
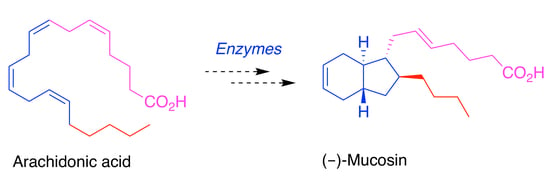
Some Biogenetic Considerations Regarding the Marine Natural Product (−)-Mucosin †



Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
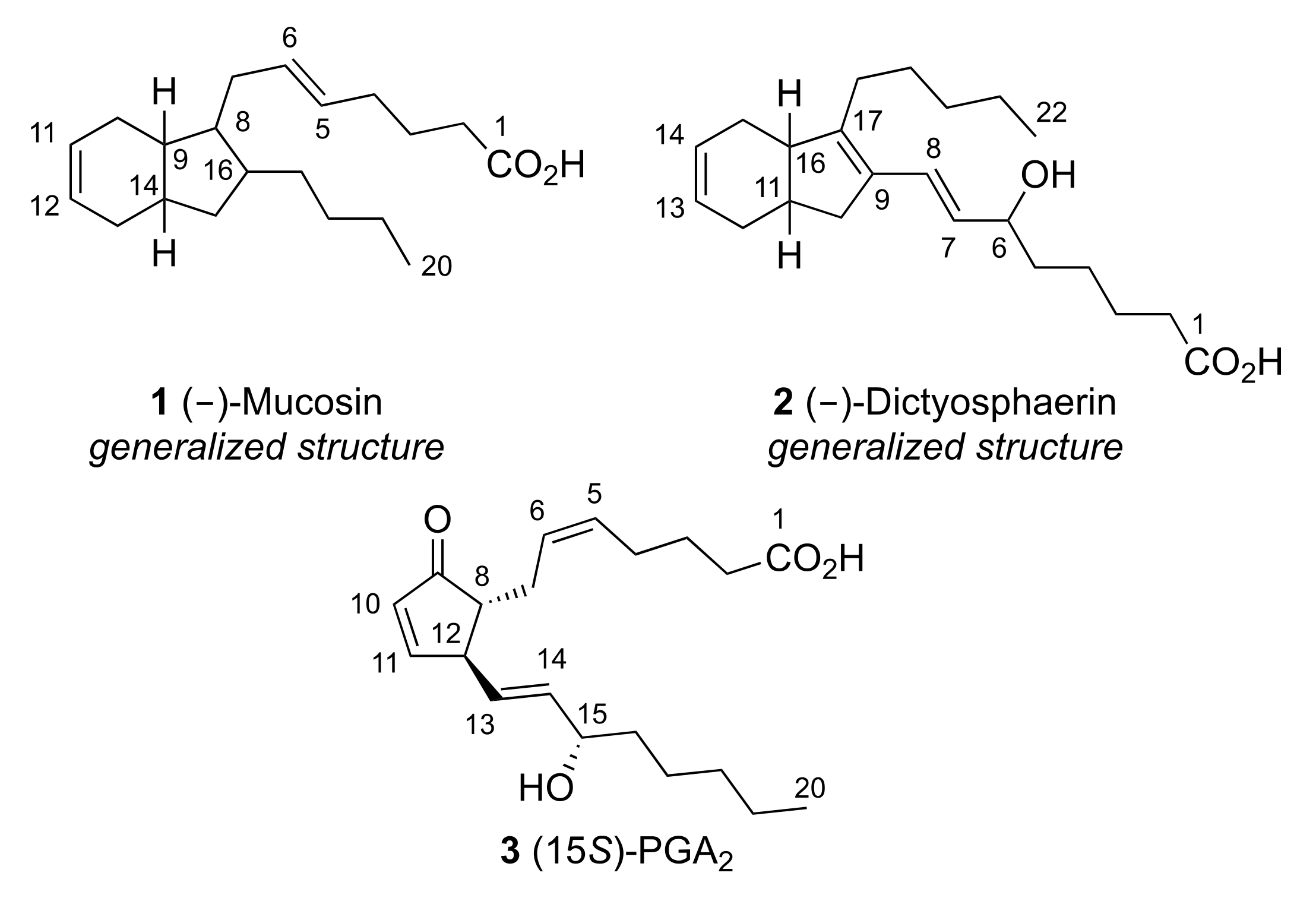
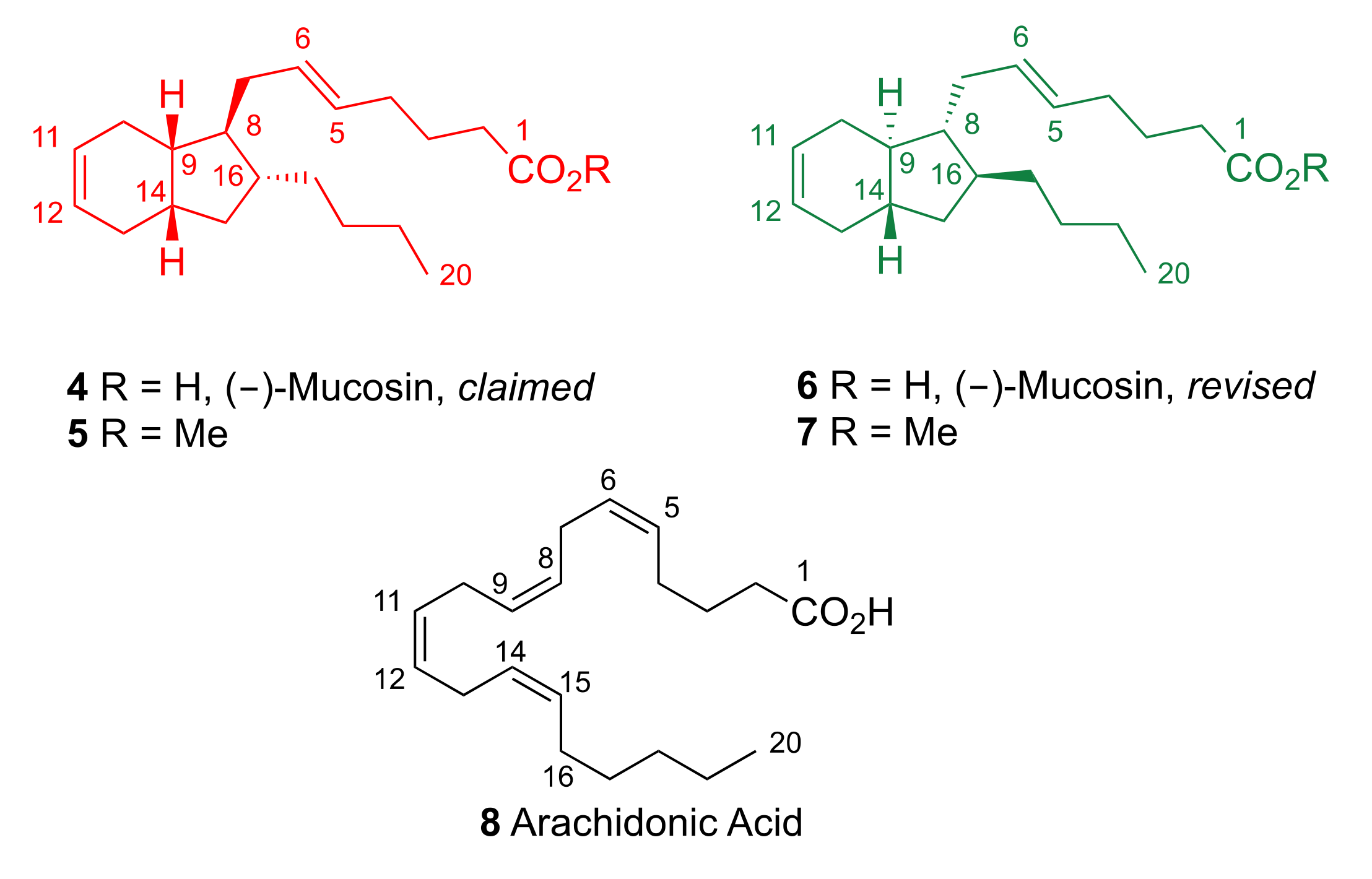
2. Results and Discussion
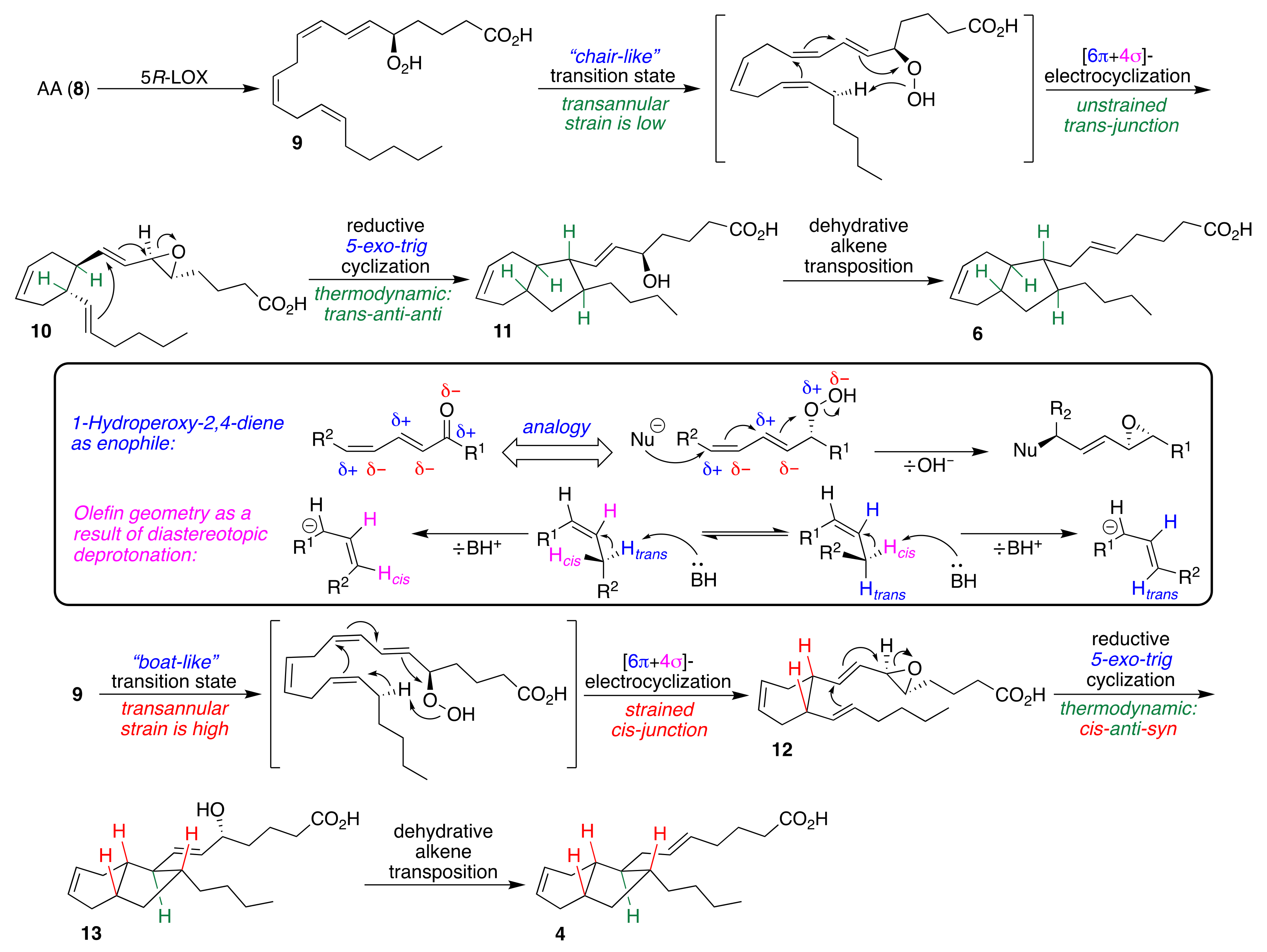
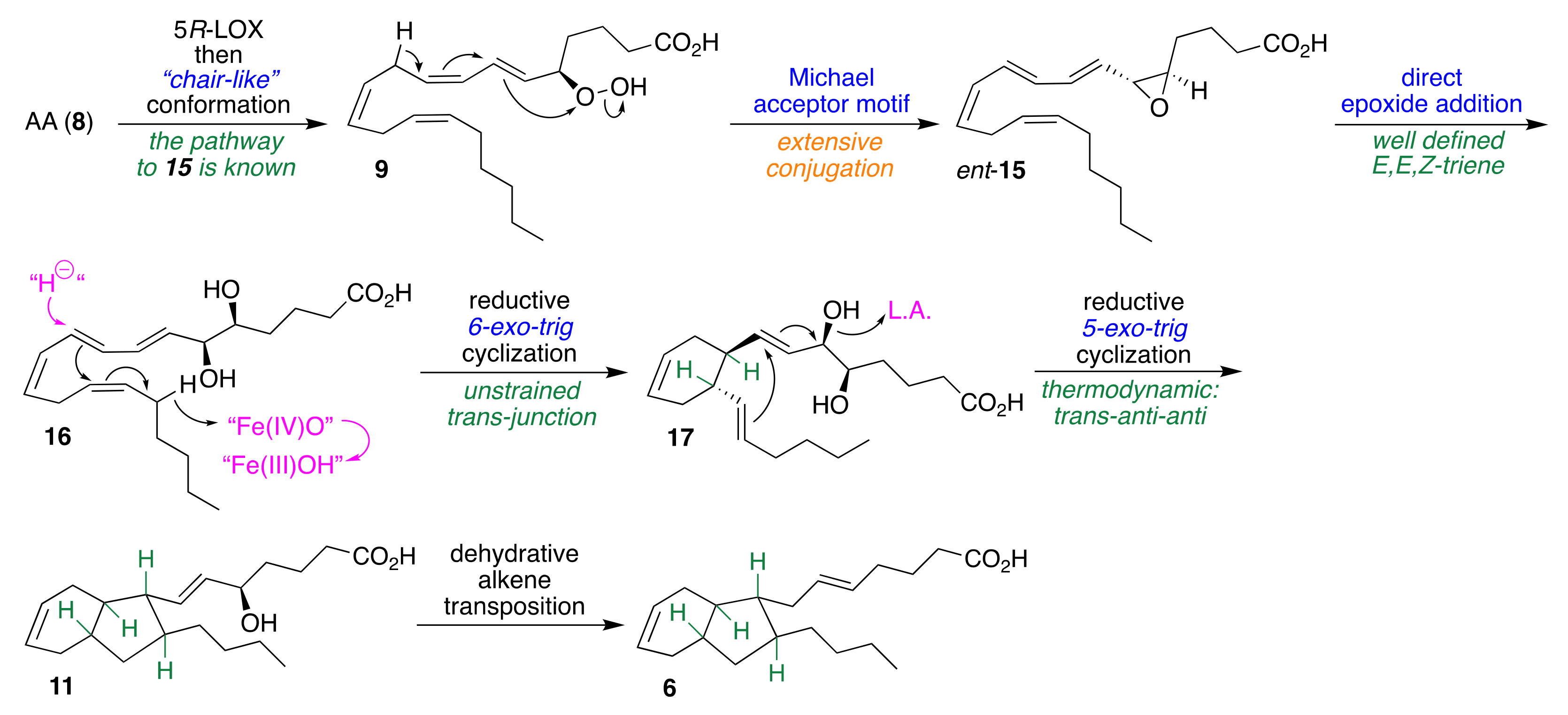
2.1. Pericyclic Pathway Involving AA (8)
2.2. Cationic Pathway Involving AA (8)
2.3. Anionic Pathway Involving AA (8) via ent-LTA4 (ent-15)
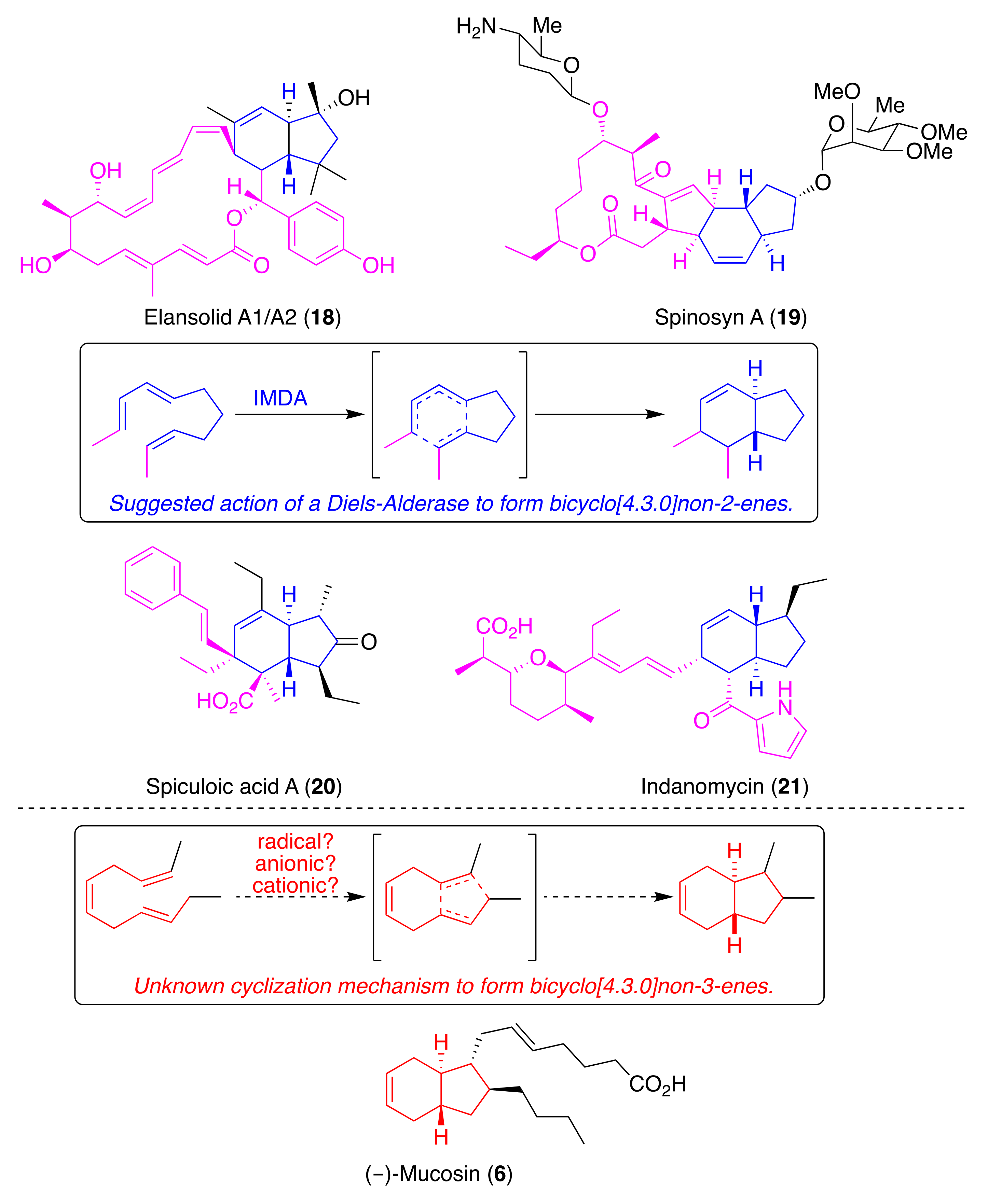
2.4. Polyketide Pathways
3. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Saini, R.K.; Keum, Y.-S. Omega-3 and omega-6 polyunsaturated fatty acids: Dietary sources, metabolism, and significance-A review. Life Sci. 2018, 203, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Wiktorowska-Owczarek, A.; Berezinńska, M.; Nowak, J.Z. PUFAs: Structures, Metabolism and Functions. Adv. Clin. Exp. Med. 2015, 24, 931–941. [Google Scholar] [CrossRef] [PubMed]
- An, J.-U.; Song, Y.-S.; Kim, K.-R.; Ko, Y.-J.; Yoon, D.-Y.; Oh, D.-K. Biotransformation of polyunsaturated fatty acids to bioactive hepoxilins and trioxilins by microbial enzymes. Nat. Commun. 2018, 9, 128. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.V.; Vik, A.; Serhan, C.N. The Protectin Family of Specialized Pro-resolving Mediators: Potent Immunoresolvents Enabling Innovative Approaches to Target Obesity and Diabetes. Front. Pharmacol. 2019, 9, 1582. [Google Scholar] [CrossRef] [PubMed]
- Garbade, G.J.; Gurina, T.S.; Gruhonjic, I.; Gunderson, C.A.; Merkler, D.J. Glycine N-acyltransferase-like 3 is responsible for long-chain N-acylglycine formation in N18TG2 cells. J. Lipid Res. 2016, 57, 781–790. [Google Scholar]
- Gerwick, W.H. Carbocyclic Oxylipins of Marine Origin. Chem. Rev. 1993, 93, 1807–1823. [Google Scholar] [CrossRef]
- Gerwick, W.H.; Singh, I.P. Structural Diversity of Marine Oxylipins. In Lipid Biotechnology; Kuo, T.M., Gardner, H.W., Eds.; Marcel Dekker: New York, NY, USA, 2002; pp. 249–275. [Google Scholar]
- Gerhart, D. Prostaglandin A2: An Agent of Chemical Defense in the Caribbean Gorgonian Plexaura homomalla. Mar. Ecol. Prog. Ser. 1984, 19, 181–187. [Google Scholar] [CrossRef]
