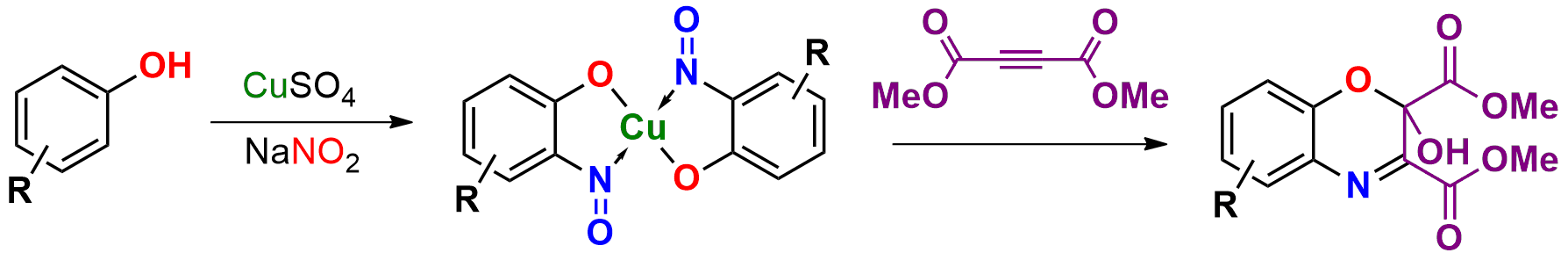
3.1. Synthesis of Copper-Nitrosophenolato Compounds
The phenolic starting material (
1a–
1o, 50 mmol) was dissolved in 25 mL of water and 15 mL acetic acid. To this mixture was added solid sodium acetate trihydrate, gradually, until the pH was 4. Copper sulfate pentahydrate (25 mmol, 6.25 g) and sodium nitrite (125 mmol, 8.65 g) were dissolved in water (250 mL) and this solution was introduced dropwise to the solution of phenolic starting material. The mixture was stirred at ambient temperature for at least 3 days and left to stand without mixing for a further 24 h. The product was collected by filtration and washed with water (3 × 50 mL), toluene (2 × 50 mL) and finally a portion of ethyl acetate (20 mL), then dried under reduced pressure. The copper complexes (
2a–
2o) were of acceptable purity at this stage and characterised by ASAP MS, IR spectroscopy and elemental analysis. For the purpose of growing crystals suitable for XRD analysis, the powder obtained was dissolved in a 1:1 mixture of boiling alcoholic and polar-organic solvents (specified), then left to cool to room temperature. The resulting crystals were collected by filtration and washed with hexane (10 mL), giving crystalline copper complexes (
2a–
2o). Yields refer to pure, washed, amorphous products, not recrystallised adducts. The spectral data is shown below and the graphical spectra and data (NMR, IR, MS and XRD) can be found in the
supplementary information.
Copper(II) bis(4-methyl-2-nitrosophenolato) (2a) [
23]
: Recrystallised from chloroform-ethanol to yield a deep purple solid. Yield: 82%, 7.2 g. C
14H
12O
4N
2Cu. ASAP-MS:
m/
z 336.0 ([M + H]
+, 100%), 138.1 ([C
7H
7O
2N + H]
+, 2) HR-MS calculated for C
14H
13N
2O
4 63Cu 336.0171, found: 336.0167. (−1.2 ppm, −0.4 mDa). IR (neat) υ
max/cm
−1 2971 (m, C-H), 1739 (s, C=O), 1365 (s, N=O), 1218 (m). Elemental (C, H, N) analysis found: C 49.75% (50.07), H 3.60% (3.66), N 8.42% (8.34).
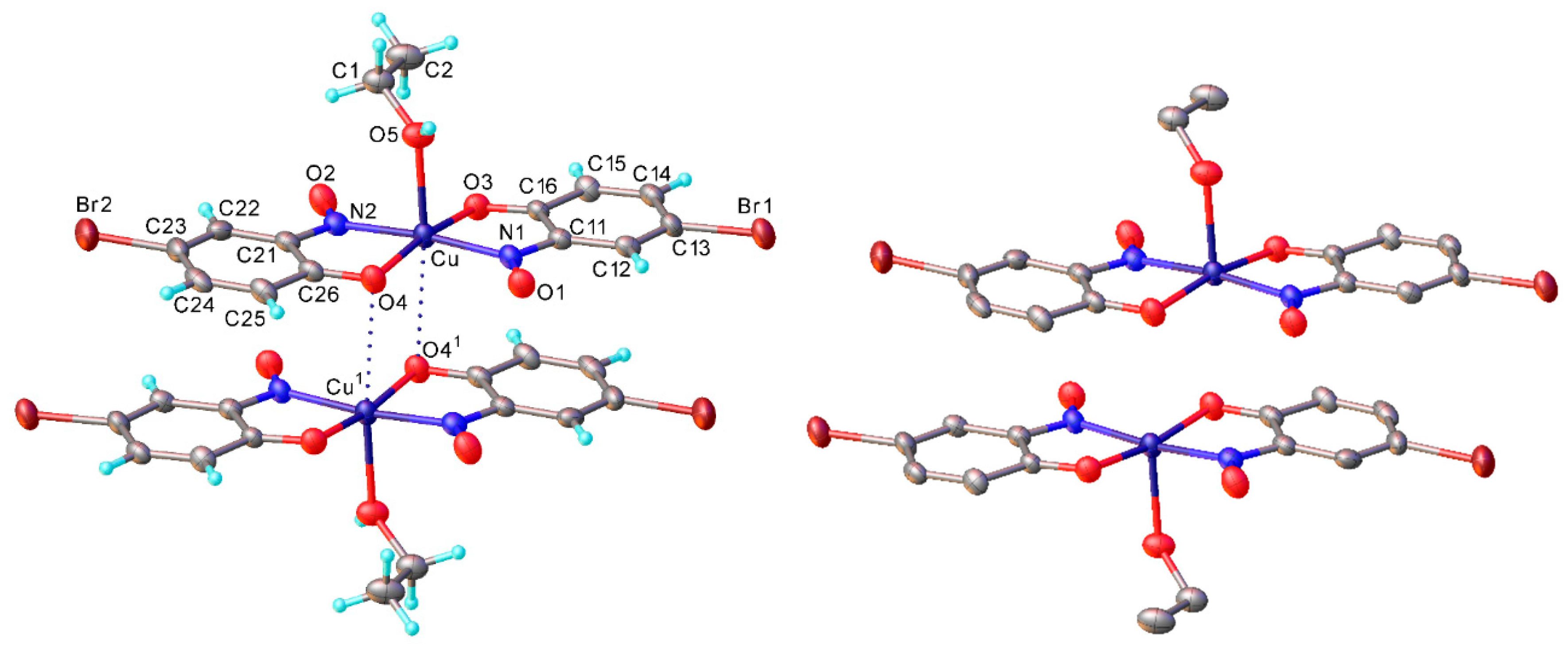
Crystal data: 1957008; C
14H
12O
4N
2Cu·C
2H
6O, f.w. 381.86, T = 120 K, monoclinic,
a = 10.1875(8),
b = 6.9915(5),
c = 22.2994(17) Å β = 91.781(3)°,
V = 1587.5(2) Å
3, space group
P2
1/
c (no. 14),
Z = 4, 22237 reflections (3840 unique,
Rint = 0.050),
R1 = 0.034,
wR2 = 0.081 The structure is essentially identical with the earlier room-temperature determination [
22]. M.p. 218.1–218.8 °C (decomposition).
