
Isolation, Structure Elucidation, and Antiproliferative Activity of Butanolides and Lignan Glycosides from the Fruit of Hernandia nymphaeifolia

 , , , and
, , , and
Abstract
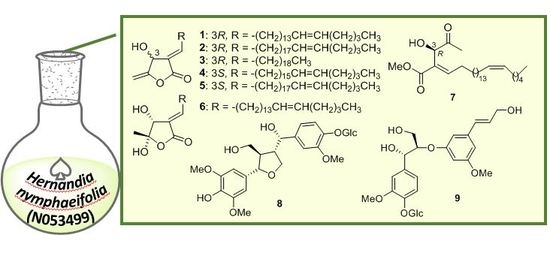
1. Introduction
2. Results and Discussion
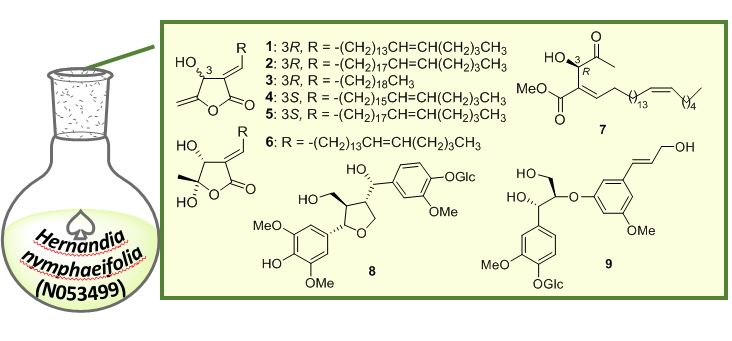
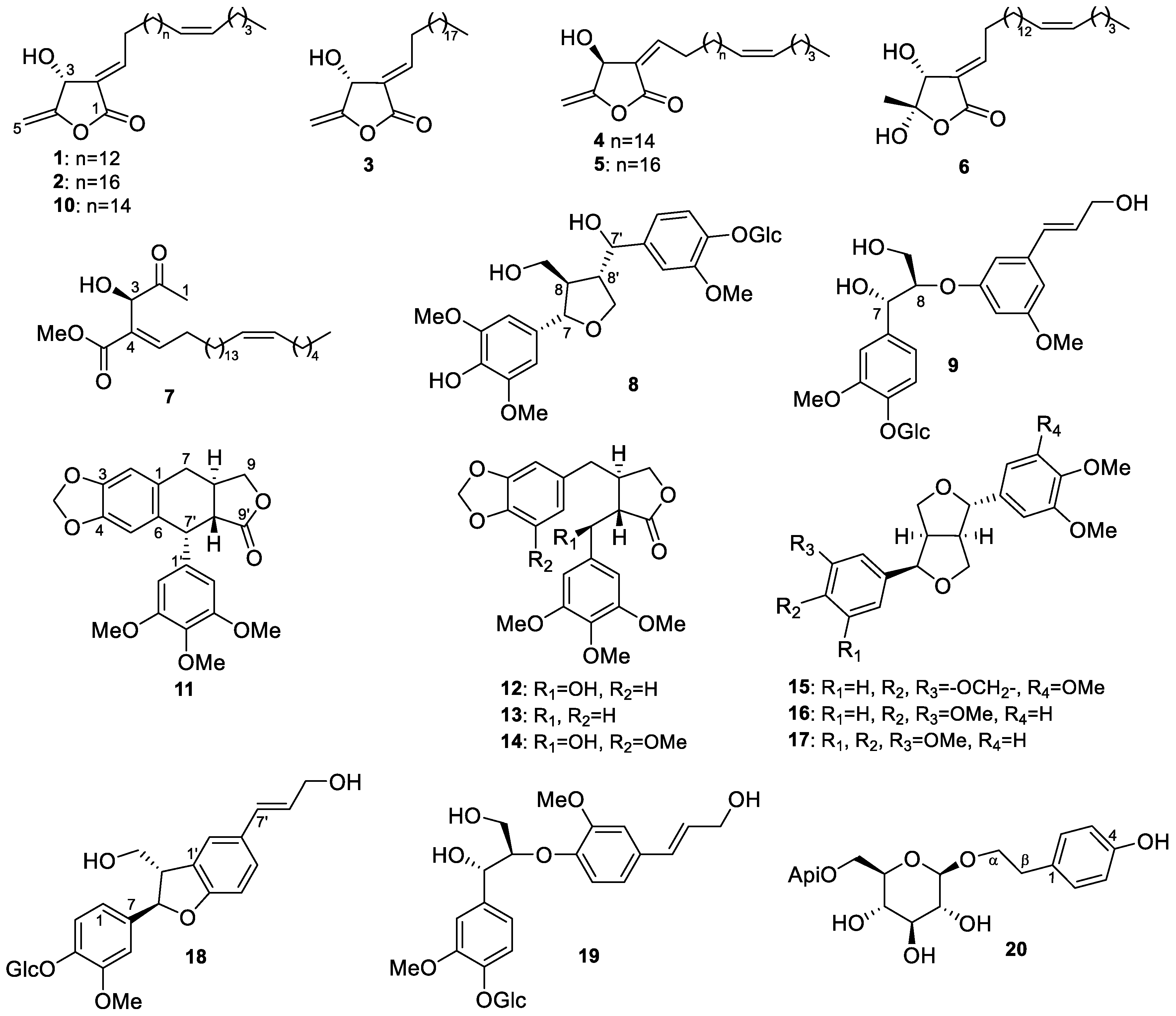
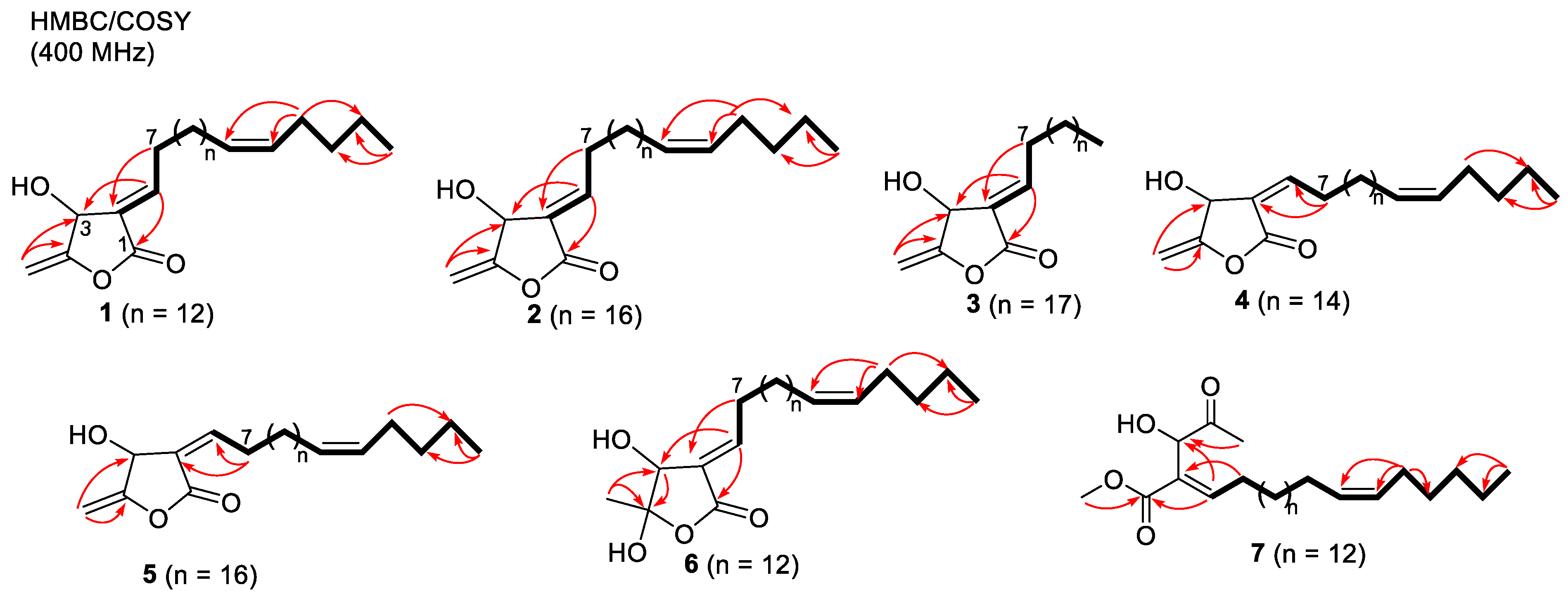
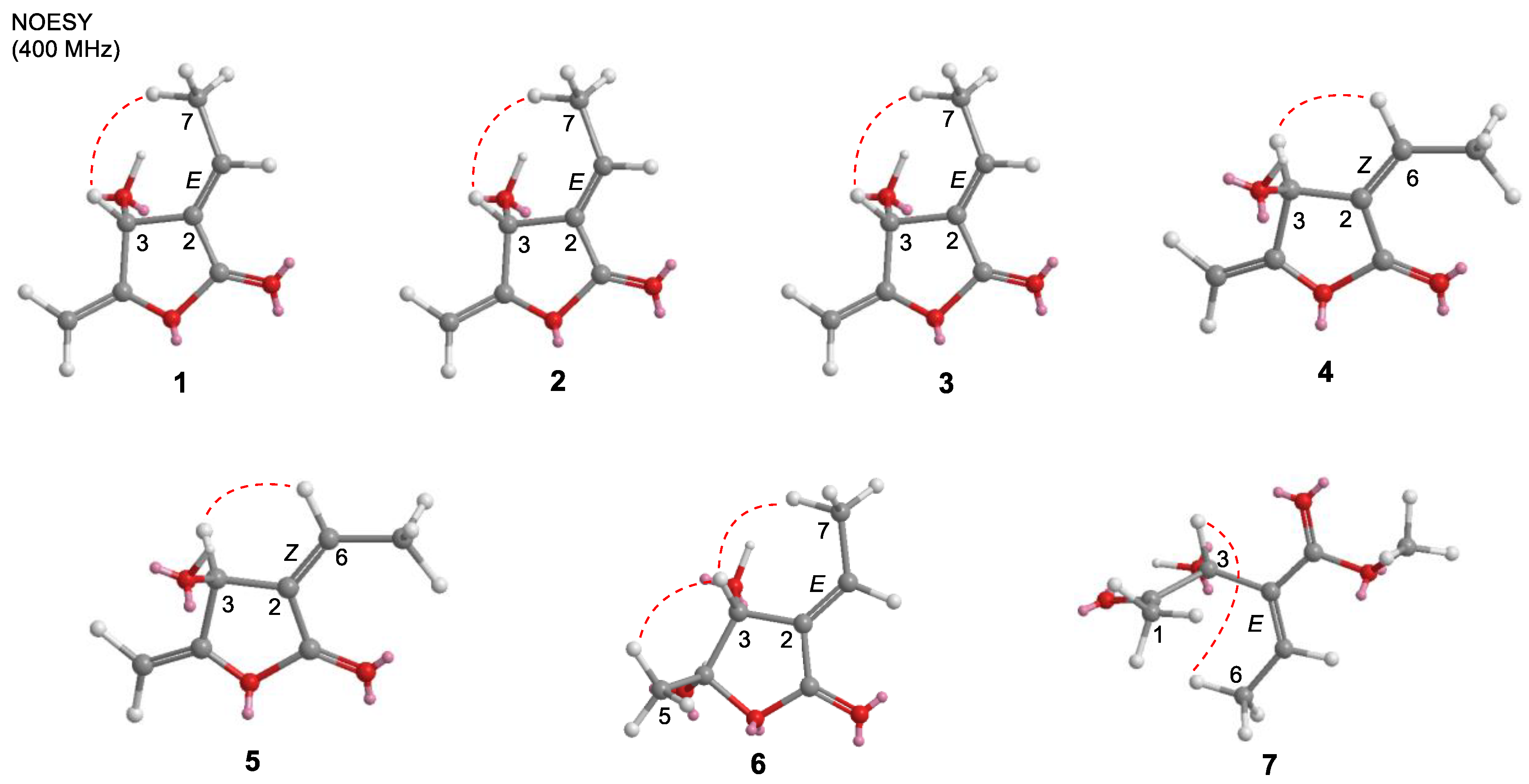
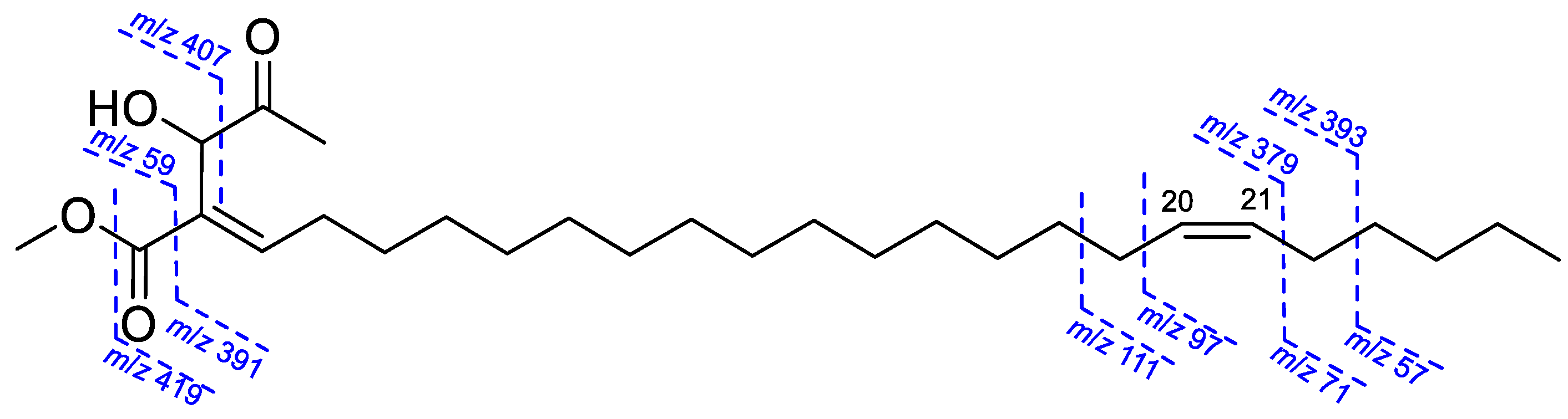
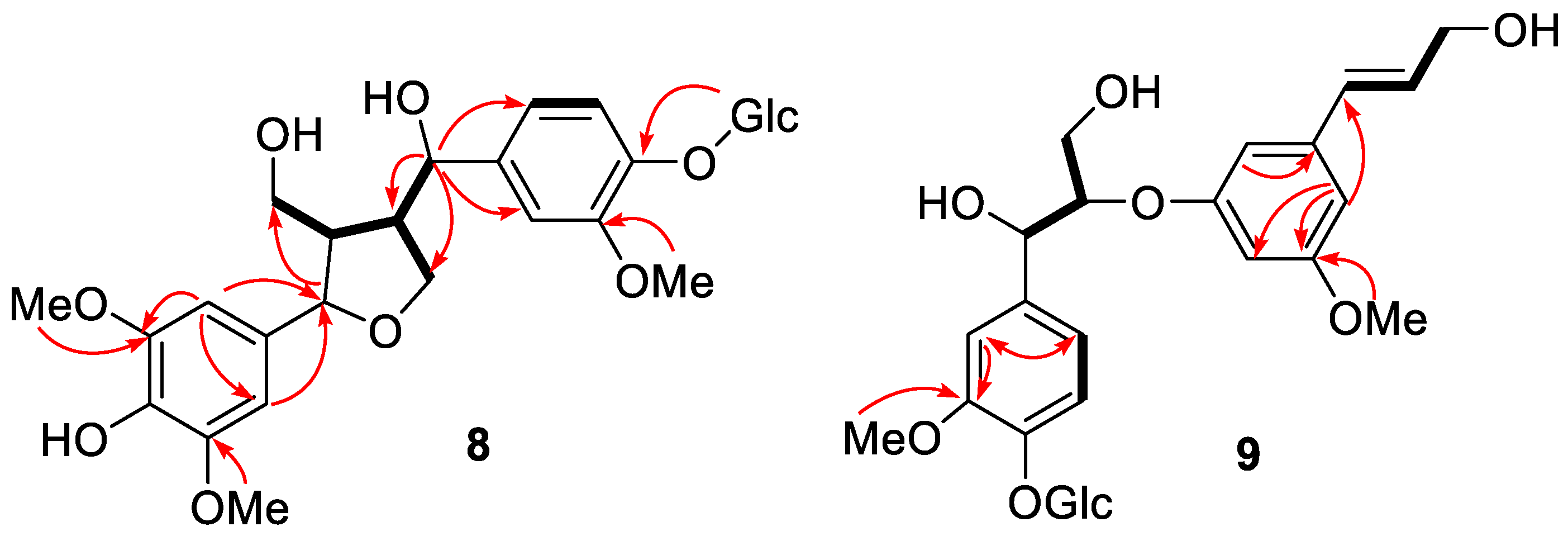
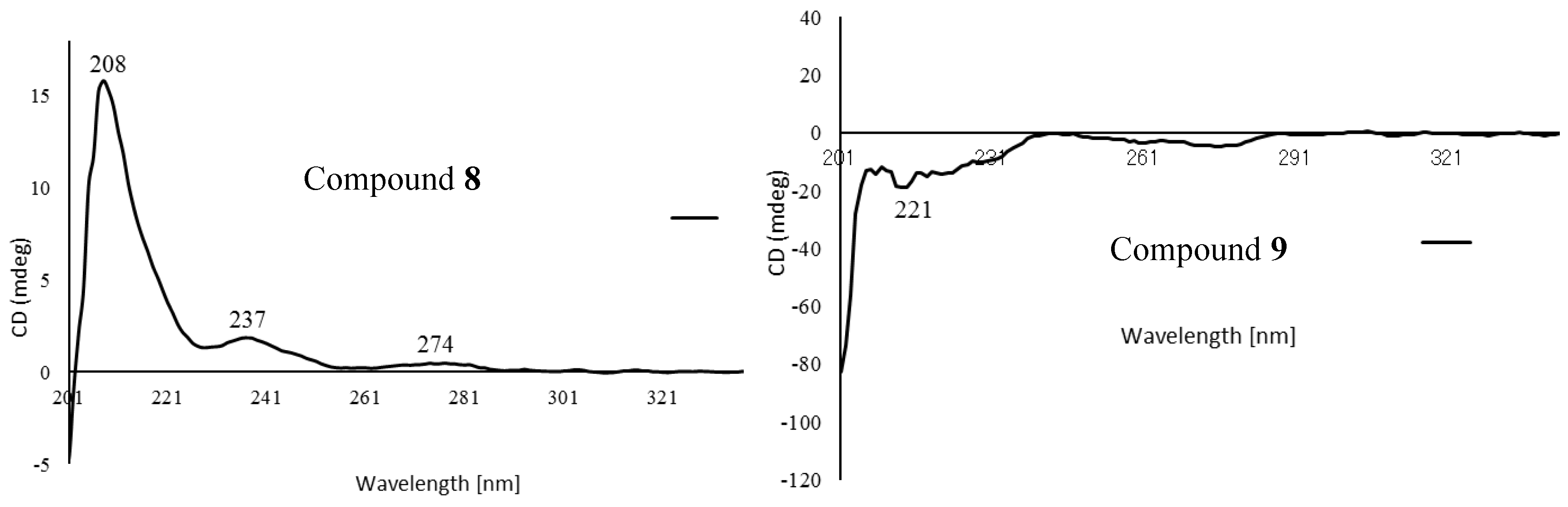
2.1. Structure Elucidation of Isolated Compounds from H. nymphaeifolia
2.2. Antiproliferative Activity of Isolated Compounds from H. nymphaeifolia
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Plant Material
3.3. Extraction and Isolation
3.3.1. Peltanolide A (1)
3.3.2. Peltanolide B (2)
3.3.3. Peltanolide C (3)
3.3.4. Peltanolide D (4)
3.3.5. Peltanolide E (5)
3.3.6. Peltanolide F (6)
3.3.7. Peltanolide G (7)
3.3.8. Peltaside A (8)
3.3.9. Peltaside B (9)
3.4. Calculation of ECD Spectra
3.5. Assay for Antiproliferative Activity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lakshmi, V.; Pandey, K.; Mishra, S.K.; Srivastava, S.; Mishra, M.; Agarwa, S.K. An overview of family Hernandiaceae. Rec. Nat. Prod. 2009, 3, 1–22. [Google Scholar]
- Pettit, G.R.; Meng, Y.H.; Gearing, R.P.; Herald, D.L.; Pettit, R.K.; Doubek, D.L.; Chapuis, J.C.; Tackett, L.P. Antineoplastic Agents. 522. Hernandia peltata (Malaysia) and Hernandia nymphaeifolia (Republic of Maldives). J. Nat. Prod. 2004, 67, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Udino, L.; Abaul, J.; Bourgeois, P.; Corrichon, L.; Duran, H.; Zedde, C. Lignans from the seeds of Hernandia sonora. Planta Med. 1999, 65, 279–281. [Google Scholar] [CrossRef] [PubMed]
- Chalandre, M.C.; Bruneton, J.; Cabalion, P.; Guinaudeau, H. Hernandiaceae. XII. Aporphine-benzylisoquinoline dimers isolated from Hernandia peltata. Can. J. Chem. 1986, 64, 123–126. [Google Scholar] [CrossRef]