- Serhan, C.N. Pro-resolving lipid mediators are leads for resolution physiology. Nature 2014, 510, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N. The resolution of inflammation: The devil in the flask and in the details. FASEB J. 2011, 25, 1441–1448. [Google Scholar] [CrossRef]
- Serhan, C.N. Treating inflammation and infection in the 21st century: New hints from decoding resolution mediators and mechanisms. FASEB J. 2017, 31, 1273–1288. [Google Scholar] [CrossRef]
- Basil, M.C.; Levy, B.D. Specialized pro-resolving mediators: Endogenous regulators of infection and inflammation. Nat. Rev. Immunol. 2016, 16, 51–67. [Google Scholar] [CrossRef]
- Aursnes, M.; Tungen, J.E.; Vik, A.; Colas, R.; Cheng, C.-Y.C.; Dalli, J.; Serhan, C.N.; Hansen, T.V. Total synthesis of the lipid mediator PD1n-3 DPA: Configurational assignments and anti-inflammatory and pro-resolving actions. J. Nat. Prod. 2014, 77, 910–916. [Google Scholar] [CrossRef] [PubMed]
- Tungen, J.E.; Aursnes, M.; Vik, A.; Ramon, S.; Colas, R.A.; Dalli, J.; Serhan, C.N.; Hansen, T.V. Synthesis and anti-inflammatory and pro-resolving activities of 22-OH-PD1, a monohydroxylated metabolite of protectin D1. J. Nat. Prod. 2014, 77, 2241–2247. [Google Scholar] [CrossRef]
- Serhan, C.N.; Chiang, N.; Dalli, J. The resolution code of acute inflammation: Novel pro-resolving lipid mediators in resolution. Semin. Immunol. 2015, 27, 200–215. [Google Scholar] [CrossRef] [PubMed]
- Primdahl, K.G.; Aursnes, M.; Walker, M.E.; Colas, R.A.; Serhan, C.N.; Dalli, J.; Hansen, T.V.; Vik, A. Synthesis of 13(R)-hydroxy-7Z,10Z,13R,14E,16Z,19Z docosapentaenoic acid (13R-HDPA) and its biosynthetic conversion to the 13-Series Resolvins. J. Nat. Prod. 2016, 79, 2693–2702. [Google Scholar] [CrossRef] [PubMed]
- Ramon, S.; Dalli, J.; Sanger, J.M.; Winkler, J.W.; Aursnes, M.; Tungen, J.E.; Hansen, T.V.; Serhan, C.N. The protectin PCTR1 is produced by human M2 macrophages and enhances resolution of infectious inflammation. Am. J. Pathol. 2016, 186, 962–973. [Google Scholar] [CrossRef] [PubMed]
- Goldblatt, M.W. A Depressor substance in seminal fluid. Chem. Ind. 1933, 52, 1056–1057. [Google Scholar]
- Von Euler, U.S. UÜber die spezifische blutdrucksenkende substanz des menschlichen prostata- und samenblasensekretes. Klin. Wochenschr. 1935, 14, 1182–1183. [Google Scholar] [CrossRef]