Copper(II) bis(4-Chloro-2-nitrosophenolato) (2b) [
24]
: Recrystallised from chloroform-ethanol to yield a purple solid. Yield: 55%, 5.4 g. C
12H
6N
2O
4Cl
2Cu. ASAP-MS:
m/
z 377.9 ([M + H]
+, 95%), 158.1 ([C
6H
3O
2NCl H]
+, 25). HR-MS calculated for C
12H
6N
2O
435Cl
263Cu 374.9012, found: 374.9001. (−2.9 ppm, −1.1 mDa). IR (neat) υ
max/cm
−1 3017 (br, C-H). 1521 (s, N=O), 1489 (s, N=O), 1332 (m, N=O), 1181 (m, C-O). Elemental (C, H, N) analysis found: C 38.37% (38.25), H 1.65% (1.61), N 7.41% (7.44). M.p. 219.3–220.5 °C (decomposition).
Copper(II) bis(4-Bromo-2-nitrosophenolato) (2c) [
8]
: Recrystallised from chloroform-ethanol to yield a purple solid. Yield: 41%, 4.8 g. C
12H
7N
2O
4Br
2Cu ASAP-MS:
m/
z 463.8 ([M + H (
79Br)]
+ 100%) 203.9 ([(C
6H
4O
2N
179Br) + H]
+, 8). HR-MS calculated for C
12H
7N
2O
479Br
263Cu 463.8069, found: 463.8076. (+1.5 ppm, +0.7 mDa). IR (neat) υ
max/cm
−1 3087 (m, C-H), 1601 (m, N=O), 1491 (s, N=O), 1315 (m, N=O), 1166 (m, C-O). Elemental (C, H, N) analysis found: 29.57% (30.06), 1.27% (1.30), 5.67% (6.02).
Crystal data: CCDC 1957005; C
12H
6O
4N
2CuBr
2·C
2H
6O, f.w. 511.62, T = 100 K, monoclinic, a = 7.0128(16),
b = 10.020(2),
c = 23.311(5) Å, β = 93.502(5)°,
V = 1635.0(6) Å
3, space group
P2
1/
n (no. 14),
Z = 4, 19075 reflections (4121 unique,
Rint = 0.081),
R1 = 0.084,
wR2 = 0.240. M.p. 241–255 °C (decomposition).
Copper(II) bis(2-methoxy-6-nitroso-4-formylphenolato) (2d): Recrystallised from chloroform-methanol to yield a dark purple solid. Yield: 63%, 7.0 g. C16H12O8N2Cu ASAP-MS: m/z 424.0 ([M + H]+, 100%) 203.9 ([(C8H7O4N1) + H]+, 8). HR-MS calculated for C16H12N2O863Cu 423.9968, found: 423.9965. (−0.7 ppm, −0.3 mDa). IR (neat) υmax/cm−1 3463 (w, br), 3062 (w, C-H), 1687 (s, C=O), 1607 (w, N=O), 1519 (s, N=O), 1388 (m, N=O), 1273 (s, N=O), 1141 (s), 1091 (m), 654 (m). Elemental (C, H, N) Analysis found: 42.86% (43.30), 3.34% (3.63), 6.31% (6.45) (compound observed was the monohydrate). Crystal data: CCDC 1957006; C16H12O8N2Cu, f.w. 423.82, T = 120 K, monoclinic, a = 6.4741(5), b = 12.0560(9), c = 10.1090(7) Å, β = 103.595(3)°, V = 766.9(1) Å3, space group P21/c (no. 14), Z = 2, 16527 reflections (2239 unique, Rint = 0.047), R1 = 0.037, wR2 = 0.101. M.p. = 269.2–271.7 °C (decomposition).
Copper(II) bis(2-methoxy-6-nitroso-4-ethenylphenolato) (2e): Dark-green solid that failed to crystallise from various solvent mixtures. Yield: 72%, 7.7 g. CuC18H16N2O8. IR (neat) υmax/cm−1 3397 (w, br), 2754 (w, C-H), 1596 (m, N=O), 1535 (m, N=O), 1380 (s, N=O), 1227 (s, N=O), 1112 (m, C-O), 800 (s), 693 (m), 504 (m). Elemental (C, H, N) Analysis found: 53.17% (53.39), 4.64% (4.93), 6.46% (6.23). M.p. 161–172 °C (decomposition).
Copper(II) bis(2-nitroso-4-carboxylphenolato)(NOH)2 (2f): Pale-purple solid that failed to crystallise from various solvent mixtures. Yield: 71%, 8.4 g. CuC14H8N4O10. IR (neat) υmax/cm−1 3247 (m, br, O-H), 1598 (s, N=O), 1474 (m, N=O), 1294 (m, N=O), 1148 (s, C-O), 993 (s), 836 (s), 648 (m), 416 (s). Elemental (C, H, N) Analysis found: 37.28% (36.73), 2.69% (2.20), 12.67% (12.24). m.p. 161–172 °C (decomposition).
