New Hydroxydecanoic Acid Derivatives Produced by an Endophytic Yeast Aureobasidium pullulans AJF1 from Flowers of Aconitum carmichaeli



Abstract
:1. Introduction
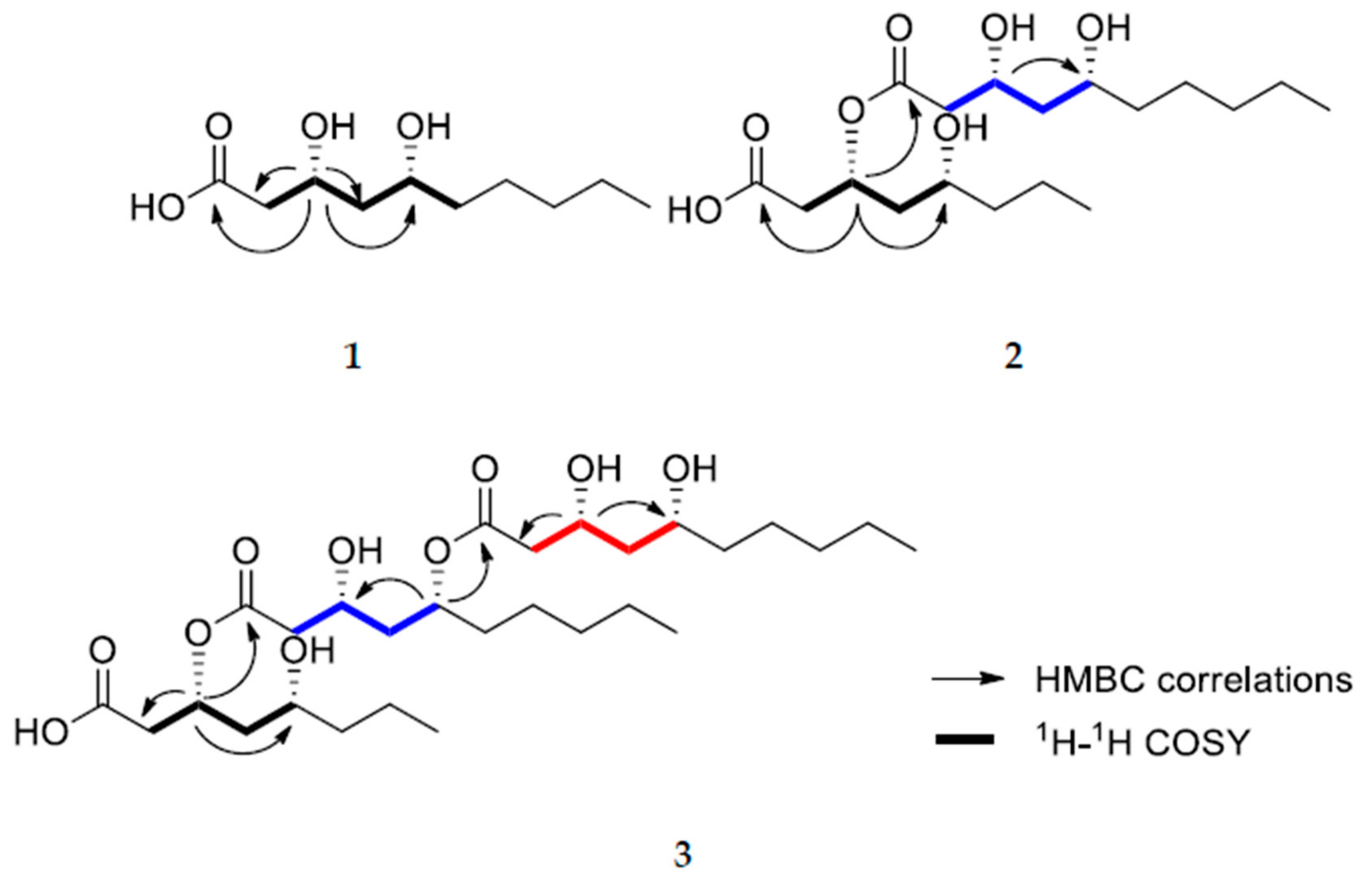
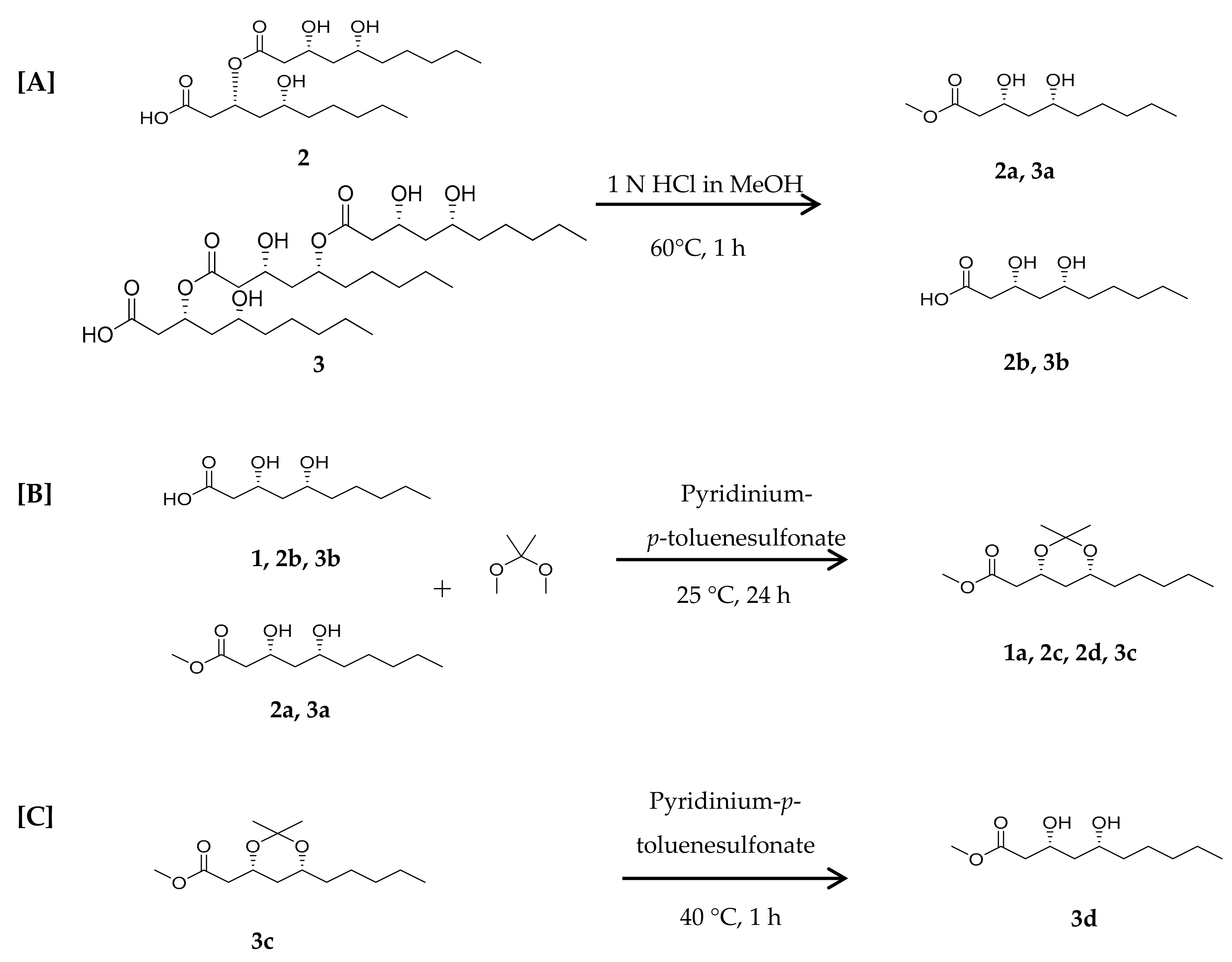
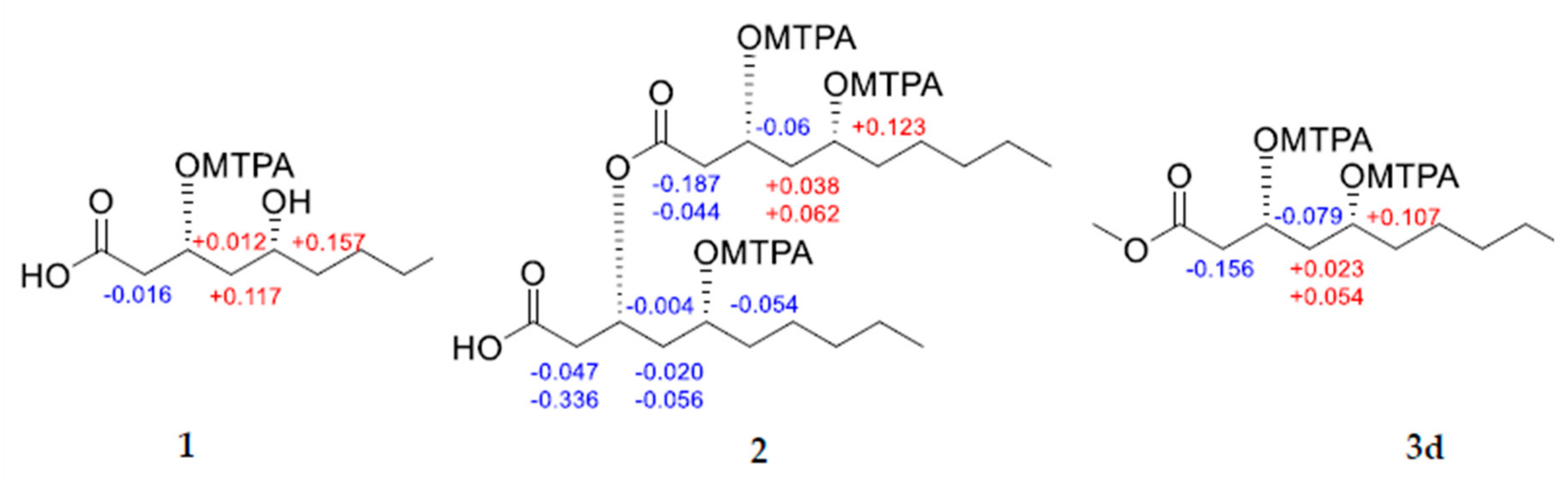
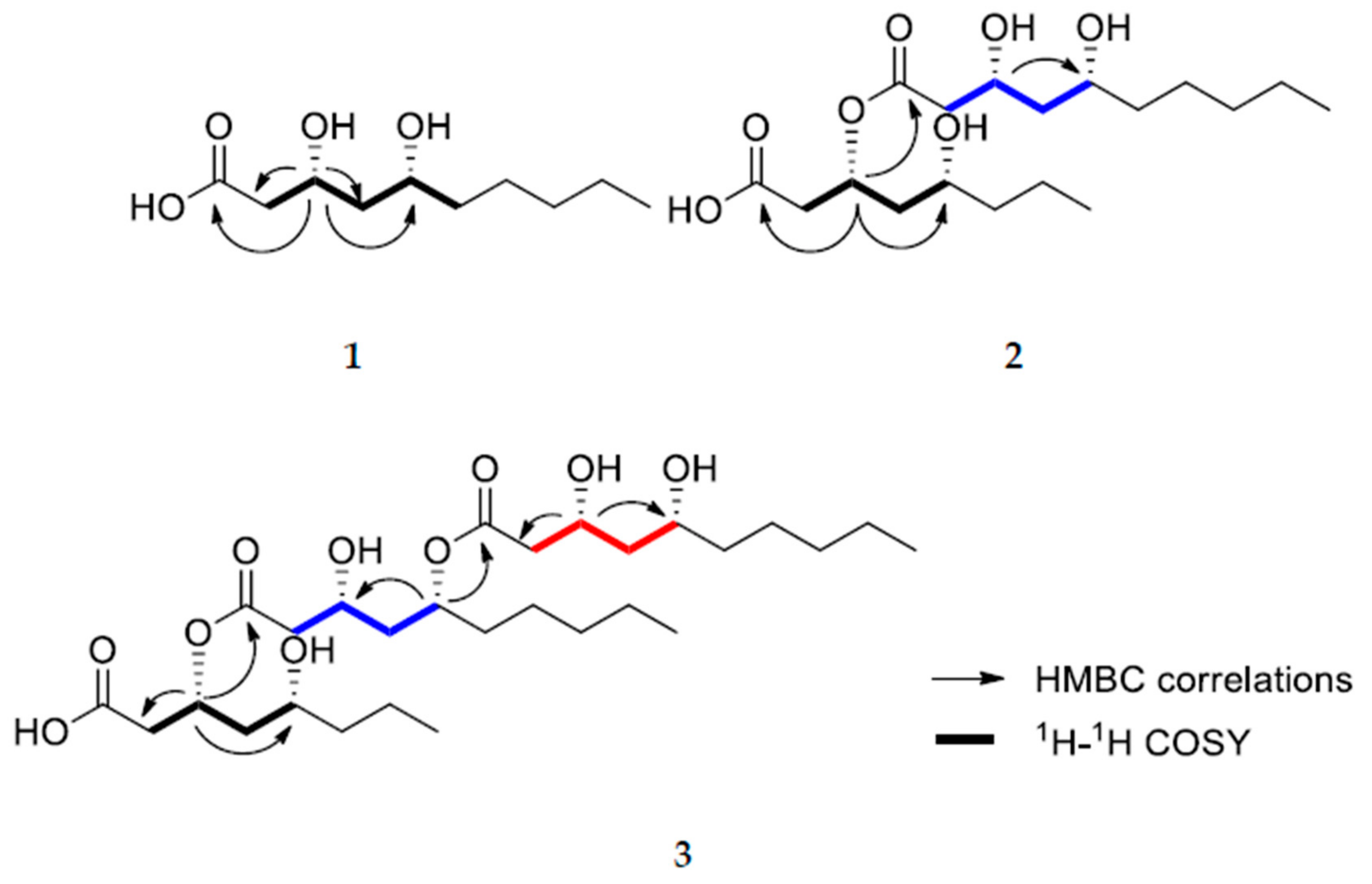
2. Results and Discussion
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Fungal Materials
3.3. Extraction and Isolations
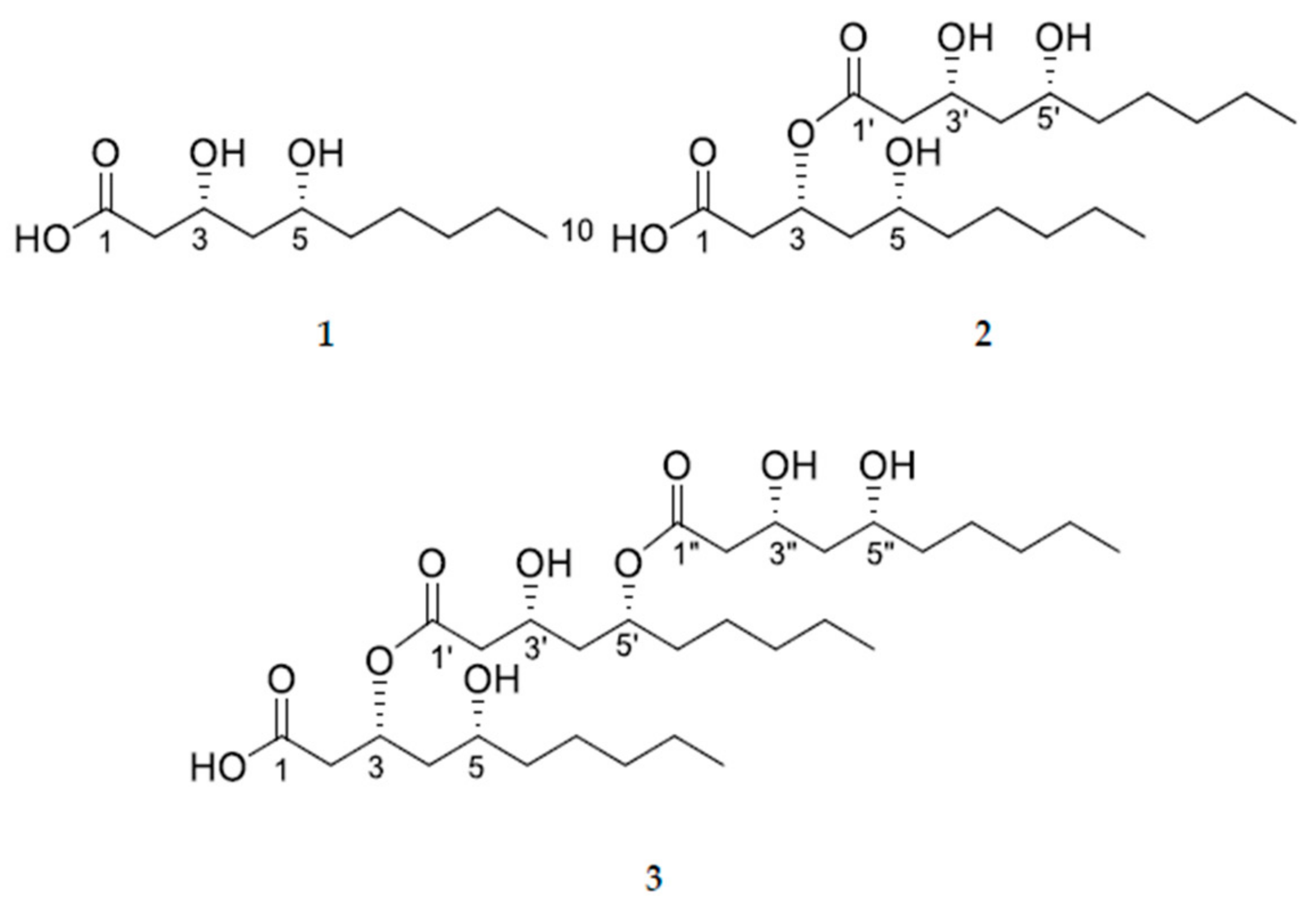
3.3.1. (3R,5R)-3,5-dihydroxydecanoic acid (1)
3.3.2. (3R,5R)-3-(((3R,5R)-3,5-dihydroxydecanoyl)oxy)-5-hydroxydecanoic acid (2)
3.3.3. (3R,5R)-3-(((3R,5R)-5-(((3R,5R)-3,5-dihydroxydecanoyl)oxy)-3-hydroxydecanoyl)oxy)-5-hydroxydecanoic acid (3)
3.4. Chemical Synthesis for Structure Determination
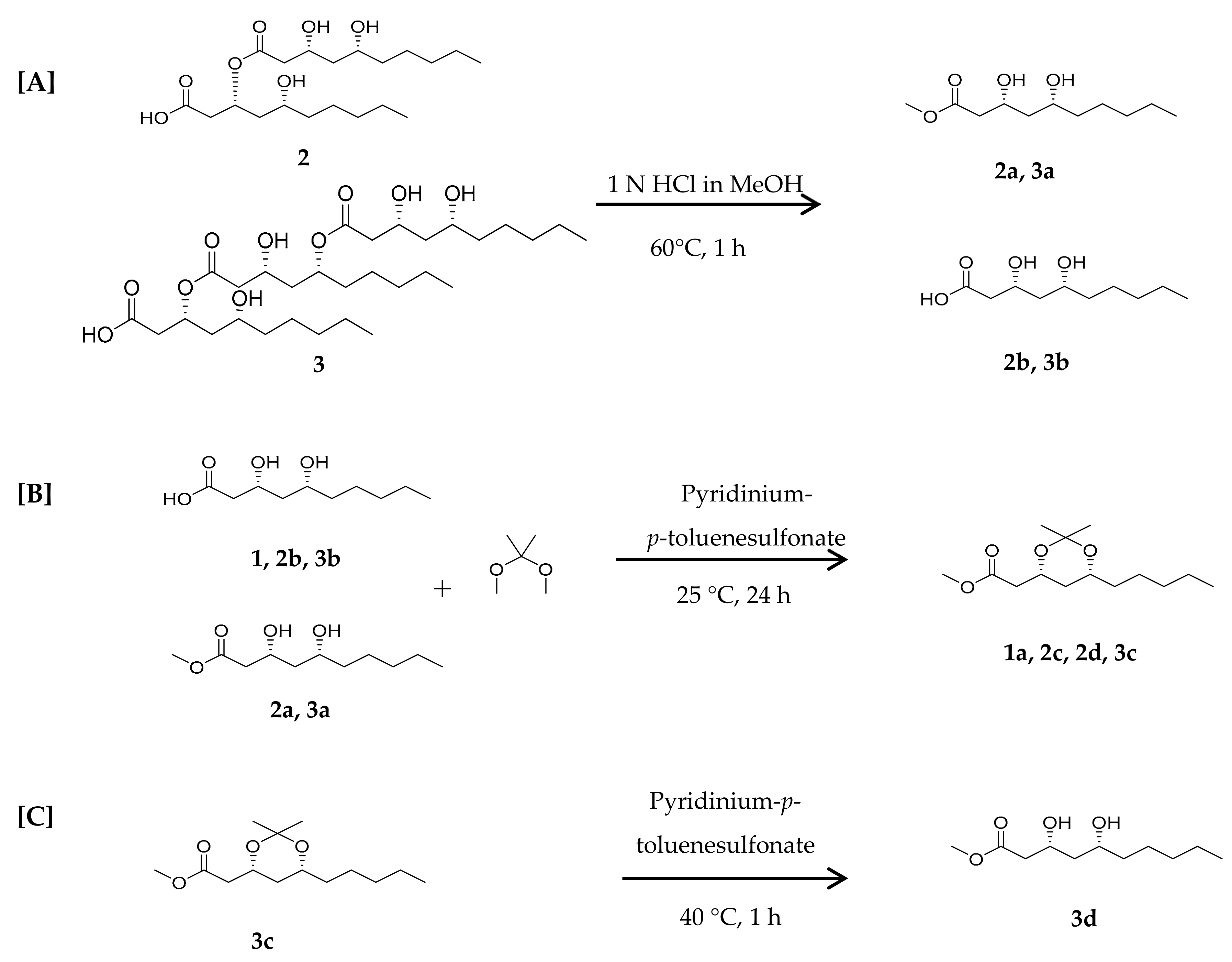
3.4.1. Acid Hydrolysis of Compounds 2 and 3
3.4.2. Synthesis of 1,3-diol-acetonides for Compounds 1, 2, and 3
3.4.3. Deprotection of 1,3-diol-acetonide for Compound 3c
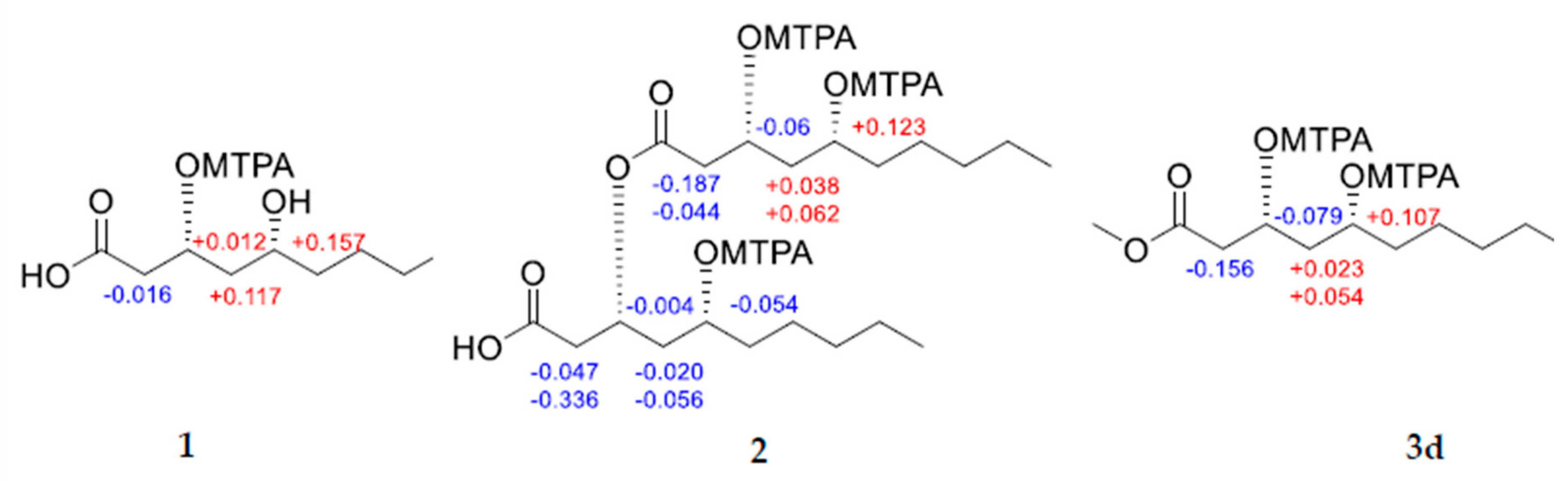
3.4.4. Mosher’s Esterification of 1, 2, and 3d