- Bergström, S.; Sjövall, J. The isolation of prostaglandin. Acta Chem. Scand. 1957, 11, 1086. [Google Scholar] [CrossRef]
- Bergström, S.; Ryhage, R.; Samuelsson, B.; Sjövall, J. Prostaglandins and related factors: 15. The structures of prostaglandin E1, F1α and F1β. J. Biol. Chem. 1963, 238, 3555–3564. [Google Scholar]
- Hamberg, M.; Samuelsson, B. On the mechanism of the biosynthesis of prostaglandins E1 and F1α. J. Biol. Chem. 1967, 242, 5336–5343. [Google Scholar]
- Smith, W.L.; Lands, W.E.M. Stimulation and blockade of prostaglandin biosynthesis. J. Biol. Chem. 1971, 246, 6700–6702. [Google Scholar] [PubMed]
- Dewitt, D.L.; El-Harith, E.A.; Kraemer, S.A.; Andrews, M.J.; Yao, E.F.; Armstrong, R.L.; Smith, W.L. The aspirin and heme-binding sites of ovine and murine prostaglandin endoperoxide synthases. J. Biol. Chem. 1990, 265, 5192–5198. [Google Scholar]
- Flower, R.J. Prostaglandins, bioassay and inflammation. Br. J. Pharmacol. 2006, 147, S182–S192. [Google Scholar] [CrossRef]
- Noverr, M.C.; Erb-Downward, J.R.; Huffnagle, G.B. Production of eicosanoids and other oxylipins by pathogenic eukaryotic microbes. Clin. Microbiol. Rev. 2003, 16, 517–533. [Google Scholar] [CrossRef] [PubMed]
- Ricciotti, E.; FitzGerald, G.A. Prostaglandins and inflammation. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 986–1000. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.; Vito, S.; Inceoglu, B.; Hammock, B.D. The role of long chain fatty acids and their epoxide metabolites in nociceptive signaling. Prostaglandins Other Lipid Mediat. 2014, 113-115, 2–12. [Google Scholar] [CrossRef]
- Rossi, A.; Kapahi, P.; Natoli, G.; Takahashi, T.; Chen, Y.; Karin, M.; Santoro, M.G. Anti-inflammatory cyclopentenone prostaglandins are direct inhibitors of IκB kinase. Nature 2000, 403, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Scher, J.U.; Pillinger, M.H. The Anti-Inflammatory Effects of Prostaglandins. J. Investig. Med. 2009, 57, 703–708. [Google Scholar] [CrossRef] [PubMed]
- Schröder, R.; Xue, L.; Konya, V.; Martini, L.; Kampitsch, N.; Whistler, J.L.; Ulven, T.; Heinemann, A.; Pettipher, R.; Kostenis, E. PGH1, the Precursor for the Anti-Inflammatory Prostaglandins of the 1-series, Is a Potent Activator of the Pro-Inflammatory Receptor CRTH2/DP2. PLoS ONE 2012, 7, e33329. [Google Scholar] [CrossRef]
