideau, S.; Deffieux, D.; Douat-Casassus, C.; Pouységu, L. Plant Polyphenols: Chemical Properties, Biological Activities, and Synthesis. Angew. Chem. Int. Ed. 2011, 50, 586–621. [Google Scholar] [CrossRef] [PubMed]
- Kshirsagar, U.A.; Regev, C.; Parnes, R.; Pappo, D. Iron-Catalyzed Oxidative Cross-Coupling of Phenols and Alkenes. Org. Lett. 2013, 15, 3174–3177. [Google Scholar] [CrossRef] [PubMed]
- Gelalcha, F.G.; Anilkumar, G.; Tse, M.K.; Brückner, A.; Beller, M. Biomimetic Iron-Catalyzed Asymmetric Epoxidation of Aromatic Alkenes by Using Hydrogen Peroxide. Chem. A Eur. J. 2008, 14, 7687–7698. [Google Scholar] [CrossRef] [PubMed]
- Tsuzuki, W.; Ue, A.; Kitamura, Y. Effect of Dimethylsulfoxide on hydrolysis of lipase. Biosci. Biotechnol. Biochem. 2001, 65, 2078–2082. [Google Scholar] [CrossRef] [PubMed]
- Brady, L.; Brzozokowsi, A.; Derewenda, Z.; Dodson, E.; Dodson, G.; Tolley, S.; Turkenburg, J.; Christiansen, L.; Huge-Jensen, B.; Norskov, L.; et al. A serine protease triad forms the center of of a triacylglycerol lipase. Nature 1990, 343, 767–770. [Google Scholar] [CrossRef]
- Schrag, J.D.; Li, Y.; Wu, S.; Cygler, M. Ser-His-Glu triad forms the catalytic site of the lipase from Geotrichum candidum. Nature 1991, 351, 761–764. [Google Scholar] [CrossRef]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Dockingwith Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated Docking Using a Lamarckian Genetic Algorithm and an Empirical Binding Free Energy Function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Grochulski, P.; Li, Y.; Schrag, J.D.; Cygler, M. Two conformational states of Candida rugosa lipase. Protein Sci. 1994, 3, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Derewenda, U.; Derewenda, Z.S.; Brzozowski, A.M.; Lawson, D.M. Catalysis at the Interface: The Anatomy of a Conformational Change in a Triglyceride Lipase. Biochemistry 1992, 31, 1532–1541. [Google Scholar] [CrossRef] [PubMed]
- Kohno, M.; Funatsu, J.; Mikami, B.; Kugimiya, W.; Matsuo, T.; Morita, Y. The crystal structure of ribonuclease Rh from Rhizopus niveus at 2.0 Å resolution. J. Biochem. 1996, 120, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Schrag, J.D.; Li, Y.; Cygler, M.; Lang, D.; Burgdorf, T.; Hecht, H.J.; Schmid, R.; Schomburg, D.; Rydel, T.J.; Oliver, J.D.; et al. The open conformation of a Pseudomonas lipase. Structure 1997, 5, 187–202. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lipase | TAG 13 | Biphenyl Ester 2 | Biphenyl Ester 3 |
|---|---|---|---|
| ANL | + | – | – |
| CALB | + | + | + |
| CRL | + | – | – |
| MML | + | – | – |
| ROL | + | – | – |
| RNL | + | – | – |
| PPL | + | – | – |
| PCL | + | – | – |
| PFL | + | – | – |
| Predicted Affinity of 3 to Lipase | CRL | RNL | MML | PFL |
|---|---|---|---|---|
| ΔG0 (kcal · mol−1) | −3.23 | −5.64 | −6.66 | −6.79 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ehlert, J.; Kronemann, J.; Zumbrägel, N.; Preller, M. Lipase-Catalyzed Chemoselective Ester Hydrolysis of Biomimetically Coupled Aryls for the Synthesis of Unsymmetric Biphenyl Esters. Molecules 2019, 24, 4272. https://doi.org/10.3390/molecules24234272
Ehlert J, Kronemann J, Zumbrägel N, Preller M. Lipase-Catalyzed Chemoselective Ester Hydrolysis of Biomimetically Coupled Aryls for the Synthesis of Unsymmetric Biphenyl Esters. Molecules. 2019; 24(23):4272. https://doi.org/10.3390/molecules24234272
Chicago/Turabian StyleEhlert, Janna, Jenny Kronemann, Nadine Zumbrägel, and Matthias Preller. 2019. "Lipase-Catalyzed Chemoselective Ester Hydrolysis of Biomimetically Coupled Aryls for the Synthesis of Unsymmetric Biphenyl Esters" Molecules 24, no. 23: 4272. https://doi.org/10.3390/molecules24234272
APA StyleEhlert, J., Kronemann, J., Zumbrägel, N., & Preller, M. (2019). Lipase-Catalyzed Chemoselective Ester Hydrolysis of Biomimetically Coupled Aryls for the Synthesis of Unsymmetric Biphenyl Esters. Molecules, 24(23), 4272. https://doi.org/10.3390/molecules24234272