
Optimizing the Synthetic Route of Chromone-2-carboxylic Acids: A Step forward to Speed-up the Discovery of Chromone-based Multitarget-directed Ligands




Abstract
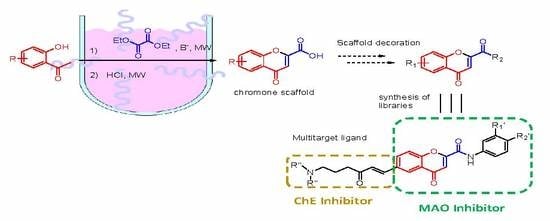
:
1. Introduction
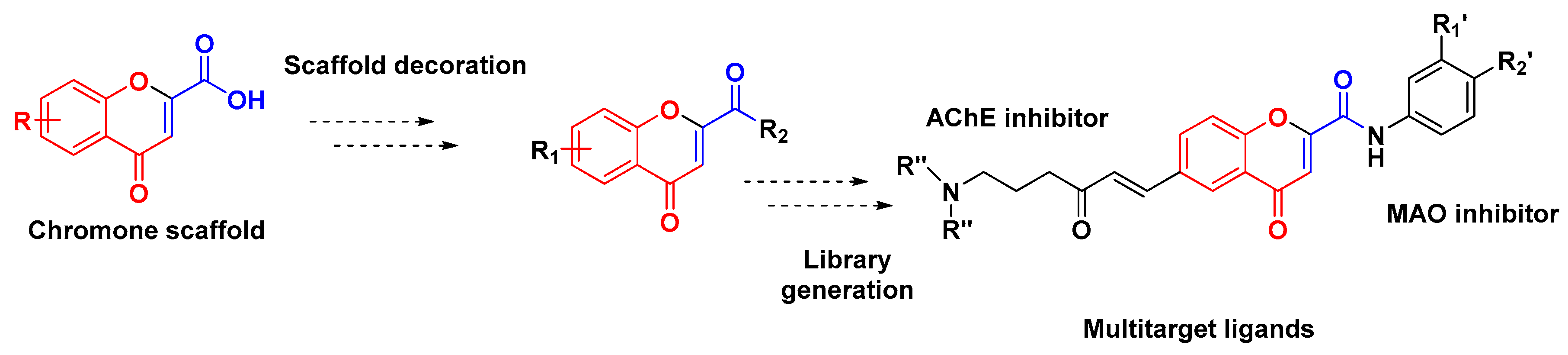
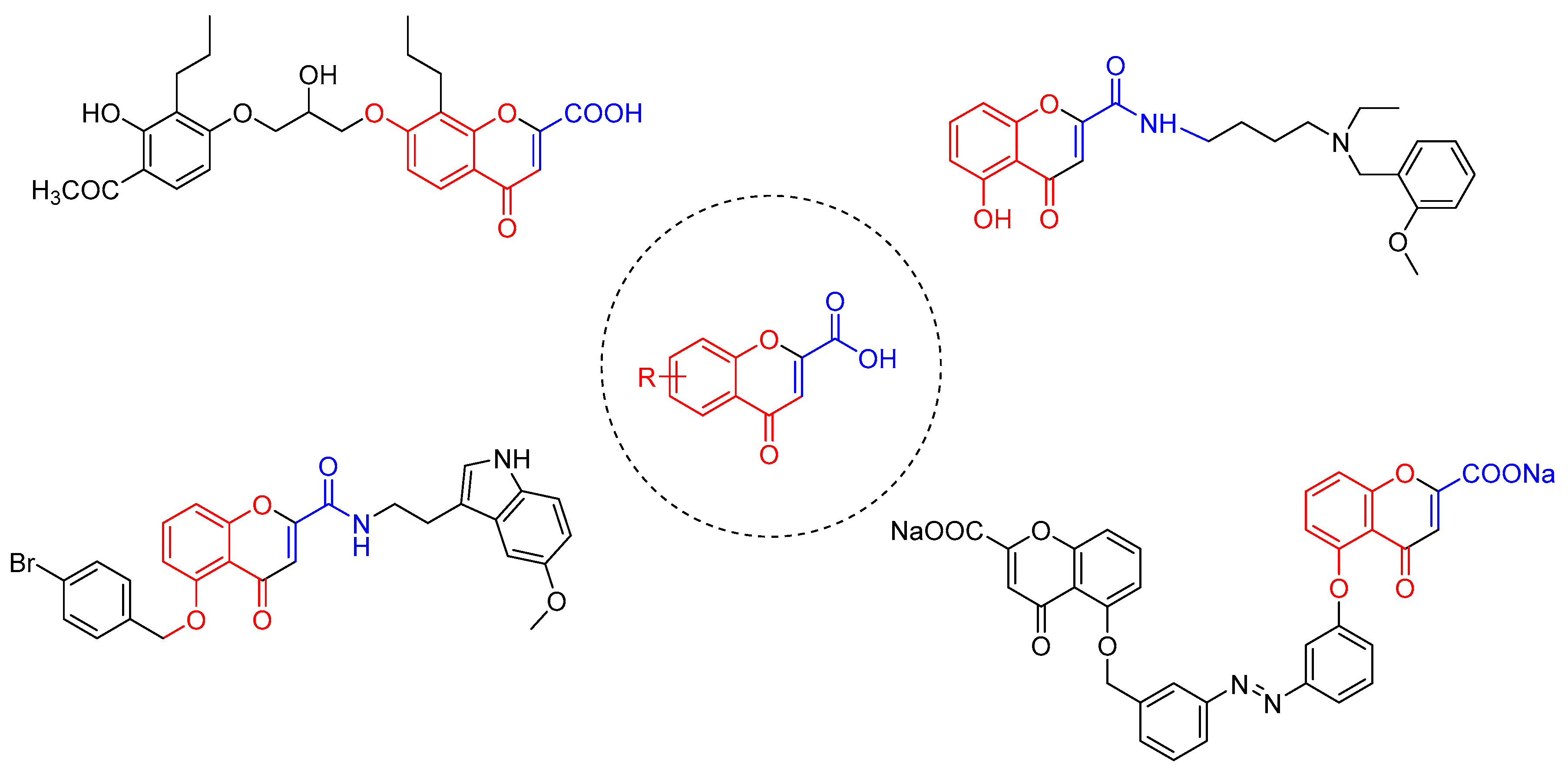
2. Results and Discussion
3. Material and Methods
3.1. Apparatus
3.2. Reagents and General Conditions
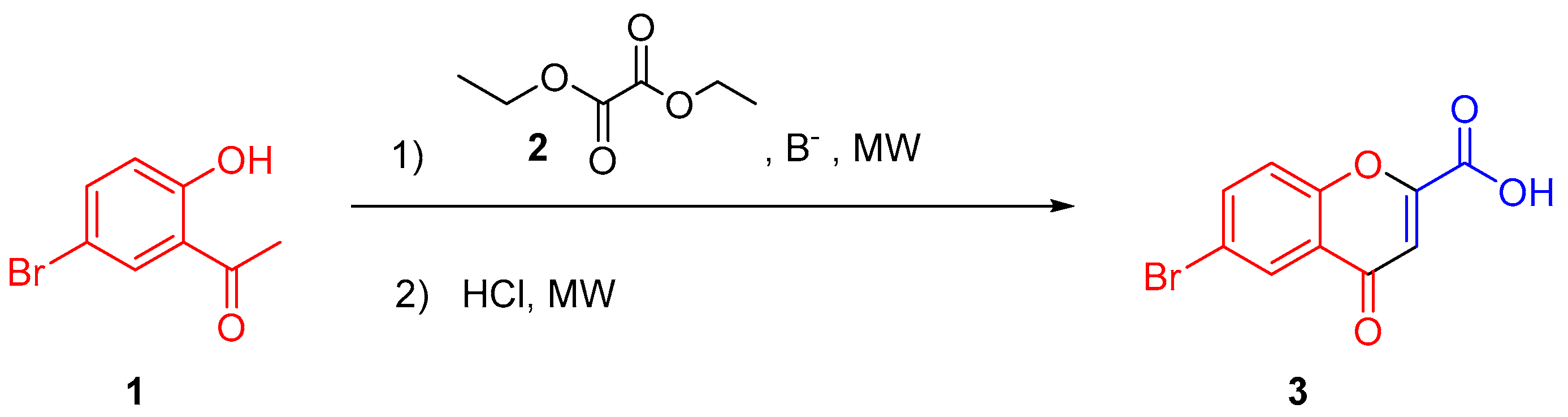
3.3. Synthesis of 4-Oxo-4H-chromene-2-carboxylic acids 3–11
General Procedure
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AChE | Acetylcholinesterase |
| MAO | Monoamine oxidase |
| MW | Microwave |
| TLC | Thin-layer chromatography |
References
- Gaspar, A.; Matos, M.J.; Garrido, J.; Uriarte, E.; Borges, F. Chromone: A Valid Scaffold in Medicinal Chemistry. Chem. Rev. 2014, 114, 4960–4992. [Google Scholar] [CrossRef] [PubMed]
- Reis, J.; Gaspar, A.; Milhazes, N.; Borges, F. Chromone as a Privileged Scaffold in Drug Discovery: Recent Advances. J. Med. Chem. 2017, 60, 7941–7957. [Google Scholar] [CrossRef] [PubMed]
- Cagide, F.; Reis, J.; Gaspar, A.; Borges, F. Accelerating lead optimization of chromone carboxamide scaffold throughout microwave-assisted organic synthesis. Tetrahedron Lett. 2011, 52, 6446–6449. [Google Scholar] [CrossRef]