- Sykes, L.; MacIntyre, D.A.; Teoh, T.G.; Bennett, P.R. Anti-inflammatory prostaglandins for the prevention of preterm labour. Reproduction 2014, 148, R29–R40. [Google Scholar] [CrossRef]
- Hein, A.M.; O’Banion, M.K. Neuroinflammation and Memory: The Role of Prostaglandins. Mol. Neurobiol. 2009, 40, 15–32. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo-Pereira, M.E.; Rockwell, P.; Schmidt-Glenewinkel, T.; Serrano, P. Neuroinflammation and J2 prostaglandins: Linking impairment of the ubiquitin-proteasome pathway and mitochondria to neurodegeneration. Front. Mol. Neurosci. 2015, 7, 104. [Google Scholar] [CrossRef] [PubMed]
- Samuelsson, B. Role of basic science in the development of new medicines: Examples from the eicosanoid field. J. Biol. Chem. 2012, 287, 10070–10080. [Google Scholar] [CrossRef]
- Dias, D.A.; Urban, S.; Roessner, U. A historical overview of natural products in drug discovery. Metabolites 2012, 2, 303–306. [Google Scholar] [CrossRef]
- Gerwick, W.H.; Moore, B.S. Lessons from the past and charting the future of marine natural products drug discovery and chemical biology. Chem. Biol. 2012, 19, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Leal, M.C.; Puga, J.; Serôdio, J.; Gomes, N.C.M.; Calado, R. Trends in the discovery of new marine natural products from invertebrates over the last two decades-Where and what are we bioprospecting? PLoS ONE 2012, 7, e30580. [Google Scholar] [CrossRef]
- Montaser, R.; Luesch, H. Marine natural products: A new wave of drugs? Future Med. Chem. 2011, 3, 1475–1489. [Google Scholar] [CrossRef] [PubMed]
- Blunt, J.W.; Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2018, 35, 8–53. [Google Scholar] [CrossRef]
- Blunt, J.W.; Copp, B.R.; Keyzers, R.A.; Munro, M.H.G.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2016, 33, 382–431. [Google Scholar] [CrossRef]
- Blunt, J.W.; Copp, B.R.; Keyzers, R.A.; Munro, M.H.G.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2015, 32, 116–211. [Google Scholar] [CrossRef]
- Blunt, J.W.; Copp, B.R.; Keyzers, R.A.; Munro, M.H.G.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2014, 31, 160–258. [Google Scholar] [CrossRef] [PubMed]
