
A Molecular Electron Density Theory Study of the Synthesis of Spirobipyrazolines through the Domino Reaction of Nitrilimines with Allenoates




Abstract
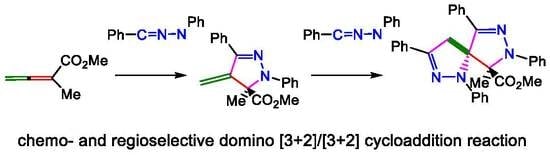
1. Introduction
2. Computational Methods
3. Results and Discussion
3.1. Topological Analysis of the ELF of Diphenyl NI 1 and Methyl 1-Methyl-Allenoate 4
3.2. Analysis of the CDFT Reactivity Indices of the Reagents
3.3. Study of the Reaction Paths Associated with the Domino Reaction of Diphenyl NI 1 with Allenoate 4
3.4. ELF Characterisation of the Single Bond Formation along the Most Favourable Reaction Path Associated with the 32CA Reaction of Diphenyl NI 1 with Allenoate 4
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Sannigrahi, M. Stereocontrolled Synthesis of Spirocyclics. Tetrahedron 1999, 55, 9007–9071. [Google Scholar] [CrossRef]
- Kotha, S.; Deb, A.C.; Lahiri, K.; Manivannan, E. Selected Synthetic Strategies to Spirocyclics. Synthesis 2009, 2, 165–193. [Google Scholar] [CrossRef]
- Grygorenko, O.O.; Radchenko, D.S.; Volochnyuk, D.M.; Tolmachev, A.A.; Komarov, I.V. Bicyclic Conformationally Restricted Diamines. Chem. Rev. 2011, 111, 5506–5568. [Google Scholar] [CrossRef] [PubMed]
- Berquist, P.R.; Wells, R.J. Marine Natural Products; Scheuer, P.J., Ed.; Academic Press: New York, NY, USA, 1983; Volume V, Chapter 1. [Google Scholar]
- Mashelkar, U.C.; Rane, D.M. Synthesis of Some Isatin Based Novel Spiroheterocycles and their Biological Activity Studies. Indian J. Chem. 2005, 44B, 1937–1939. [Google Scholar] [CrossRef]
- Lukyanov, B.S.; Lukyanova, M.B. Spiropyrans: Synthesis, Properties, and Application (Review). Chem. Heterocycl. Compd. 2005, 41, 281–311. [Google Scholar] [CrossRef]
- Güzel, O.; Ilhan, E.; Salman, A. Synthesis and Antimycobacterial Activity of New 2-Hydroxy-N-(3-oxo-1-Thia-4-Azaspiro[4.4]non-4-yl)/(3-Oxo-1-Thia-4-Azaspiro[4.5]dec-4-yl)-2,2-Diphenylacetamide Derivatives. Monatsh. Chem. 2006, 137, 795–801. [Google Scholar] [CrossRef]
- Joshi, N.S.; Karale, B.K.; Gill, C.H. Synthesis of some Thiadiazoles, Selenadiazoles and Spiroheterocyclic Compounds from Their 2,2-Dimethyl-Benzopyran Precursor. Chem. Heterocycl. Compd. 2006, 42, 681–685. [Google Scholar] [CrossRef]
- Habib-Zahmani, H.; Viala, J.; Hacini, S.; Rodriguez, J. Synthesis of Functionalized Spiroheterocycles by Sequential Multicomponent Reaction/Metal-Catalyzed Carbocylizations from Simple β-Ketoesters and Amides. Synlett 2007, 1037–1042. [Google Scholar]
- Arai, M.A.; Arai, T.; Sasai, H. Design and Synthesis of the First Spiro Bis(isoxazoline) Derivatives as Asymmetric Ligands. Org. Lett. 1999, 1, 1795–1797. [Google Scholar] [CrossRef]
- Arai, M.A.; Kuraishi, M.; Arai, T.; Sasai, H. A New Asymmetric Wacker-Type Cyclization and Tandem Cyclization Promoted by Pd(II)-Spiro Bis(isoxazoline) Catalyst. J. Am. Chem. Soc. 2001, 123, 2907–2908. [Google Scholar] [CrossRef]
- Na, R.; Li, Z.; Liu, H.; Liu, J.; Wang, M.-A.; Wang, M.; Zhong, J.; Guo, H. Thermal [3+2] Cycloaddition Reaction of Azomethine Imines with Allenoates for Dinitrogen-fused Heterocycles. Tetrahedron 2012, 68, 2349–2356. [Google Scholar] [CrossRef]