- Oliveira, C.; Cagide, F.; Teixeira, J.; Amorim, R.; Sequeira, L.; Mesiti, F.; Silva, T.; Garrido, J.; Remião, F.; Vilar, S.; et al. Hydroxybenzoic Acid Derivatives as Dual-Target Ligands: Mitochondriotropic Antioxidants and Cholinesterase Inhibitors. Front. Chem. 2018, 6, 126. [Google Scholar] [CrossRef] [PubMed]
- Reis, J.; Cagide, F.; Valencia, M.E.; Teixeira, J.; Bagetta, D.; Perez, C.; Uriarte, E.; Oliveira, P.J.; Ortuso, F.; Alcaro, S.; et al. Multi-target-directed ligands for Alzheimer’s disease: Discovery of chromone-based monoamine oxidase/cholinesterase inhibitors. Eur. J. Med. Chem. 2018, 158, 781–800. [Google Scholar] [CrossRef] [PubMed]
- Kappe, C.O.; Stadler, A. Microwaves in Organic and Medicinal Chemistry. Microwaves in Organic Synthesis, 2nd ed.; Loupy, A., Ed.; Wiley-VCH: Weinheim, Germany, 2006; pp. 134–218. [Google Scholar]
- Wathey, B.; Tierney, J.; Lidström, P.; Westman, J. The impact of microwave-assisted organic chemistry on drug discovery. Drug Discov. Today 2002, 7, 373–380. [Google Scholar] [CrossRef]
- Shipe, W.D.; Wolkenberg, S.E.; Lindsley, C.W. Accelerating lead development by microwave-enhanced medicinal chemistry. Drug Discov. Today Technol. 