Copper(II) bis(2-nitroso-5-(diethylamino)phenolato) (2g) [
25]
: Purple solid that failed to crystallise in a range of solvent mixtures. Yield: 40%, 4.1 g. CuC
20H
26N
4O
4. ASAP-MS:
m/
z 195.1 ([C
10H
14O
2N
2]
+ 100%). IR (neat) υ
max/cm
−1 3057 (w, C-H), 1588 (m, N=O), 1442 (m, N=O), 1372 (s, N=O), 1227 (s, N=O), 1217 (s, N=O), 1100 (m, C-O), 1014 (m), 925 (m), 653 (m), 500 (m). M.p. > 300 °C.
Copper(II) bis(3-methoxy-6-nitrosophenolato) (2h) [
26]
: Orange solid that failed to crystallise in various solvent mixtures. Yield: 86%, 8.3 g. CuC
14H
12N
2O
6 IR (neat) υ
max/cm
−1 3405 (m, br), 3072 (w, C-H), 1595 (m, N=O), 1421 (m, N=O), 1379 (s, N=O), 1227 (s, N=O), 1206 (s, N=O), 1112 (m, C-O), 1029 (m), 856 (m), 693 (m), 503 (m). ASAP-MS:
m/
z 368.0 ([M + H]
+, 100%), 154.0 ([C
7H
7O
3N + H]
+, 8). HR-MS calculated for C
14H
13N
2O
663Cu 367.0009, found: 366.9991. (−4.9 ppm, mDa −1.8). Elemental Analysis found: C 43.46% (43.57), H 3.75% (3.66), 7.34% (7.26). M.p. 137–141 °C (decomposition).
Copper(II) bis(3-chloro-6-nitrosophenolato) (2i): Red solid that failed to crystallise in various solvent mixtures. Yield: 73%, 7.2 g. C12H7N2O4Cl2Cu. ASAP-MS: m/z 377.9 ([M + H]+, 100), 158.0 ([C6H3O2NCl + H]+, 91). HR-MS calculated for C12H7N2O435Cl263Cu 375.9076, found: 375.9081 (+1.3 ppm, mDa +0.5). IR (neat) υmax/cm−1 3351 (br, C-H), 1624 (m, N=O), 1394 (s, N=O), 1248 (m, N=O), 1112 (m, C-O). Elemental (C, H, N) Analysis found: C 39.91% (38.25), H 2.36% (1.61), N 7.48% (7.44). M.p. 153.2–154.0 °C (decomposition).
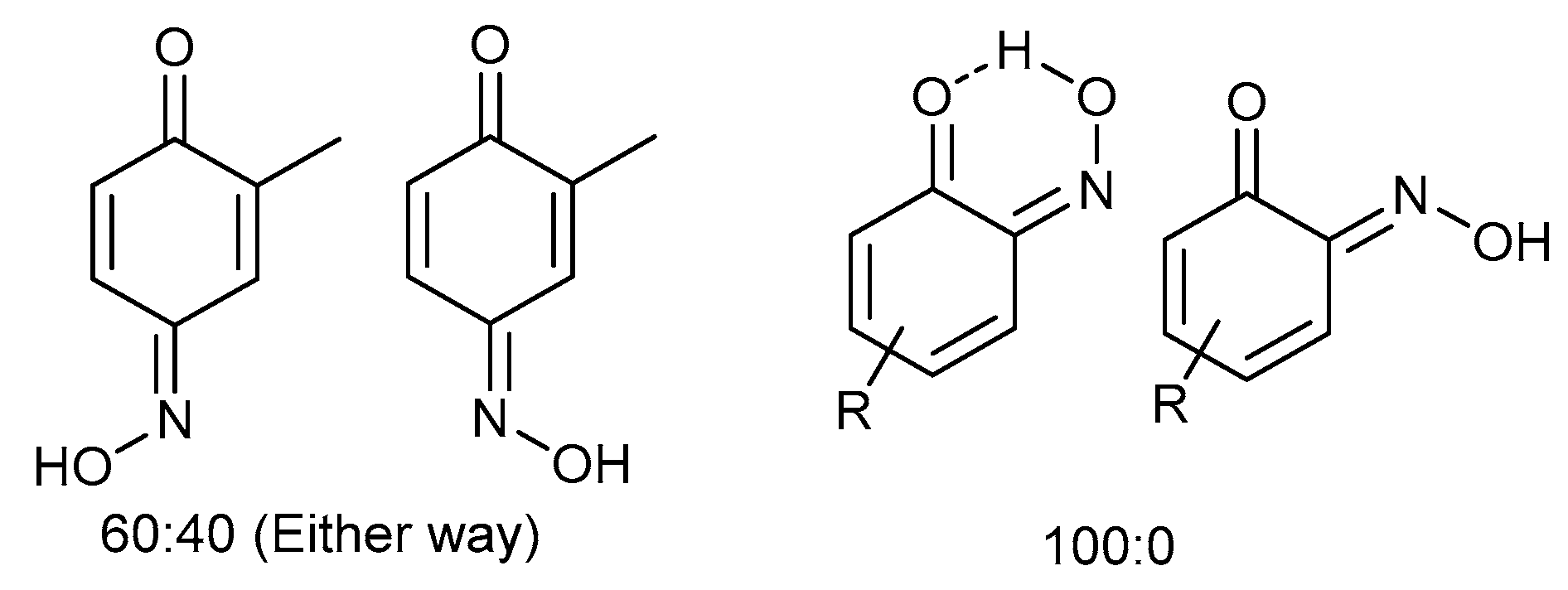
Copper(II) bis(5-methyl-2-nitrosophenolato) (2j): Isolated mixture with 3-methyl-1,4- benzoquinone monoxime (3j). Brown solid that failed to crystallise. Conversion >% estimated by derivatisation to the heterocyclic product (4j). C14H12O4N2Cu. ASAP-MS: m/z 334.99 ([M]+, 100%). IR (neat) υmax/cm−1 2773 (m, C-H), 1632 (m, N=O), 1401 (s, N=O), 1116 (m, C-O).