- Zhang, X.; Yuan, C.; Zhang, C.; Gao, X.; Wang, B.; Sun, Z.; Xiao, Y.; Guo, H. Thermal [3+2] Cycloaddition of N-Iminoquiazoline Ylides with Allenoates for Synthesis of Tetrahydropyrazoloquinazoline Derivatives. Tetrahedron 2016, 72, 8274–8281. [Google Scholar] [CrossRef]
- Li, F.; Chen, J.; Hou, Y.; Li, Y.; Wu, X.; Tong, X. 1,3-Dipolar Cycloadditions of 4-Acetoxy Allenoates: Access to 2,3-Dihydropyrazoles, 2,3-Dihydroisoxazoles, and Indolizines. Org. Lett. 2015, 17, 5376–5379. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Yu, H.; Feng, Y.; Hou, Z.; Zhang, L.; Yang, W.; Wu, Y.; Xiao, Y.; Guo, H. Phosphine-Catalyzed [4+3] Cycloaddition reaction of Aromatic Azomethine Imines with Allenoates. RSC Adv. 2015, 5, 34481–34485. [Google Scholar] [CrossRef]
- Li, Z.; Yu, H.; Liu, Y.; Zhou, L.; Sun, Z.; Guo, H. Phosphane-Catalyzed [3+3] Annulation of C,N-Cyclic Azomethine Imines with Ynones: A Practical Method for Tricyclic Dinitrogen-Fused Heterocycles. Adv. Synth. Catal. 2016, 358, 1880–1885. [Google Scholar] [CrossRef]
- Yuan, C.; Zhou, L.; Xia, M.; Sun, Z.; Wang, D.; Guo, H. Phosphine-Catalyzed Enantioselective [4+3] Annulation of Allenoates with C,N-Cyclic Azomethine Imines: Synthesis of Quinazoline-Based Tricyclic Heterocycles. Org. Lett. 2016, 18, 5644–5647. [Google Scholar] [CrossRef]
- Zhou, L.; Yuan, C.; Zhang, C.; Zhang, L.; Gao, Z.; Wang, C.; Liu, H.; Wu, Y.; Guo, H. Enantioselective Synthesis of Quinazoline-Based Heterocycles through Phosphine-Catalyzed Asymmetric [3+3] Annulation of Morita-Baylis-Hillman Carbonates with Azomethine Imines. Adv. Synth. Catal. 2017, 359, 2316–2321. [Google Scholar] [CrossRef]
- Dadiboyena, S. Cycloadditions and Condensations as Essential Tools in Spiropyrazoline Synthesis. Eur. J. Med. Chem. 2013, 63, 347–377. [Google Scholar] [CrossRef]
- Kerbal, A.; Vebrel, J.; Roche, M.; Laude, B. Reaction of Diarylnitrilimines with 3-Arylidene-4-Isothiochromanones. A General Approach to the First Thiadiazaphenanthrene. Tetrahedron Lett. 1990, 31, 4145–4146. [Google Scholar] [CrossRef]
- Dawood, K.; Fuchigami, T. Electrolytic Partial Fluorination of Organic Compounds. 79. Anodic Fluorination of Spiropyrazoline-5,3‘-chroman-4-ones and Thiochromanone Analogues. A Route to Aroyl Fluoride Derivatives. J. Org. Chem. 2005, 70, 7537–7541. [Google Scholar] [CrossRef]
- Singh, V.; Singh, V.; Batra, S. Straightforward Strategy for the Stereoselective Synthesis of Spiro-Fused (C-5)Isoxazolino- or (C-3)Pyrazolino-(C-3)quinolin-2-ones from Baylis–Hillman Adducts by 1,3-Dipolar Cycloaddition and Reductive Cyclization. Eur. J. Org. Chem. 2008, 5446–5460. [Google Scholar] [CrossRef]
- Mernyak, E.; Kozma, E.; Hetenyi, A.; Mark, L.; Schneider, G.; Wolfling, J. Stereoselective Synthesis of Spiro and Condensed Pyrazolines of Steroidal α,β-Unsaturated Ketones and Nitrilimines by 1,3-Dipolar Cycloaddition. Steroids 2009, 74, 520–525. [Google Scholar] [CrossRef] [PubMed]
- Dadiboyena, S.; Hamme, A.T., II. One Pot Spiropyrazoline Synthesis via Intramolecular Cyclization/Methylation. Tetrahedron Lett. 2011, 52, 2536–2539. [Google Scholar] [CrossRef] [PubMed]