2005, 2, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Stadler, A.; Kremsner, J.M. Microwave-assisted processing techniques in medicinal chemistry. Microwaves in Drug Discovery and Development: Recent Advances, 1st ed.; Spencer, J., Bagley, M.C., Eds.; Future Medicine Ltd.: London, U.K, 2014; pp. 6–19. [Google Scholar]
- Ellis, G.P. Chromenes, Chromanones, and Chromones. the Chemistry of Heterocyclic Compounds, 1st ed.; Ellis John, G.P., Ed.; Wiley & Sons: New York, NY, USA, 1977. [Google Scholar]
- Friden-Saxin, M.; Pemberton, N.; Andersson, K.; Dyrager, C.; Friberg, A.; Grotli, M.; Luthman, K. Synthesis of 2-Alkyl-Substituted Chromone Derivatives Using Microwave Irradiation. J. Org. Chem. 2009, 74, 2755–2759. [Google Scholar] [CrossRef] [PubMed]
- Goel, S.; Ritu, R.; Makrandi, J.K. Microwave assisted synthesis of 1-(2-hydroxyphenyl)-5-phenylpent-4-ene-1,3-diones and their conversion to 2-styrylchromones. Indian J. Chem. Sect. B 2006, 45, 1278–1281. [Google Scholar] [CrossRef]
- Balakrishna, C.; Kandula, V.; Gudipati, R.; Yennam, S.; Devi, P.U.; Behera, M. An Efficient Microwave-Assisted Propylphosphonic Anhydride (T3P)-Mediated One Chromone Synthesis via Enaminones. Synlett 2018, 29, 1087–1091. [Google Scholar]
- Zagorevskii, V.A.; Glozman, S.M.; Klyuev, S.M. Researches on pyranes, their analogs, and related compounds. Chem. Heterocycl. Compd. 1967, 3, 592–595. [Google Scholar] [CrossRef]