- Wei, C.Y.; Wang, S.W.; Ye, J.W.; Hwang, T.L.; Cheng, M.J.; Sung, P.J.; Chang, T.H.; Chen, J.J. New anti-inflammatory aporphine and lignan derivatives from the root wood of Hernandia nymphaeifolia. Molecules 2018, 23, 2286. [Google Scholar] [CrossRef]
- Lavault, M.; Cabalion, P.; Bruneton, J. Study on Hernandiaceae. IV. Alkaloids of Hernandia peltata. Planta Med. 1982, 46, 119–121. [Google Scholar] [CrossRef]
- Chen, J.J.; Tsai, I.L.; Chen, I.S. New Oxoaporphine Alkaloids from Hernandia nymphaeifolia. J. Nat. Prod. 1996, 59, 156–158. [Google Scholar] [CrossRef]
- Chen, J.J.; Ishikawa, T.; Duh, C.Y.; Tsai, I.L.; Chen, I.S. New dimeric aporphine alkaloids and cytotoxic constituents of Hernandia nymphaefolia. Planta Med. 1996, 62, 528–533. [Google Scholar] [CrossRef]
- Angerhofer, C.K.; Guinaudeau, H.; Wongpanich, V.; Pezzuto, J.M.; Cordell, G.A. Antiplasmodial and cytotoxic activity of natural bisbenzylisoquinoline alkaloids. J. Nat. Prod. 1999, 62, 59–66. [Google Scholar] [CrossRef]
- Rasoanaivo, R.; Urverg, R.; Rafatro, H.; Ramanitrahasimbola, D.; Palazzino, G.; Galeffi, C.; Nicoletti, M. Alkaloids of Hernandia voyronii. Chloroquine-potentiating activity and structure elucidation of herveline D. Planta Med. 1998, 64, 58–62. [Google Scholar] [CrossRef]
- Dittmar, A. The effectiveness of Hernandia spp. (Hernandiaceae) in traditional Samoan medicine and according to scientific analyses. J. Ethnopharmacol. 1991, 33, 243–251. [Google Scholar] [CrossRef]
- Bruneton, J.; Shamma, M.; Minard, R.D.; Freyer, A.J.; Guinaudeau, H. Novel biogenetic pathways from (+)-reticuline. Three dimeric alkaloids: (+)-vanuatine, (+)-vateamine, and (+)-malekulatine. J. Org. Chem. 1983, 48, 3957–3960. [Google Scholar] [CrossRef]
- Chen, I.S.; Chen, J.J.; Duh, C.Y.; Tsai, I.L. Cytotoxic lignans from Formosan Hernandia nymphaeifolia. Phytochemistry 1997, 45, 991–996. [Google Scholar] [CrossRef]