Copper(II) bis(2,4-dichloro-6-nitrophenolato) (2k): Recrystallised from chloroform-acetonitrile to yield a black solid. Yield: 55%, 6.56 g. IR (neat) υmax/cm−1 3082 (w, C-H), 1607 (m, N=O), 1536 (s, N=O), 1447 (m, N=O), 1336 (m, N=O), 1243 (s, N=O), 1151 (s, O-H), 890 (m), 773 (m), 682 (s). Crystal data: CCDC 1957007; C12H4O4N2Cl4Cu·2C2H3N, f.w. 559.62, T = 120 K, orthorhombic, a = 20.491(2), b = 7.5407(7), c = 13.2872(14) Å, V = 2053.1(4) Å3, space group Cmce (no. 64), Z = 4, 8029 reflections (922 unique, Rint = 0.073), R1 = 0.045, wR2 = 0.118. M.p. = 223.3–225.0 °C (decomposition).
Copper(II) bis(2-nitroso-6-methylphenolato)(nitroso) (2l): Brown solid that failed to crystallise from various solvent mixtures. Yield: 57%, 5.3 g. CuC14H12O5N3. ASAP-MS: m/z 124.0 ([C6H5O2N + H]+, 100%). IR υmax/cm−1 3264 (m, br, C-H), 1595 (m, N=O), 1429 (s, N=O), 1282 (s), 1111 (m, C-O). Elemental (C, H, N) analysis found: C 45.16% (45.96), H 3.67% (3.31), N 12.28% (11.49). M.p. = 161.5–163.8 °C (decomposition).
Copper(II) bis(2-chloro-4-nitrosophenolato)di(nitroso) (2m): Brown solid that failed to crystallise from various solvent mixtures. Yield: 72%, 7.3 g. CuC12Cl2H6O5N3. ASAP-MS: m/z 158.0 ([C6H3O2NCl + H]+, 94). IR (neat) υmax/cm−1 2817 (m, C-H). 1623 (m, N=O), 1448 (s, N=O), 1302 (m, N=O), 1116 (m, C-O). d.p. 154–158 °C. Elemental (C, H, N) analysis found: C 33.16 (32.86), H 1.84 (1.83), N 11.29 (12.77). M.p. = 154.7–156.2 °C (decomposition).
Copper(II) bis(4-nitroso-2-carboxylphenolato)(nitroso) isolated mixture (2n): Beige solid that failed to crystallise. Yield: 9.6 g. Formula not assigned. ASAP-MS: m/z 168.0 ([C7H5O3N + H]+, 29). IR υmax/cm−1 3382 (w, br, O-H), 1601 (m, N=O), 1455 (s, N=O), 1151 (s, C-O), 828 (m), 644 (s) 575 (s). Elemental (C, H, N) analysis found: C 37.78%, H 2.93%, N 15.59%. M.p. = 243–251 °C (decomposition).
Copper(II) bis(4-nitrosophenolato)di(nitroso) (2o): Red solid that failed to crystallise in various solvent mixtures. Yield: 72%, 6.1g. CuC12H8O6N4. ASAP-MS: m/z 124.0 ([C6H5O2N + H]+, 100%). IR (neat) υmax/cm−1 3248 (s, br, O-H). 1598 (m, N=O), 1506 (m, N=O), 1293 (m, N=O), 1147 (m, br, C-O). Elemental (C, H, N) analysis found: C 37.94% (39.19), H 2.72% (2.19), N 13.52% (15.23). M.p. = 163–170 °C (decomposition).
3.2. Isolation of Organic Ligands from Complexes
The copper-nitrosophenolato starting material (2a–2i) (1.0 g) was dissolved in acetone (40 mL) and the resulting mixture was stirred with polymer-supported thiourea (10 g) and 4 Å molecular sieve (0.5 g) under a nitrogen atmosphere at ambient temperature for 24 h. The solid polymer-supports were removed by filtration and the filtrate passed-through a bed of silica (1 cm depth), to remove any dissolved copper-containing species. The resultant filtrate solution was evaporated to dryness. Pure nitroso products were obtained in the case of 3h, 3i, 3j and 3o (with a 10% 4-nitrophenol impurity), for others, the organic residue was purified by column chromatography (hexane-ethyl acetate mixtures, various ratios), giving low yields of stated aromatic products. For nitroso products that were too unstable for characterisation after attempting the procedure, the isolation was instead performed in THF (40 mL) saturated with O2 gas, rather than acetone, to give low yields of oxidised nitro products (3a, 3b, 3c, 3d) and decomposition products.
4-Methyl-2-nitrophenol (3a) [
27]
: Eluent: hexane-ethyl acetate (4:1) to yield a brown solid. Yield: 14%, 6.1 g.
1H NMR (400 MHz; CDCl
3) 10.46 (1H, s, O
H), 7.92 (1H, m, Aryl-
H), 7.42 (1H, dd, J 8.0, 2.0, Aryl-
H), 7.08 (1H, d, J 8.8, Aryl-
H), 2.37 (3H, s, C
H3). δ
C(400 MHz; DMSO) 150.6 (
C1), 136.7 (
C2), 136.5 (
C4), 129.1 (
C5), 125.0 (
C3), 119.5 (
C6), 19.7 (
CH
3). ASAP-MS:
m/
z 153.1 (M
●+, 100%). HR-MS calculated for C
7H
7NO
3 153.0405, found: 153.0400. (−3.3 ppm, −0.5 mDa).
4-Chloro-2-nitrophenol (3b) [
27]
: Eluent: hexane-ethyl acetate (4:1) to yield a beige solid. Yield: 5%, g.
1H NMR (400 MHz; CDCl
3) 10.49 (1H, s, O
H) 8.11 (1H, d, J 2.8, Aryl-
H), 7.54 (1H, dd, J 8.8, 2.8, Aryl-
H), 7.15 (1H, d, J 8.8, Aryl-
H).