3.5. Biological Activity Evaluation
3.5.1. Antibacterial Assay
3.5.2. Cytotoxicity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Hirsch, G.; Braun, U. Communities of Parasitic Microfungi: Handbook of Vegetation Science; Kluwer Academic Publisher: Dordrecht, The Netherlands, 1992; pp. 225–250. [Google Scholar]
- Rosenblueth, M.; Martínez-Romero, E. Bacterial Endophytes and Their Interactions with Hosts. Mol. Plant Microbe Interact. 2006, 19, 827–837. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, J.L.; Maccheroni, W.; Pereira, J.O.; Araujo, W.L. Endophytic microorganisms: A review on insect control and recent advances on tropical plants. Electron. J. Biotechnol. 2000, 31, 15–16. [Google Scholar] [CrossRef]
- Muramatsu, D.; Iwai, A.; Aoki, S.; Uchiyama, H.; Kawata, K.; Nakayama, Y.; Nikawa, Y.; Kusano, K.; Okabe, M.; Miyazaki, T. β-Glucan Derived from Aureobasidium pullulans Is Effective for the Prevention of Influenza in Mice. PLoS ONE 2012, 7, e41399. [Google Scholar] [CrossRef] [PubMed]
- Price, N.P.J.; Manitchotpisit, P.; Vermillion, K.E.; Bowman, M.J.; Leathers, T.D. Structural characterization of novel extracellular liamocins (mannitol oils) produced by Aureobasidium pullulans strain NRRL 50380. Carbohydr. Res. 2013, 370, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Qi, B.; Zhao, J.; Qiao, C.; Su, Y.; Wan, Y.J. Control strategy of pH, dissolved oxygen concentration and stirring speed for enhancing β-poly (malic acid) production by Aureobasidium pullulans ipe-1. Chem. Technol. Biotechnol. 2013, 88, 808–817. [Google Scholar] [CrossRef]
- Zain, M.E.; Awaad, A.S.; Razak, A.A.; Maitland, D.J.; Khamis, N.E.; Sakhawy, M.A. Secondary Metabolites of Aureobasidium Pullulans Isolated from Egyptian Soil and Their Biological Activity. J. Appl. Sci. Res. 2009, 5, 1582–1591. [Google Scholar]
- Zhou, K.; Tang, L.; Zhou, X.; Wang, T.; Kou, Z.; Wang, Z. A review on phytochemistry and pharmacological activities of the processed lateral root of Aconitum carmichaelii Debeaux. J. Ethnopharmacol. 2015, 160, 173–193. [Google Scholar] [CrossRef] [PubMed]