- Chen, I.J.; Chang, Y.L.; Teng, C.M.; Chen, I.S. Anti-platelet aggregation alkaloids and lignans from Hernandia nymphaefolia. Planta Med. 2000, 66, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Yoder, B.J.; Cao, S.; Norris, A.; Miller, J.S.; Ratovoson, F.; Andriantsiferana, R.; Rasamison, V.E.; Kingston, D.G.I. Tambouranolide, a new cytotoxic hydroxybutanolide from a Tambourissa sp. (Monimiaceae). Nat. Prod. Res. 2007, 21, 37–41. [Google Scholar] [CrossRef] [PubMed]
- Muto, N.; Tomokuni, T.; Haramoto, M.; Tatemoto, H.; Nakanishi, T.; Inatomi, Y.; Murata, H.; Inada, A. Isolation of apoptosis- and differentiation-inducing substances toward human promyelocytic leukemia HL-60 cells from leaves of Juniperus taxifolia. Biosci. Biotechnol. Biochem. 2008, 72, 477–484. [Google Scholar] [CrossRef] [PubMed]
- Trazzi, G.; André, M.F.; Coelho, F.J. Diastereoselective synthesis of β-piperonyl-γ-butyrolactones from Morita-Baylis-Hillman adducts. Highly efficient synthesis of (±)-yatein, (±)-podorhizol and (±)-epi-podorhizol. Braz. Chem. Soc. 2010, 21, 2327–2339. [Google Scholar] [CrossRef]
- Okunishi, T.; Umezawa, T.; Shimada, M. Enantiomeric compositions and biosynthesis of Wikstroemia sikokiana lignans. J. Wood Sci. 2000, 46, 234–242. [Google Scholar] [CrossRef]
- Li, N.; Wu, J.L.; Sakai, J.I.; Ando, M. Dibenzylbutyrolactone and Dibenzylbutanediol Lignans from Peperomia duclouxii. J. Nat. Prod. 2003, 66, 1421–1426. [Google Scholar] [CrossRef]
- Ahmed, A.A.; Mahmoud, A.A.; Ali, E.T.; Tzakou, O.; Couladis, M.; Mabry, T.J.; Gáti, T.; Tóth, G. Two highly oxygenated eudesmanes and ten lignans from Achillea holosericea. Phytochemistry 2002, 59, 851–856. [Google Scholar] [CrossRef]
- Iida, T.; Nakano, M.; Ito, K. Hydroperoxysesquiterpene and lignan constituents of Magnolia kobus. Phytochemistry 1982, 21, 673–675. [Google Scholar] [CrossRef]
- Asikin, Y.; Takahashi, M.; Mizu, M.; Takara, K.; Okua, H.; Wada, K. DNA damage protection against free radicals of two antioxidant neolignan glucosides from sugarcane molasses. J. Sci. Food Agric. 2016, 96, 1209–1215. [Google Scholar] [CrossRef] [PubMed]
- Calis, I.; Kirmizibekmez, H.; Beutler, J.A.; Donmez, A.A.; Yalc, F.N.; Kilic, I.E.; Ozlap, M.; Ruedi, P.; Tasdemir, D. Secondary metabolites of Phlomis viscosa and their biological activities. Turk. J. Chem. 2005, 29, 71–81. [Google Scholar]