- Patani, G.A.; LaVoie, E.J. Bioisosterism: A rational approach in drug design. Chem. Rev. 1996, 96, 3147–3176. [Google Scholar] [CrossRef] [PubMed]
- Lemke, T.L.; Williams, D.A.; Roche, V.F.; Zito, S.W. Foye’s Principles of Medicinal Chemistry, 7th ed.; Lippincott Williams & Wilkins: Baltimore, MD, USA, 2013. [Google Scholar]
- Bratulescu, G. Excellentes voies de synthese des derives des 4-oxo-4H-1-benzopyranes avec le concours de micro-ondes. Acta Chim. Slov. 2002, 49, 173–180. [Google Scholar]
Sample Availability: Samples of the compounds are available from the authors. |

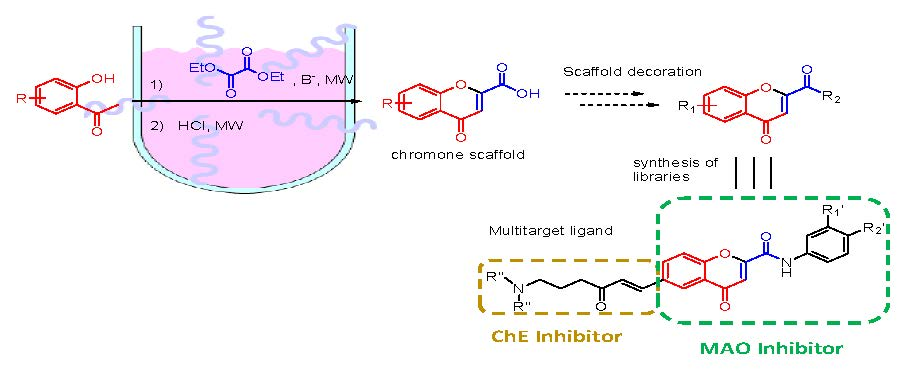{kind=link}
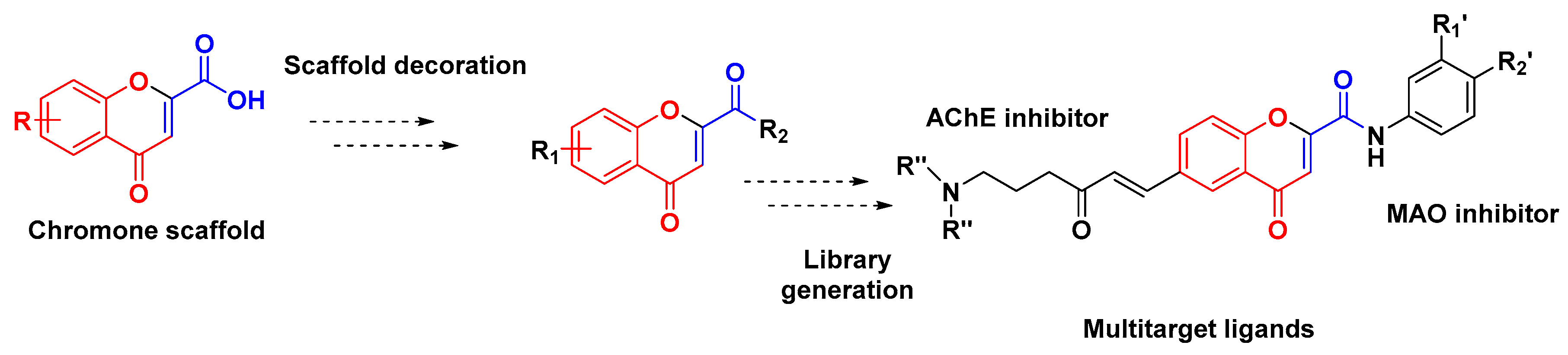{kind=link}
{kind=link}
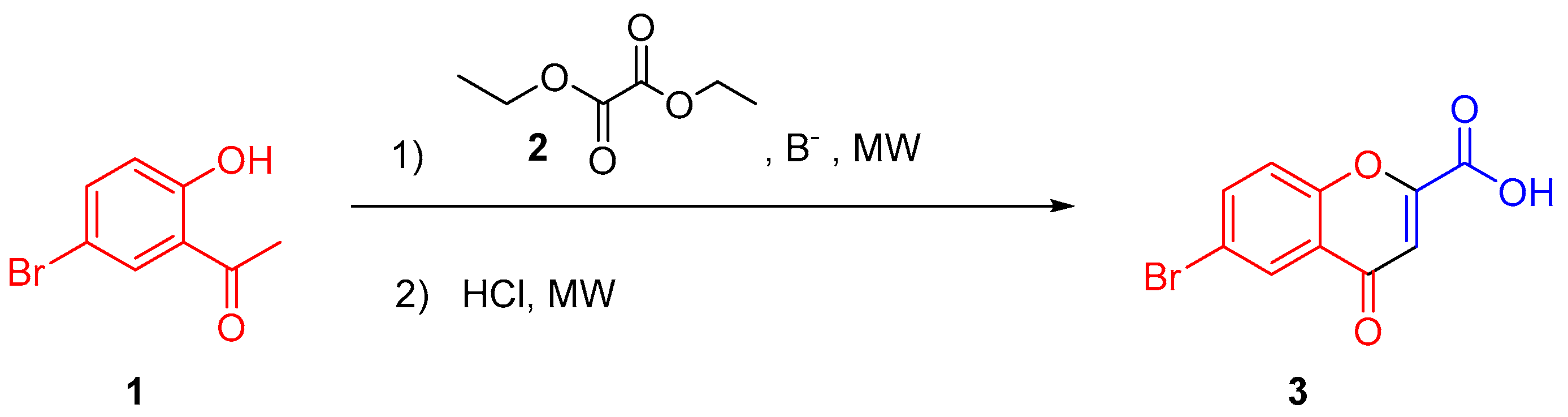{kind=link}
| Entry | Base (Equiv) | 2 (Equiv) | Solvent | T (°C) | Time 1 * (min) | Acid (V = 3 mL) | Time 2 * (min) | Yield (%) |
|---|---|---|---|---|---|---|---|---|
| 1 | EtONa (1) | 1 | EtOH | 120 | 10 | HCl (1 M) | 10 | 14 |
| 2 | MeONa (1) | 1 | MeOH | 120 | 10 | HCl (1 M) | 10 | 15 |
| 3 | MeONa (2) | 1 | MeOH | 120 | 10 | HCl (1 M) | 10 | 19 |
| 4 | MeONa (3) | 1 | MeOH | 120 | 10 | HCl (1 M) | 10 | 17 |
| 5 | MeONa (2) | 2 | MeOH | 120 | 10 | HCl (1 M) | 10 | 19 |
| 6 | MeONa (2) | 3 | MeOH | 120 | 10 | HCl (1 M) | 10 | 21 |
| 7 | MeONa (2) | 3 | MeOH | 120 | 10 | HCl (3 M) | 10 | 23 |