- Strobel, G.; Daisy, B.; Castillo, U.; Harper, J. Natural Products from Endophytic Microorganisms. J. Nat. Prod. 2004, 67, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Stierle, A.; Strobel, G.; Stierle, D. Taxol and taxane production by Taxomyces andreanae, an endophytic fungus of Pacific yew. Science 1993, 260, 214–216. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Huang, R.; Miao, C.; Chen, Y. Two New Steroids from an Endophytic Fungus Phomopsis sp. Chem. Biodivers. 2013, 10, 1276–1283. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.; Shen, X.; Huang, R.; Wang, T.; Xie, X.; Liu, S.; Wu, S.; He, J. Bioactive Metabolites Produced by the Endophytic Fungus Phomopsis sp. YM355364. Nat. Prod. Commun. 2014, 9, 669–670. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Huang, R.; Li, F.; Wei, H.; Fang, X.; Xie, X.; Lin, D.; Wu, S.; He, J. Antiviral anthraquinones and azaphilones produced by an endophytic fungus Nigrospora sp. from Aconitum carmichaeli. Fitoterapia 2016, 112, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Kurosawa, T.; Sakai, K.; Nakahara, T.; Oshima, Y.; Tabuchi, T. Extracellular Accumulation of the Polyol Lipids, 3,5-Dihydroxydecanoyl and 5-Hydroxy-2-decenoyl Esters of Arabitol and Mannitol, by Aureobasidium sp. Biosci. Biotech. Biochem. 1994, 58, 2057–2060. [Google Scholar] [CrossRef]
- Rychnovsky, S.D.; Rogers, B.N.; Richardson, T.I. Configurational Assignment of Polyene Macrolide Antibiotics Using the [13C]Acetonide Analysis. Acc. Chem. Res. 1998, 31, 9–17. [Google Scholar] [CrossRef]
- Su, B.N.; Park, E.J.; Mbwambo, Z.H.; Santarsiero, B.D.; Mesecar, A.D.; Fong, H.H.S.; Pezzuto, J.M.; Kinghorn, A.D. New Chemical Constituents of Euphorbia quinquecostata and Absolute Configuration Assignment by a Convenient Mosher Ester Procedure Carried Out in NMR. J. Nat. Prod. 2002, 65, 1278–1282. [Google Scholar] [CrossRef] [PubMed]
- Salunke, G.B.; Shivakumar, I.; Gurjar, M.K. Total synthesis of verbalactone: An efficient, carbohydrate-based approach. Tetrahedron Lett. 2009, 50, 2048–2049. [Google Scholar] [CrossRef]
- Doshida, J.; Hasegawa, H.; Onuki, H.; Shimidzu, N. Exophilin A, a new antibiotic from a marine microorganism Exophiala pisciphila. J. Antibiot. 1996, 49, 1105–1109. [Google Scholar] [CrossRef] [PubMed]