- Sugiyama, M.; Kikuchi, M. Phenylethanoid glycosides from Osmanthus asiaticus. Phytochemistry 1993, 32, 1553–1555. [Google Scholar] [CrossRef]
- Seki, K.; Sasaki, T.; Wano, S.; Haga, K.; Kaneko, R. Linderanolides and isolinderanolides, ten butanolides from Lindera glauca. Phytochemistry 1995, 40, 1175–1181. [Google Scholar] [CrossRef]
- Anderson, J.E.; Ma, W.; Smith, D.L.; Chang, C.J.; Mclaughlin, J.L. Biologically active γ-lactones and methylketoalkenes from Lindera benzoin. J. Nat. Prod. 1992, 55, 71–83. [Google Scholar] [CrossRef]
- Tamura, S.; Doke, S.; Murakami, N. Total synthesis of peumusolide A, NES non-antagonistic inhibitor for nuclear export of MEK. Tetrahedron 2010, 66, 8476–8480. [Google Scholar] [CrossRef]
- Tamura, S.; Tonokawa, M.; Murakami, N. Stereo-controlled synthesis of analogs of peumusolide A, NES non-antagonistic inhibitor for nuclear export of MEK. Tetrahedron Lett. 2010, 51, 3134–3137. [Google Scholar] [CrossRef]
- Tsenga, M.; Su, Y.S.; Cheng, M.J.; Liu, T.W.; Chen, I.S.; Wu, M.D.; Chang, H.S.; Yuan, G.F. Chemical constituents from a soil-derived actinomycete, Actinomadura miaoliensis BCRC 16873, and their inhibitory activities on lipopolysaccharide-induced tumor necrosis factor production. Chem. Biodivers. 2013, 10, 303–312. [Google Scholar] [CrossRef]
- Tanaka, H.; Takaya, Y.; Toyoda, J.; Yasuda, T.; Sato, M.; Murata, J.; Murata, H.; Kaburagi, K.; Iida, O.; Sugiyama, K.; et al. Two new butanolides from the roots of Litsea acuminate. Phytochemistry Lett. 2015, 11, 32–36. [Google Scholar] [CrossRef]
- Cheng, H.I.; Lin, W.Y.; Duh, C.Y.; Lee, K.H.; Tsai, I.L.; Chen, I.S. New cytotoxic butanolides from Litsea acutivena. J. Nat. Prod. 2001, 64, 1502–1505. [Google Scholar] [CrossRef] [PubMed]
- Juan, C.; Martinez, V.; Yoshida, M.; Gottlieb, O.R. The chemistry of Brazilian Lauraceae. Part LXI. ω-Ethyl, ω-ethenyl and ω-ethynyl-α-alkylidene-γ-lactones from Clinostemon mahuba. Phytochemistry 1981, 20, 459–464. [Google Scholar] [CrossRef]