- Ong, C.W.; Chien, T.-L. 1,3-Dipolar Cycloaddition to Tricarbonyl[(1−4-η)-2- Methoxy-5-Methylenecyclohexa-1,3-Diene]iron: Rapid Construction of a Spiro[4.5]decane System. Organometallics 1996, 15, 1323–1326. [Google Scholar] [CrossRef]
- Bartels, A.; Jones, P.G.; Liebscher, J. Stereoselective Diazoalkane Cycloadditions to Chiral 5-Alkylidene-1, 3-Dioxan-4-Ones and 3-Benzylidene-β-lactones. Synthesis 1998, 11, 1645–1654. [Google Scholar] [CrossRef]
- Levai, A. Synthesis of Spiro-1-Pyrazolines by the Reaction of z-3-Arylidene-1-Thioflavanones with Diazomethane. Org. Prep. Proced. Int. 2002, 34, 425–429. [Google Scholar] [CrossRef]
- Verma, D.; Mobin, S.; Namboothiri, I.N.N. Highly Selective Synthesis of Pyrazole and Spiropyrazoline Phosphonates via Base-Assisted Reaction of the Bestmann–Ohira Reagent with Enones. J. Org. Chem. 2011, 76, 4764–4770. [Google Scholar] [CrossRef]
- Santos, B.S.; Gomes, C.S.B.; Pinho e Melo, T.M.V.D. Synthesis of Chiral Spiropyrazoline-β-Lactams and Spirocyclopropyl-β-Lactams from 6-Alkylidenepenicillanates. Tetrahedron 2014, 70, 3812–3821. [Google Scholar] [CrossRef]
- Ortiz-Leon, A.; TorresValencia, J.M.; Manriquez-Torres, J.J.; Alvarado-Rodriguez, J.G.; Cerda-Garcia-Rojas, C.M.; Joseph-Nathan, P. The Stereochemistry of the 1,3-Dipolar Cycloadditions of Diazomethane to Pseudoguaianolides. Tetrahedron Asymmetry 2017, 28, 367–373. [Google Scholar] [CrossRef]
- Budzisz, E.; Paneth, P.; Geromino, I.; Muziol, T.; Rozalski, M.; Krajewska, U.; Pipiak, P.; Ponczek, M.B.; Malecka, M.; Kupcewicz, B. The Cytotoxic Effect of Spiroflavanone Derivatives, their Binding Ability to Human Serum Albumin (HSA) and a DFT Study on the Mechanism of their Synthesis. J. Mol. Struct. 2017, 1137, 267–276. [Google Scholar] [CrossRef]
- Nabeya, A.; Tamura, Y.; Kodama, I.; Iwakura, Y. Diaziridines. J. Org. Chem. 1973, 38, 3758–3762. [Google Scholar] [CrossRef]
- Makhova, N.N.; Petukhova, V.Y.; Kuznetsov, V.V. Synthesis of Monocyclic Diaziridines and their Fused Derivatives. ARKIVOC 2008, 128–152. [Google Scholar]
- Capretto, D.A.; Brouwer, C.; Poor, C.B.; He, C. Gold(I)-Catalyzed Formation of 3-Pyrazolines through Cycloaddition of Diaziridine to Alkynes. Org. Lett. 2011, 13, 5842–5845. [Google Scholar]
- Bird, C.W.; Cheesemen, G.W.H.; Brown, R.C.; Dyke, S.F. Aromatic and Heteroaromatic Chemistry; Bird, C.W., Cheesemen, G.W.H., Eds.; RSC: London, UK, 1973; pp. 98–122. [Google Scholar]
- Li, J.J. Name Reactions: A Collection of Detailed Mechanisms and Synthetic Applications, 4th ed.; Springer: New York, NY, USA, 2009. [Google Scholar]
- Li, J.J.; Corey, E.J. Name Reactions in Heterocyclic Chemistry II (Comprehensive Name Reactions); Wiley: New York, NY, USA, 2011. [Google Scholar]
- Shawali, A.S.; Abdelhamid, A.O. Synthesis of Spiro-heterocycles via 1,3-Dipolar Cycloadditions of Nitrilimines to Exoheterocyclic Enones. Site-, Regio- and Stereo-Selectivities Overview. Curr. Org. Chem. 2012, 16, 2673–2689. [Google Scholar] [CrossRef]