- Wiegand, I.; Hilpert, K.; Hancock, R.E. Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat. Protoc. 2008, 3, 163–175. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | 2 | 3 | ||
|---|---|---|---|---|
| δH (m, J in Hz) b) | δC c) | δH (m, J in Hz) b) | δC c) | |
| 1 | 169.2 | 169.0 | ||
| 2a2b | 2.77 (dd, J = 18.1, 5.4 Hz) 2.69 (ddd, J = 18.1, 3.5, 1.6 Hz) | 35.3 | 2.76 (dd, 18.1, 5.3), 2.68 (ddd, 18.1, 3.5, 1.6) | 35.3 |
| 3 | 5.27 (dt, J = 7.0, 3.5 Hz) | 65.7 | 5.26 (dq, 7.2, 3.5) | 66.1 |
| 4a4b | 2.07 (dtd, J = 14.9, 3.5, 1.6 Hz) 1.77 (ddd, J = 14.9, 11.5, 3.5 Hz) | 32.7 | 2.07 (dtd, 14.9, 3.5, 1.6) 1.76 (ddd, 14.9, 12.3, 4.0) | 32.8 |
| 5 | 4.53 (dtd, J = 11.5, 5.0, 2.9 Hz) | 76.1 | 4.53 (dtd, 11.1, 4.9, 3.1) | 76.2 |
| 6a6b | 1.69 (dddd, J = 13.5, 10.2, 7.5, 5.0 Hz) 1.57 (m) | 35.2 | 0.55 (m) | 34.7 |
| 7a7b | 1.46 (m) 1.37 (m) | 24.3 | 1.47 (m) 1.36 (m) | 24.4 |
| 8 | 1.27 (m) | 31.7 | 1.25 (m) | 31.8 |
| 9 | 1.35 (m) | 22.5 | 1.35 (m) | 22.5 |