- Takeda, K.I.; Sakurawi, K.; Ishi, H. Components of the Lauracea family. I. New lactonic compounds from Litsea japonica. Tetrahedron 1972, 28, 3757–3766. [Google Scholar] [CrossRef]
- Li, X.J.; Dong, J.W.; Cai, L.; Wang, J.P.; Yu, N.X.; Ding, Z.T. Illigerones A and B, two new long-chain secobutanolides from Illigera henryi W. W. Sm. Phytochem. Lett. 2017, 19, 181–186. [Google Scholar] [CrossRef]
- Yang, Y.N.; Huang, X.Y.; Feng, Z.M.; Jiang, J.S.; Zhang, P.C. Hepatoprotective Activity of Twelve Novel 7′-Hydroxy Lignan Glucosides from Arctii Fructus. J. Agric. Food Chem. 2014, 62, 9095–9102. [Google Scholar] [CrossRef] [PubMed]
- Greca, M.D.; Molinaro, A.; Monaco, P.; Previtera, L. Neolignans from Arum italicum. Phytochemistry 1994, 35, 777–779. [Google Scholar] [CrossRef]
- Arnoldi, A.; Merlini, L. Asymmetric synthesis of 3-methyl-2-phenyl-1,4-benzodioxanes. Absolute configuration of the neolignans eusiderin and eusiderin C and D. J. Chem. Soc. Perkin Trans. I 1985, 2555–2557. [Google Scholar] [CrossRef]
- Gan, M.; Zhang, Y.L.; Lin, S.; Liu, M.T.; Song, W.X.; Zi, J.C.; Yang, Y.C.; Fan, X.N.; Shi, J.G.; Hu, J.F.; et al. Glycosides from the root of Iodes cirrhosa. J. Nat. Prod. 2008, 71, 647–654. [Google Scholar] [CrossRef]
- Pascitelli, G.; Bruhn, T. Good Computational Practice in the Assignment of Absolute Configurations by TDDFT Calculations of ECD Spectra. Chirality 2016, 28, 466–474. [Google Scholar] [CrossRef]
- Nakagawa-Goto, K.; Oda, A.; Hamel, E.; Ohkoshi, E.; Lee, K.H.; Goto, M. Development of a novel class of tubulin inhibitors from desmosdumotin B with a hydroxylated bicyclic B-ring. J. Med. Chem. 2015, 58, 2378–2389. [Google Scholar] [CrossRef]
Sample Availability: Not available. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1a (CDCl3) | 2b (CDCl3) | 3b (CDCl3) | 4b (CDCl3) | 5b (CDCl3) | 6a (CDCl3) | 7b (CDCl3) | |
|---|---|---|---|---|---|---|---|
| Position | δH (J in Hz) | δH (J in Hz) | δH (J in Hz) | δH (J in Hz) | δH (J in Hz) | δH (J in Hz) | δH (J in Hz) |
| 1 | 2.15 s | ||||||