| 8 | MeONa (2) | 3 | MeOH | 120 | 10 | HCl (6 M) | 10 | 30 |
| 9 | MeONa (2) | 3 | MeOH | 140 | 10 | HCl (6 M) | 10 | 19 |
| 10 | MeONa (2) | 3 | MeOH | 120 | 20 | HCl (6 M) | 10 | 34 |
| 11 | MeONa (2) | 3 | MeOH | 120 | 30 | HCl (6 M) | 10 | 34 |
| 12 | MeONa (2) | 3 | iPrOH | 120 | 20 | HCl (6 M) | 10 | 51 |
| 13 | MeONa (2) | 3 | Dioxane | 120 | 20 | HCl (6 M) | 10 | 68 |
| 14 | MeONa (2) | 3 | Dioxane | 120 | 20 | HCl (6 M) | 30 | 81 |
| 15 | MeONa (2) | 3 | Dioxane | 120 | 20 | HCl (6 M) | 40 | 87 |
| 16 | MeONa (2) | 3 | Dioxane | 120 | 20 | HCl (1 M) | 50 | 85 |
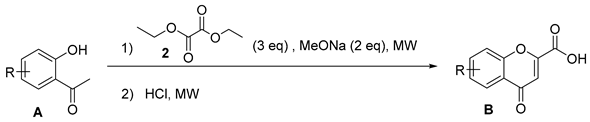
| Compound | 2′-Hydroxyacetophenone (A) | Chromone-2-Carboxylic Acid (B) | Yield (%) |
|---|---|---|---|
| 4 | 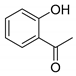 | 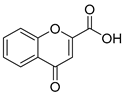 | 54 a |
| 5 | 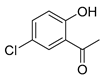 | 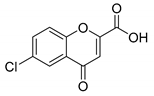 | 71 |
| 6 | 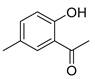 | 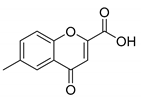 | 64 |
| 7 | 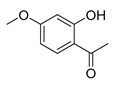 | 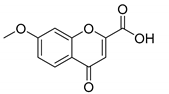 | 83 |
| 8 |  | 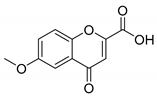 | 81 |
| 9 | 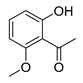 | 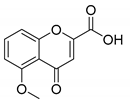 | 93 |
| 10 | 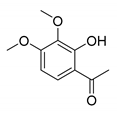 |  | 62 |