- Monteiro, A.; Gonçalves, L.M.; Santos, M.M.M. Synthesis of Novel Spiropyrazoline Oxindoles and Evaluation of Cytotoxicity in Cancer Cell Lines. Eur. J. Med. Chem. 2014, 79, 266–272. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Jia, H.; Wang, B.; Xiao, Y.; Guo, H. Synthesis of Spirobidihydropyrazole through Double 1,3-Dipolar Cycloaddition of Nitrilimines with Allenoates. Org. Lett. 2017, 19, 4714–4717. [Google Scholar] [CrossRef] [PubMed]
- Domingo, L.R. Molecular Electron Density Theory: A Modern View of Reactivity in Organic Chemistry. Molecules 2016, 21, 1319. [Google Scholar] [CrossRef]
- Ríos-Gutiérrez, M.; Domingo, L.R. The Carbenoid-Type Reactivity of Simplest Nitrile Imine from a Molecular Electron Density Theory Perspective. Tetrahedron 2019, 75, 1961–1967. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. Hybrid Meta Density Functional Theory Methods for Thermochemistry, Thermochemical Kinetics, and Noncovalent Interactions: The MPW1B95 and MPWB1K Models and Comparative Assessments for Hydrogen Bonding and van der Waals interactions. J. Phys. Chem. A 2004, 108, 6908–6918. [Google Scholar] [CrossRef]
- Hehre, M.J.; Radom, L.; Schleyer, P.v.R.; Pople, J. Ab initio Molecular Orbital Theory; Wiley: New York, NY, USA, 1986. [Google Scholar]
- Schlegel, H.B. Optimization of Equilibrium Geometries and Transition Structures. J. Comput. Chem. 1982, 3, 214–218. [Google Scholar] [CrossRef]
- Schlegel, H.B. Modern Electronic Structure Theory; Yarkony, D.R., Ed.; World Scientific Publishing: Singapore, 1994. [Google Scholar]
- Fukui, K. Formulation of the Reaction Coordinate. J. Phys. Chem. 1970, 74, 4161–4163. [Google Scholar] [CrossRef]
- González, C.; Schlegel, H.B. Reaction Path Following in Mass-Weighted Internal Coordinates. J. Phys. Chem. 1990, 94, 5523–5527. [Google Scholar] [CrossRef]
- González, C.; Schlegel, H.B. Improved Algorithms for Reaction Path Following: Higher-order Implicit Algorithms. J. Chem. Phys. 1991, 95, 5853–5860. [Google Scholar] [CrossRef]
- Tomasi, J.; Persico, M. Molecular Interactions in Solution: And Overview of Methods Based on Continuous Distributions of the Solvent. Chem. Rev. 1994, 94, 2027–2094. [Google Scholar] [CrossRef]
- Simkin, B.Y.; Sheikhet, I.I. Quantum Chemical and Statistical Theory of Solutions–Computational Approach; Ellis Horwood: London, UK, 1995. [Google Scholar]
- Cossi, M.; Barone, V.; Cammi, R.; Tomasi, J. Ab Initio Study of Solvated Molecules: A New Implementation of the Polarizable Continuum Model. Chem. Phys. Lett. 1996, 255, 327–335. [Google Scholar] [CrossRef]
- Cances, E.; Mennucci, B.; Tomasi, J. A New Integral Equation Formalism for the Polarizable Continuum Model: Theoretical Background and Applications to Isotropic and Anisotropic Dielectrics. J. Chem. Phys. 1997, 107, 3032–3041. [Google Scholar] [CrossRef]
- Barone, V.; Cossi, M.; Tomasi, J. Geometry Optimization of Molecular Structures in Solution by the Polarizable Continuum Model. J. Comput. Chem. 1998, 19, 404–417. [Google Scholar] [CrossRef]
- Domingo, L.R. A New C-C Bond Formation Model Based on the Quantum Chemical Topology of Electron Density. RSC Adv. 2014, 4, 32415–32428. [Google Scholar] [CrossRef]