| 3 | 5.26 brs | 5.26 brs | 5.26 brd (5.6) | 5.11 m | 5.10 brd (7.3) | 4.82 brs | 4.89 d (4.2) |
| 5a | 4.72 dd (2.8, 1.4) | 4.72 dd (2.8, 1.4) | 4.72 dd (2.8, 1.4) | 4.67 dd (2.8, 1.4) | 4.66 dd (2.8, 1.4) | 1.62 s | 7.07 t (8.0) |
| 5b | 4.96 dd (2.8, 1.4) | 4.96 dd (2.8, 1.4) | 4.96 dd (2.8, 1.4) | 4.89 dd (2.8, 1.4) | 4.89 dd (2.8, 1.4) | ||
| 6 | 7.09 td (7.8, 2.2) | 7.09 td (7.8, 2.2) | 7.10 td (7.8, 2.2) | 6.69 td (7.8, 2.2) | 6.67 td (7.8, 2.2) | 7.04 td (7.8, 2.2) | 2.34 td (14.8, 8.0) |
| 7 | 2.48 m | 2.48 m | 2.48 m, 2.43 m | 2.78 m | 2.76 m | 2.38 m | 1.50 m |
| 8 | 1.52 m | 1.52 m | 1.53 m | 1.46 m | 1.45 m | 1.52 m | 1.25 mp |
| 9–18 | 1.26 mc | 1.26 me | 1.25 mg | 1.25 mh | 1.25 mj | 1.25 mm | 1.25 mp |
| 19 | 2.00 m | 1.26 me | 1.25 mg | 1.25 mh | 1.25 mj | 2.01 mn | 2.00 mq |
| 20 | 5.36 md | 1.26 me | 1.25 mg | 1.25 mh | 1.25 mj | 5.34 t (4.8)o | 5.34 t (4.8)r |
| 21 | 5.36 md | 1.26 me | 1.25 mg | 2.02 m | 1.25 mj | 5.34 t (4.8)o | 5.34 t (4.8)r |
| 22 | 2.02 m | 1.26 me | 1.25 mg | 5.35 mi | 1.25 mj | 2.01 mn | 2.00 mq |
| 23 | 1.26 mc | 2.00 m | 1.25 mg | 5.35 mi | 2.01 mk | 1.25 mm | 1.25 mp |
| 24 | 1.32 m | 5.36 mf | 1.25 mg | 2.02 | 5.33 ml | 1.31 | 1.25 mp |
| 25 | 0.89 t (6.9) | 5.36 mf | 0.89 br t (7.3) | 1.25 mh | 5.33 ml | 0.89 t (6.0) | 1.31 m |
| 26 | 2.02 m | 1.33 m | 2.01 mk | 0.89 t (6.0) | |||
| 27 | 1.26 me | 0.88 t (6.9) | 1.25 mj | ||||
| 28 | 1.32 m | 1.32 m | |||||
| 29 | 0.89 t (7.3) | 0.87 t (6.9) | |||||
| 2-OH | 2.26 m | ||||||
| 3-OH | 4.00 brs | ||||||
| OCH3 | 3.72 s |
| 1a | 2b | 3b | 4b | 5b | 6a | 7b | |
|---|---|---|---|---|---|---|---|
| Position | δc | δc | δc | δc | δc | δc | δc |
| 1 | 166.5 | 166.5 | 166.3 | 163.1 | 166.4 | 24.8 | |
| 2 | 127.3 | 127.3 | 127.2 | 127.4 | 127.3 | 125.2 | 206.3 |
| 3 | 66.5 | 66.5 | 66.6 | 68.9 | 68.9 | 70.9 | 73.4 |
| 4 | 157.6 | 157.6 | 157.5 | 160.1 | 160.2 | 100.1 | 129.8 |
| 5 | 91.4 | 91.4 | 91.5 | 90.3 | 90.4 | 26.8 | 149.0 |
| 6 | 150.3 | 150.3 | 150.3 | 151.4 | 151.4 | 151.9 | 28.7 |
| 7 | 29.8 | 29.8 | 29.8 | 29.8 | 29.8 | 30.1 | 29.3 |