| 10 | 0.86 (t, J = 6.9 Hz) | 13.9 | 0.86 (t, 6.9) | 14.0 |
| 1′ | 171.1 | 171.5 | ||
| 2′a2′b | 2.48 (m) 2.48 (m) | 42.2 | 2.52 (dd, 16.1, 3.6) 2.45 (m) | 41.7 |
| 3′ | 4.25 (ddt, 9.4, 7.5, 5.1) | 68.9 | 4.10 (tt, 8.2, 4.0) | 66.3 |
| 4′a4′b | 1.56 (m) 1.54 (m) | 42.0 | 1.83 (m) 1.64 (m) | 41.0 |
| 5′ | 3.85 (ddd, 12.1, 7.5, 4.5) | 72.3 | 5.04 (tt, 8.9, 4.8) | 72.9 |
| 6′a6′b | 1.45 (m) 1.41 (m) | 37.8 | 1.68 (m) 1.57 (m) | 35.4 |
| 7′a7′b | 1.46 (m) 1.37 (m) | 24.9 | 1.47 (m) 1.36 (m) | 24.8 |
| 8′ | 1.25 (m) | 31.4 | 1.25 (m) | 31.5 |
| 9′ | 1.27 (m) | 22.4 | 1.35 (m) | 22.5 |
| 10′ | 0.86 (t, 6.9) | 13.9 | 0.86 (t, 6.9) | 14.0 |
| 1′′ | 172.3 | |||
| 2′′a2′′b | 2.45 (m) 2.40 (m) | 42.7 | ||
| 3′′ | 4.25 (tt, 8.5, 4.0) | 69.8 | ||
| 4′′a4′′b | 1.55 (m) | 42.4 | ||
| 5′′ | 3.84 (tt, 8.1, 4.1) | 72.4 | ||
| 6′′a6′′b | 1.44 (m) 1.55 (m) | 37.9 | ||
| 7′′a7′′b | 1.37 (m) 1.26 (m) | 25.0 | ||
| 8′′ | 1.25 (m) | 31.5 | ||
| 9′′ | 1.27 (m) | 22.5 | ||