- Reed, A.E.; Weinstock, R.B.; Weinhold, F. Natural Population Analysis. J. Chem. Phys. 1985, 83, 735–746. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular Interactions from a Natural Bond Orbital, Donor-Acceptor Viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Geerlings, P.; De Proft, F.; Langenaeker, W. Conceptual Density Functional Theory. Chem. Rev. 2003, 103, 1793–1873. [Google Scholar] [CrossRef] [PubMed]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Pérez, P. Applications of the Conceptual Density Functional Indices to Organic Chemistry Reactivity. Molecules 2016, 21, 748. [Google Scholar] [CrossRef] [PubMed]
- Domingo, L.R.; Pérez, P.; Sáez, J.A. Understanding the Local Reactivity in Polar Organic Reactions through Electrophilic and Nucleophilic Parr Functions. RSC Adv. 2013, 3, 1486–1494. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Becke, A.D.; Edgecombe, K.E. A Simple Measure of Electron Localization in Atomic and Molecular-Systems. J. Chem. Phys. 1990, 92, 5397–5403. [Google Scholar] [CrossRef]
- Noury, S.; Krokidis, X.; Fuster, F.; Silvi, B. Computational Tools for the Electron Localization Function Topological Analysis. Comput. Chem. 1999, 23, 597–604. [Google Scholar] [CrossRef]
- Krokidis, X.; Noury, S.; Silvi, B. Characterization of Elementary Chemical Processes by Catastrophe Theory. J. Phys. Chem. A 1997, 101, 7277–7282. [Google Scholar] [CrossRef]
- GaussView, Version 6.1, R.; Dennington, T.A., Keith, J.M., Eds.; Millam, Semichem Inc.: Shawnee Mission, KS, USA, 2016. [Google Scholar]
- Ríos-Gutiérrez, M.; Domingo, L.R. Unravelling the Mysteries of the [3+2] Cycloaddition Reactions. Eur. J. Org. Chem. 2019, 267–282. [Google Scholar] [CrossRef]
- Hamza-Reguig, S.; Bentabed-Ababsa, G.; Domingo, L.R.; Ríos-Gutiérrez, M.; Philippot, S.; Fontanay, S.; Duval, R.E.; Ruchaud, S.; Bach, S.; Roisnel, T.; et al. A Combined Experimental and Theoretical Study of the Thermal [3+2] Cycloaddition of Carbonyl Ylides with Activated Alkenes. J. Mol. Struct. 2018, 1157, 276–287. [Google Scholar] [CrossRef]
- Huisgen, R. 1,3-Dipolar Cycloadditions. Proc. Chem. Soc. 1961, 357–396. [Google Scholar]
- Parr, R.G.; Yang, W. Density Functional Theory of Atoms and Molecules; Oxford University Press: New York, NY, USA, 1989. [Google Scholar]
- Parr, R.G.; Szentpaly, L.V.; Liu, S. Electrophilicity Index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Domingo, L.R.; Chamorro, E.; Pérez, P. Understanding the Reactivity of Captodative Ethylenes in Polar Cycloaddition Reactions. A Theoretical Study. J. Org. Chem. 2008, 73, 4615–4624. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.G.; Polanyi, M. Some Applications of the Transition State Method to the Calculation of Reaction Velocities, Especially in Solution. Trans. Faraday Soc. 1935, 31, 875–894. [Google Scholar] [CrossRef]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Pérez, P. How Does the Global Electron Density Transfer Diminish Activation Energies in Polar Cycloaddition Reactions? A Molecular Electron Density Theory study. Tetrahedron 2017, 73, 1718–1724. [Google Scholar] [CrossRef]