13C NMR (100 MHz; DMSO) 153.7 (
C1), 137.7 (
C2), 135.8 (
C4), 125.5 (
C5), 124.4 (
C3), 121.5 (
C6). LC-MS:
m/
z 172.0 ([M − H]
−, 100%).
4-Bromo-2-nitrophenol (3c) [
27]
: Eluent: hexane-ethyl acetate (4:1) to yield an orange solid. Yield: 9%, g.
1H NMR (400 MHz; DMSO) 8.27 (1H, d, J 2.4, Aryl-
H), 7.67 (1H, dd, J 8.8 2.4, Aryl-
H) 7.10 (1H, d, J 8.8, Aryl-
H).
13C NMR (100 MHz; CDCl
3) 154.1 (
C1), 140.4 (
C2), 134.1 (
C4), 127.3 (
C5), 121.8 (
C3), 111.7 (
C6). LC-MS
m/
z 216.3 ([M − H]
−, 100%).
2-Nitro-6-methoxy-4-formylphenol (3d) [
28]
: Recrystallised from ethanol to yield an orange solid. Yield: 21%, g.
1H NMR (400 MHz, DMSO-
d6) δ 9.87 (s, 1H,
HCO), 8.10 (d,
J = 1.8 Hz, 1H, Aryl-
H), 7.62 (d,
J = 1.8 Hz, 1H, Aryl-
H), 3.96 (s, 3H, OC
H3).
13C NMR (101 MHz, DMSO-d
6) δ 190.91, 150.54, 148.23, 137.54, 127.28, 121.33, 113.03, 57.26.
3-(Diethylamino)-6-nitrosophenol (3g) [
29]
: Brown crystals that required no further purification. Yield: %, g.
1H NMR (400 MHz, DMSO-d
6) δ 7.30 (d, J = 9.8 Hz, 2H, Aryl-
H), 6.89 (d, J = 9.8 Hz, 1H, Aryl-
H), 5.74 (s, 1H, O
H), 3.74–3.50 (m, 4H, C
H2), 1.27–1.02 (m, 6H, C
H3).
13C NMR (101 MHz, DMSO) δ 169.25, 157.58, 149.61, 134.89, 115.81, 95.52, 46.08, 30.05. LC-MS
m/
z 195.1 ([M − H]
−, 100%). HR-MS calculated for C
10H
14O
2N
2 195.1134, found: 195.1152 (9.2 ppm, 1.8 mDa).
3-Methoxy-6-nitrosophenol (3h) [
30]
: Eluent: hexane-ethyl acetate (4:1) to yield a green solid. Yield: 49%, g. LC-MS
m/
z 154.7 ([M + H]
+, 100%), 152.4 ([M − H]
−, 100).
1H NMR (400 MHz; CDCl
3) 7.66 (1H, d, J 10.0, Aryl-
H), 6.59 (1H, dd, J 10.4, 2.0, Aryl-
H), 6.09 (1H, s, Aryl-
H) 3.94 (3H, s, O-C
H3).
13C NMR (100 MHz; CDCl
3) 171.3 (
C6), 170.1 (
C1), 150.7 (
C3), 135.3 (
C4), 117.93 (
C2), 100.56 (
C5), 56.66 (
CH
3). HR-MS calculated for C
7H
8N
1O
3 154.0504, found: 154.0504. (0.0 ppm, 0.0 mDa).
3-Nitroso-4-methylphenol (3j): Eluent: hexane-ethyl acetate (4:1) to yield an orange solid. Yield: % not applicable as starting material was an isolated mixture, 715 mg. 1H NMR (400 MHz; DMSO) 8.27 (1H, d, J 2.4, Aryl-H), 7.67 (1H, dd, J 8.8 2.4, Aryl-H) 7.10 (1H, d, J 8.8, Aryl-H). 13C NMR (100 MHz; CDCl3) 154.1 (C1), 140.4 (C2), 134.1 (C4), 127.3 (C5), 121.8 (C3), 111.7 (C6). LC-MS m/z 138.1 ([M + H]+, 100%). HR-MS: calculated for C7H8NO2 138.0555, found: 138.0555 (0.0 ppm, 0.0 mDa). Crystal data: C7H7O2N, f.w. 137.14, studied as two polymorphs, viz. α-3j (from hexane/ethylacetate), CCDC 1957003, T = 100 K, triclinic, a = 7.2648(8), b = 7.3201(8), c = 12.8572(14) Å, α = 88.333(2), β = 78.713(2), γ = 84.279(2)°, V = 667.1(1) Å3, space group P (no. 2), Z = 4, 9388 reflections (3893 unique, Rint = 0.050), R1 = 0.056, wR2 = 0.159; β-3j (from chloroform/methanol), CCDC 1957009, T = 120 K, monoclinic, a = 12.8007(13), b = 7.7538(8), c = 13.8767(14) Å, β = 107.805(4)°, V = 1311.4(2) Å3, space group P21/c (no. 14), Z = 8, 16949 reflections (3000 unique, Rint = 0.036), R1 = 0.037, wR2 = 0.102.
3-Nitro-4-methylphenol (3j(i)) [
27]
: Eluent: hexane-ethyl acetate (9:1) to yield a brown solid. Yield: 9%, g.
1H NMR (400 MHz; CDCl
3) 9.45 (1H, s, O
H) 8.08 (1H, d, J 2.8, Aryl-
H), 8.02 (1H, dd, J 8.8, 2.8, Aryl-
H), 6.88 (1H, d, J 8.8,
HC). LC-MS
m/
z 153.1 (M
●+, 100%). HR-MS calculated for C
7H
7NO
3 153.0405, found: 153.0400. (−3.3 ppm, mDa −0.5).
2,4-Dichloro-6-nitrophenol (3k) [
31]
: Recrystallised from ethanol to yield an orange solid. Yield: 49%, g.
1H NMR (400 MHz; CDCl
3) 10.96 (1H, s, O
H), 7.92 (1H, s, Aryl-
H), 7.62 (1H, s, Aryl-
H),
13C NMR (100 MHz, CDCl
3) 150.1, 137.7, 125.7, 124.5, 123.1. LC-MS
m/
z 207.9 ([M + H]
+, 100%).