| 11 | 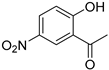 | 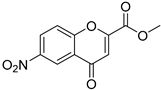 | 83 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cagide, F.; Oliveira, C.; Reis, J.; Borges, F. Optimizing the Synthetic Route of Chromone-2-carboxylic Acids: A Step forward to Speed-up the Discovery of Chromone-based Multitarget-directed Ligands. Molecules 2019, 24, 4214. https://doi.org/10.3390/molecules24234214
Cagide F, Oliveira C, Reis J, Borges F. Optimizing the Synthetic Route of Chromone-2-carboxylic Acids: A Step forward to Speed-up the Discovery of Chromone-based Multitarget-directed Ligands. Molecules. 2019; 24(23):4214. https://doi.org/10.3390/molecules24234214
Chicago/Turabian StyleCagide, Fernando, Catarina Oliveira, Joana Reis, and Fernanda Borges. 2019. "Optimizing the Synthetic Route of Chromone-2-carboxylic Acids: A Step forward to Speed-up the Discovery of Chromone-based Multitarget-directed Ligands" Molecules 24, no. 23: 4214. https://doi.org/10.3390/molecules24234214
APA StyleCagide, F., Oliveira, C., Reis, J., & Borges, F. (2019). Optimizing the Synthetic Route of Chromone-2-carboxylic Acids: A Step forward to Speed-up the Discovery of Chromone-based Multitarget-directed Ligands. Molecules, 24(23), 4214. https://doi.org/10.3390/molecules24234214