| 10′′ | 0.86 (t, 6.9) | 14.0 | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, H.G.; Kim, J.W.; Choi, H.; Kang, K.S.; Shim, S.H. New Hydroxydecanoic Acid Derivatives Produced by an Endophytic Yeast Aureobasidium pullulans AJF1 from Flowers of Aconitum carmichaeli. Molecules 2019, 24, 4051. https://doi.org/10.3390/molecules24224051
Choi HG, Kim JW, Choi H, Kang KS, Shim SH. New Hydroxydecanoic Acid Derivatives Produced by an Endophytic Yeast Aureobasidium pullulans AJF1 from Flowers of Aconitum carmichaeli. Molecules. 2019; 24(22):4051. https://doi.org/10.3390/molecules24224051
Chicago/Turabian StyleChoi, Hyun Gyu, Jung Wha Kim, Hyukjae Choi, Ki Sung Kang, and Sang Hee Shim. 2019. "New Hydroxydecanoic Acid Derivatives Produced by an Endophytic Yeast Aureobasidium pullulans AJF1 from Flowers of Aconitum carmichaeli" Molecules 24, no. 22: 4051. https://doi.org/10.3390/molecules24224051
APA StyleChoi, H. G., Kim, J. W., Choi, H., Kang, K. S., & Shim, S. H. (2019). New Hydroxydecanoic Acid Derivatives Produced by an Endophytic Yeast Aureobasidium pullulans AJF1 from Flowers of Aconitum carmichaeli. Molecules, 24(22), 4051. https://doi.org/10.3390/molecules24224051