4-Nitroso-2-methylphenol (2-methyl-1,4-benzoquinone, E and Z isomers (9:4)) (3l) [
32]
: Eluent: hexane-ethyl acetate (9:1) to yield a brown solid. Yield: 48%, 147 mg. LC-MS
m/
z 154.7 ([M + H]
+, 100%), 152.4 ([M − H]
−, 100).
1H NMR (400 MHz; CDCl
3) 7.66 (1H, d, J 10.0, Aryl-
H), 6.59 (1H, dd, J 10.0, 2.0, Aryl-
H), 6.09 (1H, s, Aryl-
H) 3.94 (3H, s, O-C
H3).
13C NMR (100 MHz; CDCl
3) 171.3 (
C6), 170.1 (
C1), 150.7 (
C3), 135.3 (
C4), 117.93 (
C2), 100.56 (
C5), 56.66 (
CH
3). HR-MS calculated for C
7H
8NO
3 154.0504, found: 154.0504. (0.0 ppm, 0.0 mDa).
2-Chloro-4-nitrosophenol in equilibrium with 2-chloro-1,4-benzoquinone monoxime, E and Z isomers (4:3) (3m): Eluent: hexane-ethyl acetate (4:1) to yield an orange solid. Yield: 34%, 289 mg. 1H NMR (400 MHz, Chloroform-d) δ 9.77 (s, 1H, O-H), 8.33 (d, J = 2.7 Hz, 1H, Aryl-H), 8.15 (dd, J = 9.0, 2.7 Hz, 1H), 8.01 (d, J = 2.4 Hz, 1H), 7.83 (dd, J = 10.2, 2.4 Hz, 1H), 7.48 (d, J = 2.5 Hz, 1H), 7.36–7.23 (m, 1H), 7.16 (d, J = 9.0 Hz, 1H), 6.67 (dd, J = 10.1, 3.8 Hz, 1H), 6.49 (s, 1H). 13C NMR (101 MHz, CDCl3) δ 180.46, 180.10, 156.96, 149.95, 149.25, 138.84, 137.69, 134.97, 134.65, 131.43, 129.78, 125.39, 124.63, 124.17, 122.05, 120.37, 116.30, 77.36, 77.04, 76.73. LC-MS: m/z = 158.0 ([M + H]+, 100%). HR-MS calculated for C6H8NO235Cl 158.0009, found: 158.0024 (+9.5 ppm, +1.5 mDa).
1,4-Benzoquinone monoxime (3o) [
33]
: Isolated mixture, green solid. Yield: up to 55%, 135 mg.
1H NMR (400 MHz; CDCl
3) 7.81 (1H, m, Aryl-
H), 7.28 (1H, m, Aryl-
H), 6.55 (2H, d, J 10.0, Aryl-
H).
13C NMR (100 MHz; CDCl
3) 160.5 (
C1), 143.1 (
C2), 136.0 (
C5), 134.0 (
C6), 129.2 (
C4), 120.5 (
C5). LC-MS:
m/
z 124.1 ([M + H]
+, 100%). HR-MS calculated for C
6H
6NO
2 124.0401, found: 124.0399 (+1.6 ppm, +0.2 mDa). IR (neat) υ
max/cm
−1 3316 (br, OH), 1553 (s, N=O), 1442 (s, N=O), 1330 (s, N=O), 1284 (s, N=O), 1127 (s, C-O).
4-Nitrophenol (3o(i)) [
34]
: Brown solid. Yield: 55%, 154 mg.
1H NMR (400 MHz; d
6-DMSO) 8.12 (2H, m, Aryl-
H), 6.92 (2H, m, Aryl-
H).
13C NMR (100 MHz; d
6-DMSO) 164.6 (
C), 139.9 (
C1), 128.3 (
C4) 126.7 (
C3 and
C5), 116.3 (
C2 and
C6). LC-MS
m/
z 138.1 ([M − H]
−, 100%). IR (neat) υ
max/cm
−1 3316 (br, OH), 1558 (s, N=O), 1496 (s, N=O), 1329 (s, N=O), 1284 (s, N=O), 1110 (s, C-O).
Cycloadditions of copper(II) bis(2-nitrosophenolato)complexes. As adapted from McKillop and Sayer [
22], the copper complex (
2a–2h) is purified using a Soxhlett procedure with hot ethanol-water, then the purified, solid material (1.25 mmol) dissolved in dimethoxyethane (40 mL) and water (5 mL). To this solution, dimethylacetylene dicarboxylate (5 mmol) in dimethoxyethane (2.5 mL) is added dropwise and the mixture stirred and heated (90 °C) under reflux for 4 h. After ambient cooling to room temperature, the remaining copper substances are removed by filtration through a bed of silica (2 cm depth) and then the filtrate is evaporated to dryness. The resulting solid is recrystallised into hexane-ethyl acetate (1:1) to give a crystalline solid (
4b, 4c, 4e, 4h).
4b was instead given using an improved procedure that used identical conditions but 0.25 mmol starting complex (
2b) instead of 1.25 mmol and 1 mmol of DMAD instead of 5 mmol.