- Domingo, L.R.; Sáez, J.A.; Zaragozá, R.J.; Arnó, M. Understanding the Participation of Quadricyclane as Nucleophile in Polar [2σ + 2σ + 2π] Cycloadditions toward Electrophilic π Molecules. J. Org. Chem. 2008, 73, 8791–8799. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| μ | η | ω | N | |
|---|---|---|---|---|
| diphenyl NI 1 | −3.38 | 5.05 | 1.13 | 4.89 |
| dimethyl NI 13 | −2.90 | 7.34 | 0.57 | 4.22 |
| allenoate 4 | −4.18 | 8.33 | 1.05 | 2.45 |
| Structures | S1 | S2 | TS11 | S3 | S4 | S5 | S6 | S7 | S8 | S9 |
|---|---|---|---|---|---|---|---|---|---|---|
| d(C1–C5) | 3.431 | 3.421 | 2.191 | 2.075 | 1.958 | 1.947 | 1.543 | 1.480 | 1.479 | 1.457 |
| d(N3–C4) | 3.235 | 3.206 | 2.580 | 2.540 | 2.499 | 2.495 | 2.234 | 1.928 | 1.918 | 1.486 |
| GEDT | 0.01 | 0.02 | 0.13 | 0.14 | 0.13 | 0.13 | −0.03 | −0.12 | −0.12 | −0.16 |
| IRC | −14.897 | −12.415 | 0.000 | 0.622 | 1.243 | 1.300 | 3.956 | 5.821 | 5.877 | 19.959 |
| ΔE | 0.0 | 0.3 | 14.8 | 13.8 | 10.3 | 9.8 | −19.3 | −36.1 | −36.6 | −64.7 |
| V(C1,N2) | 2.65 | 2.32 | 2.26 | 2.18 | 1.09 | 1.11 | 3.14 | 3.11 | 3.11 | 3.03 |
| V′(C1,N2) | 3.04 | 3.10 | 1.17 | 1.10 | 2.09 | 2.07 | ||||
| V(N2) | 1.72 | 1.95 | 2.13 | 2.14 | 2.57 | 2.67 | 2.67 | 2.77 | ||
| V(N2,N3) | 2.13 | 2.13 | 1.98 | 1.94 | 1.91 | 1.91 | 1.78 | 1.69 | 1.68 | 1.55 |
| V(N3) | 3.50 | 3.48 | 3.10 | 3.03 | 2.99 | 2.98 | 2.98 | 3.37 | 2.33 | 1.93 |
| V(C4,C5) | 1.83 | 1.83 | 3.49 | 3.10 | 2.78 | 2.76 | 2.27 | 2.18 | 2.17 | 2.04 |
| V′(C4,C5) | 1.86 | 1.86 | ||||||||
| V(C1) | 0.28 | 1.17 | 1.23 | 1.28 | ||||||
| V(C5) | 0.32 | 0.55 | ||||||||
| V(C4) | 0.19 | |||||||||
| V(C1,C5) | 1.85 | 2.14 | 2.22 | 2.23 | 2.28 | |||||
| V(N3,C4) | 1.06 | 1.66 | ||||||||
| V(C1,Ph) | 2.53 | 2.52 | 2.30 | 2.28 | 2.25 | 2.25 | 2.20 | 2.22 | 2.22 | 2.26 |
| V(C5,C5′) | 1.86 | 1.86 | 1.84 | 1.80 | 1.76 | 1.76 | 1.71 | 1.71 | 1.71 | 1.73 |
| V′(C5,C5′) | 1.86 | 1.85 | 1.89 | 1.88 | 1.87 | 1.87 | 1.74 | 1.72 | 1.72 | 1.72 |
| V(C4,CO) | 2.31 | 2.31 | 2.48 | 2.56 | 2.64 | 2.65 | 2.51 | 2.34 | 2.33 | 2.19 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Domingo, L.R.; Ghodsi, F.; Ríos-Gutiérrez, M. A Molecular Electron Density Theory Study of the Synthesis of Spirobipyrazolines through the Domino Reaction of Nitrilimines with Allenoates. Molecules 2019, 24, 4159. https://doi.org/10.3390/molecules24224159
Domingo LR, Ghodsi F, Ríos-Gutiérrez M. A Molecular Electron Density Theory Study of the Synthesis of Spirobipyrazolines through the Domino Reaction of Nitrilimines with Allenoates. Molecules. 2019; 24(22):4159. https://doi.org/10.3390/molecules24224159
Chicago/Turabian StyleDomingo, Luis R., Fatemeh Ghodsi, and Mar Ríos-Gutiérrez. 2019. "A Molecular Electron Density Theory Study of the Synthesis of Spirobipyrazolines through the Domino Reaction of Nitrilimines with Allenoates" Molecules 24, no. 22: 4159. https://doi.org/10.3390/molecules24224159
APA StyleDomingo, L. R., Ghodsi, F., & Ríos-Gutiérrez, M. (2019). A Molecular Electron Density Theory Study of the Synthesis of Spirobipyrazolines through the Domino Reaction of Nitrilimines with Allenoates. Molecules, 24(22), 4159. https://doi.org/10.3390/molecules24224159