| 8 | 28.4 | 28.4 | 28.4–29.6d | 28.4 | 28.4 | 29.5–29.9g | 29.4–30.0h |
| 9–18 | 29.4–30.0 | 29.4–30.0c | 28.4–29.6d | 29.4–30.0e | 29.4–30.0f | 29.5–29.9g | 29.4–30.0h |
| 19 | 27.0 | 29.4–30.0c | 28.4–29.6d | 29.4–30.0e | 29.4–30.0f | 27.0 | 27.2 |
| 20 | 129.8 | 29.4–30.0c | 28.4–29.6d | 29.4–30.0e | 29.4–30.0f | 129.8 | 129.8 |
| 21 | 129.9 | 29.4–30.0c | 28.4–29.6d | 27.0 | 29.4–30.0f | 129.9 | 129.9 |
| 22 | 27.2 | 29.4–30.0c | 28.4–29.6d | 129.8 | 29.4–30.0f | 27.7 | 27.2 |
| 23 | 32.0 | 27.0 | 32.0 | 129.9 | 27.0 | 32.1 | 29.8 |
| 24 | 22.4 | 129.8 | 22.8 | 27.2 | 129.8 | 22.5 | 31.9 |
| 25 | 14.0 | 129.9 | 14.1 | 32.0 | 129.9 | 14.1 | 22.7 |
| 26 | 27.2 | 22.3 | 27.2 | 14.1 | |||
| 27 | 32.0 | 14.0 | 32.0 | ||||
| 28 | 22.4 | 22.4 | |||||
| 29 | 14.0 | 14.0 | |||||
| COO | 166.5 | ||||||
| OCH3 | 52.0 |
| 8 (CD3OD) | 9 (CD3OD) | |||
|---|---|---|---|---|
| Position | δCa | δH (J in Hz)b | δCa | δH (J in Hz)b |
| 1 | 134.1 | 131.5 | ||
| 2 | 104.6c | 6.52 sd | 112.3 | 7.09 brs |
| 3 | 149.3 | 147.4 | ||
| 4 | 135.9 | 150.5 | ||
| 5 | 149.3 | 120.6 | 7.06 d (8.2) | |
| 6 | 104.6c | 6.52 sd | 121.1 | 6.96 dd (2.2, 8.2) |
| 7 | 85.1 | 4.63 d (7.3) | 74.9 | 4.85 overlap |
| 8 | 53.7 | 1.89 m | 85.9 | 4.36 m |
| 9 | 62.3 | 3.87 m | 62.2 | 3.82 m |
| 3.63 m | 3.47 m | |||
| 1′ | 139.5 | 137.8 | ||
| 2′ | 112.2 | 6.92 d (1.8) | 118.7g | 6.86 brsg |
| 3′ | 150.7 | 149.2 | ||
| 4′ | 147.5 | 118.7g | 6.86 brsg | |
| 5′ | 117.5 | 7.11 d (8.2) | 151.2 | |
| 6′ | 120.9 | 6.82 dd (8.2, 1.8) | 111.3 | 6.97 d (2.2) |
| 7′ | 74..8 | 4.52 d (8.2) | 132.9 | 6.52 d (15.4) |
| 8′ | 50.7 | 2.53 m | 111.3 | 6.27 dd (5.7, 15.4) |
| 9′ | 76.2 | 4.26 dd (9.0, 4.6) | 63.8 | 4.19 d (5.9) |
| 3.92 m | ||||
| 3-OMe | 56.9 | 3.84 se | 56.7 | 3.81 s |
| 5-OMe | 56.8 | 3.84 se | 56.5 | 3.79 s |
| 3′-OMe | 56.7 | 3.83 s | ||
| Glc-1 | 102.7 | 4.84 m | 103.1 | 4.81 d (6.9) |
| Glc-2 | 74.9 | 3.4–3.8 mf | 73.9 | 3.4–3.8 mh |
| Glc-3 | 78.2 | 3.4–3.8 mf | 78.2 | 3.4–3.8 mh |
| Glc-4 | 71.4 | 3.4–3.8 mf | 71.4 | 3.4–3.8 mh |
| Glc-5 | 77.9 | 3.4–3.8 mf | 77.9 | 3.4–3.8 mh |