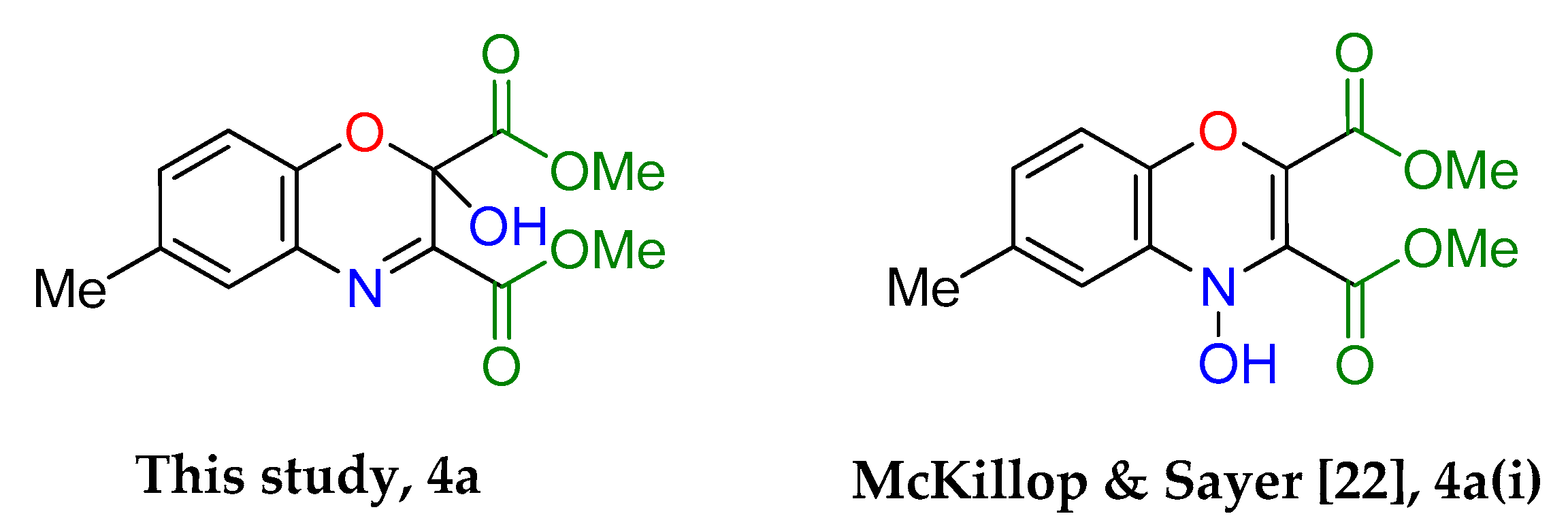
Dimethyl 2-hydroxy-6-methyl-2H-benzo[b][1,4]oxazine-2,3-dicarboxylate (4a): Recrystallised from hexane-ethyl acetate (1:1) to yield an orange solid. Yield: 59%, 154 mg. 1H NMR (400 MHz; d6-DMSO) 8.66 (1H, s, OH) 7.43 (1H, m, Aryl-H), 7.27 (1H, dd, J 8.0, 2.0 Aryl-H), 7.00 (1H, d, J 8.34, Aryl-H), 3.80 (6H, d, J 11.2, O-CH3), 2.32 (3H, s, O-CH3). 13C NMR (100 MHz; d6-DMSO) 167.4 (C=O), 162.3 (C=O), 148.9 (q-C), 142.5 (q-C), 132.6 (Aryl-CH), 130.8 (Aryl-C), 128.8 (Aryl-CH), 127.0 (Aryl-C), 119.0 (Aryl-CH), 89.7 (Aryl-C), 53.7 (O-CH3), 53.5 (O-CH3). LC-MS m/z 278.8 ([M − H]−, 100%), 280.8 ([M + H]+, 78). HR-MS calculated for C13H14NO6 280.0816, found 280.0820 (−0.4 ppm, mDa −0.4). IR (neat) υmax/cm−1 3164 (br, w, O-H), 2963 (w, C-H) 1764 (s, C=O), 1723 (s, C=O), 1439 (m, N-O), 1229 (s, N-O) 1129 (s, N-O), 1056 (s, C-O). Crystal data: CCDC 1957010, C13H13O6N, f.w. 279.24, T = 100 K, monoclinic, a = 5.861(6), b = 14.109(15), c = 7.528(8) Å, β = 90.70(3)°, V = 622.5(11) Å3, space group Pc (no. 7), Z = 2, pseudo-merohedral twinning by a 180° rotation around z axis, 3986 reflections (2028 unique, Rint = 0.111), R1 = 0.147, wR2 = 0.390. M.p = 207.2–207.8 °C.
Dimethyl 6-chloro-2-hydroxy-2H-benzo[b] [1,4]oxazine-2,3-dicarboxylate (4b): Recrystallised from hexane-ethyl acetate (1:1) to yield a yellow solid. Yield 19%, 142 mg. 1H NMR (400 MHz; CDCl3) 7.51 (1H, m, Aryl-H), 7.22 (1H, dd, J 8.0, 2.0, Aryl-H), 6.94 (1H, d, J 8.34, Aryl-H), 5.13 (1H, s, O-H), 3.97 (6H, d, J 11.2, O-CH3). 13C NMR (100 MHz; DMSO) 167.0 (C=O), 162.3 (C=O), 148.9 (q-C), 142.5 (q-C), 132.6 (Aryl-CH), 130.8 (Aryl-C), 128.8 (Aryl-CH), 127.0 (Aryl-C), 119.0 (Aryl-CH), 89.7 (Aryl-C), 53.7 (O-CH3), 53.5 (O-CH3). LC-MS m/z (300.3 [M + H]+, 100%), 302.3 ([M + H]+ (37Cl), 25). HR-MS calculated for C12H11NO635Cl 300.0273 found: 300.0274 (−0.3 ppm, mDa −0.1). IR (neat) υmax/cm−1 3189 (w, br, O-H), 2963 (w, C-H) 1768 (s, C=O), 1725 (s, C=O), 1439 (m, N-O), 1152 (s, N-O), 1000 (s, C-O). Crystal data: CCDC 1957004, C12H10O6NCl, f.w. 299.66, T = 120 K, monoclinic, a = 5.8201(12), b = 7.5030(15), c = 14.348(3) Å, β = 90.784(8)°, V = 626.5(2) Å3, space group P21 (no. 4), Z = 2, pseudo-merohedral twinning by a 180° rotation around x axis, 7346 reflections (2831 unique, Rint = 0.043), R1 = 0.085, wR2 = 0.224. M.p = 217.4–219.0 °C.