- Blunt, J.W.; Copp, B.R.; Munro, M.H.G.; Northcote, P.T.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2005, 22, 15–61. [Google Scholar] [CrossRef]
- Faulkner, D.J. Marine natural products. Nat. Prod. Rep. 2001, 18, 1–49. [Google Scholar] [CrossRef] [PubMed]
- Di Costanzo, F.; Di Dato, V.; Ianora, A.; Romano, G. Prostaglandins in Marine Organisms: A Review. Mar. Drugs 2019, 17, 428. [Google Scholar] [CrossRef]
- Casapullo, A.; Scognamiglio, G.; Cimino, G. Mucosin: A new bicyclic eicosanoid from the Mediterranean sponge Reniera mucosa. Tetrahedron Lett. 1997, 38, 3643–3646. [Google Scholar] [CrossRef]
- Rochfort, S.J.; Watson, R.; Capon, R.J. Dictyosphaerin: A Novel Bicyclic Lipid from a Southern Australian Marine Green Algae, Dictyosphaeria sericea. J. Nat. Prod. 1996, 59, 1154–1156. [Google Scholar] [CrossRef]
- Kitsche, A.; Kalesse, M. Configurational Assignment of Secondary Hydroxyl Groups and Methyl Branches in Polyketide Natural Products through Bioinformatic Analysis of the Ketoreductase Domain. ChemBioChem 2013, 14, 851–861. [Google Scholar] [CrossRef]
- Gallantree-Smith, H.C.; Antonsen, S.G.; Görbitz, C.H.; Hansen, T.V.; Nolsøe, J.M.J.; Stenstrøm, Y.H. Total synthesis based on the originally claimed structure of mucosin. Org. Biomol. Chem. 2016, 14, 8433–8437. [Google Scholar] [CrossRef]
- Antonsen, S.G.; Gallantree-Smith, H.C.; Görbitz, C.H.; Hansen, T.V.; Stenstrøm, Y.H.; Nolsøe, J.M.J. Stereopermutation on the Putative Structure of the Marine Natural Product Mucosin. Molecules 2017, 22, 1720. [Google Scholar] [CrossRef]
- Nolsøe, J.M.J.; Antonsen, S.G.; Görbitz, C.H.; Hansen, T.V.; Nesman, J.I.; Røhr, Å.K.; Stenstrøm, Y.H. Total Synthesis of (−)-Mucosin and Revision of Structure. J. Org. Chem. 2018, 83, 15066–15076. [Google Scholar] [CrossRef] [PubMed]
- Samuelsson, B. From studies of biochemical mechanism to novel biological mediators: Prostaglandin endoperoxides, thromboxanes, and leukotrienes. Biosci. Rep. 1983, 3, 791–813. [Google Scholar] [CrossRef] [PubMed]
- Lanea, A.L.; Moore, B.S. A sea of biosynthesis: Marine natural products meet the molecular age. Nat. Prod. Rep. 2011, 28, 411–428. [Google Scholar] [CrossRef]
- Jiang, Z.D.; Ketchum, S.O.; Gerwick, W.H. 5-Lipoxygenase- derived Oxylipins from the Red Alga Rhodymenia pertusa. Phytochemistry 2000, 53, 129–133. [Google Scholar] [CrossRef]