| Glc-6 | 62.5 | 3.4–3.8 mf | 62.5 | 3.4–3.8 mh |
| Compounds | Cell Linesa (IC50 μM)b | ||||
|---|---|---|---|---|---|
| A549 | MDA-MB-231 | MCF-7 | KB | KB-VIN | |
| 1 | >40 | 22.5 | >40 | 25.7 | 31.7 |
| 2 | 21.9 | 24.6 | 24.6 | 22.6 | 21.4 |
| 8 | 35.1 | 35.3 | 35.7 | 32.7 | 21.7 |
| 10 | 12.5 | 10.8 | 8.8 | 18.6 | 8.8 |
| 13 | 32.8 | 37.7 | 33.5 | >40 | 8.7 |
| 14 | 23.2 | 32.9 | 32.8 | 23.2 | 19.9 |
| 15 | 8.1 | 20.8 | 6.8 | 20.3 | 5.4 |
| 16 | 5.7 | 8.2 | 8.1 | 12.6 | 5.3 |
| 17 | 37.8 | >40 | 38.0 | >40 | 8.2 |
| 18 | >40 | >40 | >40 | >40 | >40 |
| Paclitaxel (nM) | 6.5 | 8.4 | 12.1 | 7.1 | 2213 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aimaiti, S.; Saito, Y.; Fukuyoshi, S.; Goto, M.; Miyake, K.; Newman, D.J.; O’Keefe, B.R.; Lee, K.-H.; Nakagawa-Goto, K. Isolation, Structure Elucidation, and Antiproliferative Activity of Butanolides and Lignan Glycosides from the Fruit of Hernandia nymphaeifolia. Molecules 2019, 24, 4005. https://doi.org/10.3390/molecules24214005
Aimaiti S, Saito Y, Fukuyoshi S, Goto M, Miyake K, Newman DJ, O’Keefe BR, Lee K-H, Nakagawa-Goto K. Isolation, Structure Elucidation, and Antiproliferative Activity of Butanolides and Lignan Glycosides from the Fruit of Hernandia nymphaeifolia. Molecules. 2019; 24(21):4005. https://doi.org/10.3390/molecules24214005
Chicago/Turabian StyleAimaiti, Simayijiang, Yohei Saito, Shuichi Fukuyoshi, Masuo Goto, Katsunori Miyake, David J. Newman, Barry R. O’Keefe, Kuo-Hsiung Lee, and Kyoko Nakagawa-Goto. 2019. "Isolation, Structure Elucidation, and Antiproliferative Activity of Butanolides and Lignan Glycosides from the Fruit of Hernandia nymphaeifolia" Molecules 24, no. 21: 4005. https://doi.org/10.3390/molecules24214005
APA StyleAimaiti, S., Saito, Y., Fukuyoshi, S., Goto, M., Miyake, K., Newman, D. J., O’Keefe, B. R., Lee, K.-H., & Nakagawa-Goto, K. (2019). Isolation, Structure Elucidation, and Antiproliferative Activity of Butanolides and Lignan Glycosides from the Fruit of Hernandia nymphaeifolia. Molecules, 24(21), 4005. https://doi.org/10.3390/molecules24214005