Dimethyl 6-bromo-2-hydroxy-2H-benzo[b] [1,4]oxazine-2,3-dicarboxylate (4c): Recrystallised from hexane-ethyl acetate (1:1) to yield a cream solid. Yield 10%, 86 mg. 1H NMR (400 MHz; d6-DMSO) 8.79 (1H, s, O-H), 7.86 (1H, d, J 2.4, Aryl-H), 7.64 (1H, dd, J 8.8, 2.4 Aryl-H), 7.13 (1H, d, J 8.8, Aryl-H), 3.86 (3H, s, J 11.2, O-CH3), 3.77 (3H, s, O-CH3). 13C NMR (100 MHz; d6-DMSO) 167.0 (C=O), 162.3 (C=O), 148.9 (q-C), 142.9 (q-C), 135.4 (Aryl-CH), 131.7 (Aryl-C-H), 131.2 (Aryl-C), 119.5 (Aryl-C-H), 114.5 (Aryl-C), 89.7 (Aryl-C), 53.8 (O-CH3), 53.6 (O-CH3). LC-MS: m/z 346.3 ([M + H]+ (81Br), 52%), 344.6 ([M − H]− (81Br), 100). HR-MS calculated for C12H11NO679Br 343.9770, found 343.9757. (−3.8 ppm, mDa −1.3). IR (neat) υmax/cm−1 3189 (w, br, O-H), 2963 (w, C-H) 1768 (s, C=O), 1725 (s, C=O), 1439 (m, N-O), 1152 (s, N-O), 1000 (s, C-O). M.p, = 220.9–222.7 °C.
Dimethyl 6-formyl-2-hydroxy-8-methoxy-2H-benzo[b] [1,4]oxazine-2,3-dicarboxylate (4d): Recrystallised from hexane-ethyl acetate (7:3) to yield a cream solid. Yield 45%, 190 mg. 1H NMR (400 MHz, DMSO-d6) δ 9.96 (s, 1H, ROC-H), 9.19 (s, 1H, O-H), 7.86 (d, J = 1.8 Hz, 1H, Aryl-H), 7.61 (d, J = 1.8 Hz, 1H, Aryl-H), 3.93 (s, 3H, O-CH3), 3.87 (s, 3H, O-CH3), 3.79 (s, 3H, O-CH3). 13C NMR (101 MHz, DMSO-d6) δ 191.68 (CHO), 166.76 (C=O), 162.31 (C=O), 149.1, 148.5, 138.4, 131.2 (2C), 130.1, 125.2 (Aryl-CH), 112.8 (Aryl-CH), 56.6 (O-CH3), 53.9 (O-CH3), 53.7 (O-CH3). LC-MS: m/z 324.4 ([M + H]+, 100%), 322.2 ([M − H]−, 100). HR-MS calculated for C14H11NO679Br 322.0563, found 322.0569. (+1.9 ppm, mDa +0.6). IR (neat) υmax/cm−1 2964 (w, C-H), 1765 (s, C=O), 1724 (s, C=O), 1601 (w, N-H), 1485 (m), 1284 (s) 1132 (s, C-O), 975 (s), 799 (s) 584 (m). Crystal data: CCDC 1963155, C14H13O8N, f.w. 323.25, T = 100 K, monoclinic, a = 5.8339(14), b = 10.095(3), c = 12.653(3) Å, α = 103.773(5), β = 91.131(5), γ = 96.467(5)°, V = 718.4(3) Å3, space group P (no. 2), Z = 2, 6906 reflections (2896 unique, Rint = 0.057), R1 = 0.064, wR2 = 0.195. M.p, = 212.0–213.8 °C.
Dimethyl 7-methoxy-2-hydroxy-2H-benzo[b][1,4]oxazine-2,3-dicarboxylate (4i): Recrystallised from hexane-ethyl acetate (3:2) to yield a white solid. Yield 30%, 109 mg. 1H NMR (400 MHz, DMSO) δ 8.68 (s, 1H, O-H), 7.53 (s, 1H, Ar-H), 6.76 (Aryl-H), 6.69 (Aryl-H), 3.81 (s, 3H, O-CH3), 3.76 (s, 3H, O-CH3), 3.74(s, 3H, O-CH3). 13C NMR (101 MHz, DMSO-d6) δ 167.45, 163.26, 162.84, 145.06, 144.12, 130.91, 123.95, 110.41, 101.87, 89.72, 56.38, 53.57, 53.15. IR (neat) υmax/cm−1 2970 (w, C-H), 1761 (s, C=O), 1719 (s, C=O), 1596 (w, N-H), 1438 (m), 1273 (s) 1159 (s, C-O), 1110 (s), 822 (s) 457 (m). LC-MS: m/z 296.3 ([M + H]+, 100%), 294.1 ([M − H]−, 100). HR-MS calculated for C13H14NO7 296.0770, found: 296.0790 (+6.8 ppm, +2.0 mDa). M.p, = 217.4–219.1 °C.
Dimethyl 2-hydroxy-7-methyl-2H-benzo[b][1,4]oxazine-2,3-dicarboxylate (4k): Recrystallised from hexane-ethyl acetate (1:1) to yield a yellow solid. Yield 9%, 62.8 mg. 1H NMR (400 MHz; d6-DMSO) 8.66 (1H, s, O-H) 7.43 (1H, m, Aryl-H), 7.27 (1H, dd, J 8.0, 2.0 Aryl-H), 7.00 (1H, d, J 8.34, Aryl-H), 3.80 (6H, d, J 11.2, O-CH3), 2.32 (3H, s, O-CH3). 13C NMR (100 MHz; d6-DMSO) 167.4 (C=O), 162.7 (C=O), 146.4 (q-C), 143.9 (q-C), 143.3 (Aryl-C), 129.4 (Aryl-CH), 127.8 (Aryl-C), 124.4 (Aryl-CH), 117.4 (Aryl-CH), 89.7 (Aryl-C), 53.7 (O-CH3), 53.5 (O-CH3), 21.7 (CH3). LC-MS: m/z 278.8 ([M − H]−, 100%) 280.8 ([M + ]+, 100). HR-MS calculated for C13H14NO6 280.0824, found: 280.0820. (−0.4 ppm, −0.4 mDa). IR (neat) υmax/cm−1 3164 (w, br, O-H), 2963 (w, C-H) 1764 (s, C=O), 1723 (s, C=O), 1439 (m, N-O), 1229 (s, N-O) 1129 (s, N-O), 1056 (s, C-O). M.p. = 211.8–213.4 °C.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}