- Newcomer, M.E.; Brash, A.R. The Structural Basis for Specificity in Lipoxygenase Catalysis. Protein Sci. 2015, 24, 298–309. [Google Scholar] [CrossRef] [PubMed]
- Hada, T.; Swift, L.L.; Brash, A.R. Discovery of 5R- Lipoxygenase Activity in Oocytes of the Surf Clam, Spisula solidissima. Biochim. Biophys. Acta 1997, 1346, 109–119. [Google Scholar] [CrossRef]
- Jensen, A.W.; Mohanty, D.K.; Dilling, W.L. The growing relevance of biological ene reactions. Bioorg. Med. Chem. 2019, 27, 686–691. [Google Scholar] [CrossRef]
- Snider, B.B. Lewis-Acid-Catalyzed Ene Reactions. Acc. Chem. Res. 1980, 13, 426–432. [Google Scholar] [CrossRef]
- Mikami, K.; Shimizu, M. Asymmetric Ene Reactions in Organic Synthesis. Chem. Rev. 1992, 92, 1021–1050. [Google Scholar] [CrossRef]
- Domingo, L.R.; Aurella, M.J.; Peńrez, P. Understanding the polar mechanism of the ene reaction. A DFT study. Org. Biomol. Chem. 2014, 12, 7581–7590. [Google Scholar] [CrossRef] [PubMed]
- Seeman, J.I. Effect of Conformational Change on Reactivity in Organic Chemistry. Evaluations, Applications, and Extensions of Curtin-Hammett/Winstein-Holness Kinetics. Chem. Rev. 1983, 83, 83–134. [Google Scholar] [CrossRef]
- Seeman, J.I. The Curtin-Hammett principle and the Winstein-Holness equation: New definition and recent extensions to classical concepts. J. Chem. Educ. 1986, 63, 42–48. [Google Scholar] [CrossRef]
- Gerwick, W.H. Epoxy Allylic Carbocations as Conceptual Intermediates in the Biogenesis of Diverse Marine Oxylipins. Lipids 1996, 31, 1215–1231. [Google Scholar] [CrossRef]
- Borgeat, P.; Samuelsson, B. Arachidonic acid metabolism in polymorphonuclear leukocytes: Unstable intermediate in formation of dihydroxy acids. Proc. Natl. Acad. Sci. USA 1979, 76, 3213–3217. [Google Scholar] [CrossRef]
- Rådmark, O.; Malmsten, C.; Samuelsson, B.; Goto, G.; Marfat, A.; Corey, E.J. Leukotriene A: Isolation from Human Polymorphonuclear Leukocytes. J. Biol. Chem. 1980, 255, 11828–11831. [Google Scholar]
- Zimmer, J.S.D.; Dyckes, D.F.; Bernlohr, D.A.; Murphy, R.C. Fatty acid binding proteins stabilize leukotriene A4: Competition with arachidonic acid but not other lipoxygenase products. J. Lipid Res. 2004, 45, 2138–2144. [Google Scholar] [CrossRef]
- Liang, A.M.; Claret, E.; Ouled-Diaf, J.; Jean, A.; Vogel, D.; Light, D.R.; Jones, S.W.; Guilford, W.J.; Parkinson, J.F.; Snider, R.M. Development of a Homogeneous Time-Resolved Fluorescence Leukotriene B4 Assay for Determining the Activity of Leukotriene A4 Hydrolase. J. Biomol. Screen. 2007, 12, 536–545. [Google Scholar] [CrossRef][Green Version]
- Stsiapanavaa, A.; Samuelsson, B.; Haeggström, J.Z. Capturing LTA4 hydrolase in action: Insights to the chemistry and dynamics of chemotactic LTB4 synthesis. Proc. Natl. Acad. Sci. USA 2017, 114, 9689–9694. [Google Scholar] [CrossRef]
- Fitzpatrick, F.A.; Morton, D.R.; Wynalda, M.A. Albumin Stabilizes Leukotriene A4. J. Biol. Chem. 1982, 257, 4680–4683. [Google Scholar]
- Haeggström, J.; Funk, C.D. Lipoxygenase and Leukotriene Pathways: Biochemistry, Biology, and Roles in Disease. Chem. Rev. 2011, 111, 5866–5898. [Google Scholar] [CrossRef]
- Haeggström, J.Z. Leukotriene biosynthetic enzymes as therapeutic targets. J. Clin. Invest. 2018, 128, 2680–2690. [Google Scholar] [CrossRef]
- Barbosa, M.; Valentaão, P.; Andrade, P.B. Biologically Active Oxylipins from Enzymatic and Nonenzymatic Routes in Macroalgae. Mar. Drugs 2016, 14, 23. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Ramphal, J.Y.; Palazon, J.M.; Spanevello, R.A. Stereocontrolled Total Synthesis of (5S,6R)-, (5S,6S)-, (5R,6R)-, and (5R,6S)(7E,9E,11Z,14Z)-5,6-Dihydroxy-7,9,11,14-icosatetraenoic Acid (5,6-DiHETE) Methyl Esters. Angew. Chem. Int. Ed. 1989, 28, 587–588. [Google Scholar] [CrossRef]
- Knaus, T.; Toogood, H.S.; Scrutton, N.S. Ene-reductases and their Applications. In Green Biocatalysis; Patel, R.N., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2016; pp. 473–488. [Google Scholar]
- Toogood, H.S.; Scrutton, N.S. Discovery, Characterization, Engineering, and Applications of Ene-Reductases for Industrial Biocatalysis. ACS Catal. 2018, 8, 3532–3549. [Google Scholar] [CrossRef]
- Heckenbichler, K.; Schweiger, A.; Brandner, L.A.; Binter, A.; Toplak, M.; Macheroux, P.; Gruber, K.; Breinbauer, R. Asymmetric Reductive Carbocyclization Using Engineered Ene Reductases. Angew. Chem. Int. Ed. 2018, 57, 7240–7244. [Google Scholar] [CrossRef]
- Poulos, T.L. Heme Enzyme Structure and Function. Chem. Rev. 2014, 114, 3919–3962. [Google Scholar] [CrossRef]
- Smith, S.; Tsai, S.-C. The type I fatty acid and polyketide synthases: A tale of two megasynthases. Nat. Prod. Rep. 2007, 24, 1041–1072. [Google Scholar] [CrossRef]
- Kirst, H.A. The spinosyn family of insecticides: Realizing the potential of natural products research. J. Antibiot. 2010, 63, 101–111. [Google Scholar] [CrossRef]
- Steinmetz, H.; Gerth, K.; Jansen, R.; Schläger, N.; Dehn, R.; Reinecke, S.; Kirschning, A.; Mülle, R. Elansolid A, a Unique Macrolide Antibiotic from Chitinophaga sancti Isolated as Two Stable Atropisomers. Angew. Chem. Int. Ed. 2011, 50, 532–536. [Google Scholar] [CrossRef]
- Huang, X.-H.; van Soest, R.; Roberge, M.; Andersen, R.J. Spiculoic Acids A and B, New Polyketides Isolated from the Caribbean Marine Sponge Plakortis angulospiculatus. Org. Lett. 2004, 6, 75–78. [Google Scholar] [CrossRef]
- Roege, K.E.; Kelly, W.L. Biosynthetic Origins of the Ionophore Antibiotic Indanomycin. Org. Lett. 2009, 11, 297–300. [Google Scholar] [CrossRef]
- Klas, K.; Tsukamoto, S.; Sherman, D.H.; Williams, R.M. Natural Diels−Alderases: Elusive and Irresistable. J. Org. Chem. 2015, 80, 11672–11685. [Google Scholar] [CrossRef]
- Heravi, M.M.; Vavsari, V.F. Recent Applications of Intramolecular Diels-Alder Reaction in Total Synthesis of Natural Products. RSC Adv. 2015, 5, 50890–50912. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nolsøe, J.M.J.; Aursnes, M.; Stenstrøm, Y.H.; Hansen, T.V. Some Biogenetic Considerations Regarding the Marine Natural Product (−)-Mucosin. Molecules 2019, 24, 4147. https://doi.org/10.3390/molecules24224147
Nolsøe JMJ, Aursnes M, Stenstrøm YH, Hansen TV. Some Biogenetic Considerations Regarding the Marine Natural Product (−)-Mucosin. Molecules. 2019; 24(22):4147. https://doi.org/10.3390/molecules24224147
Chicago/Turabian StyleNolsøe, Jens M. J., Marius Aursnes, Yngve H. Stenstrøm, and Trond V. Hansen. 2019. "Some Biogenetic Considerations Regarding the Marine Natural Product (−)-Mucosin" Molecules 24, no. 22: 4147. https://doi.org/10.3390/molecules24224147
APA StyleNolsøe, J. M. J., Aursnes, M., Stenstrøm, Y. H., & Hansen, T. V. (2019). Some Biogenetic Considerations Regarding the Marine Natural Product (−)-Mucosin. Molecules, 24(22), 4147. https://doi.org/10.3390/molecules24224147