Bypass of the Major Alkylative DNA Lesion by Human DNA Polymerase η



Abstract
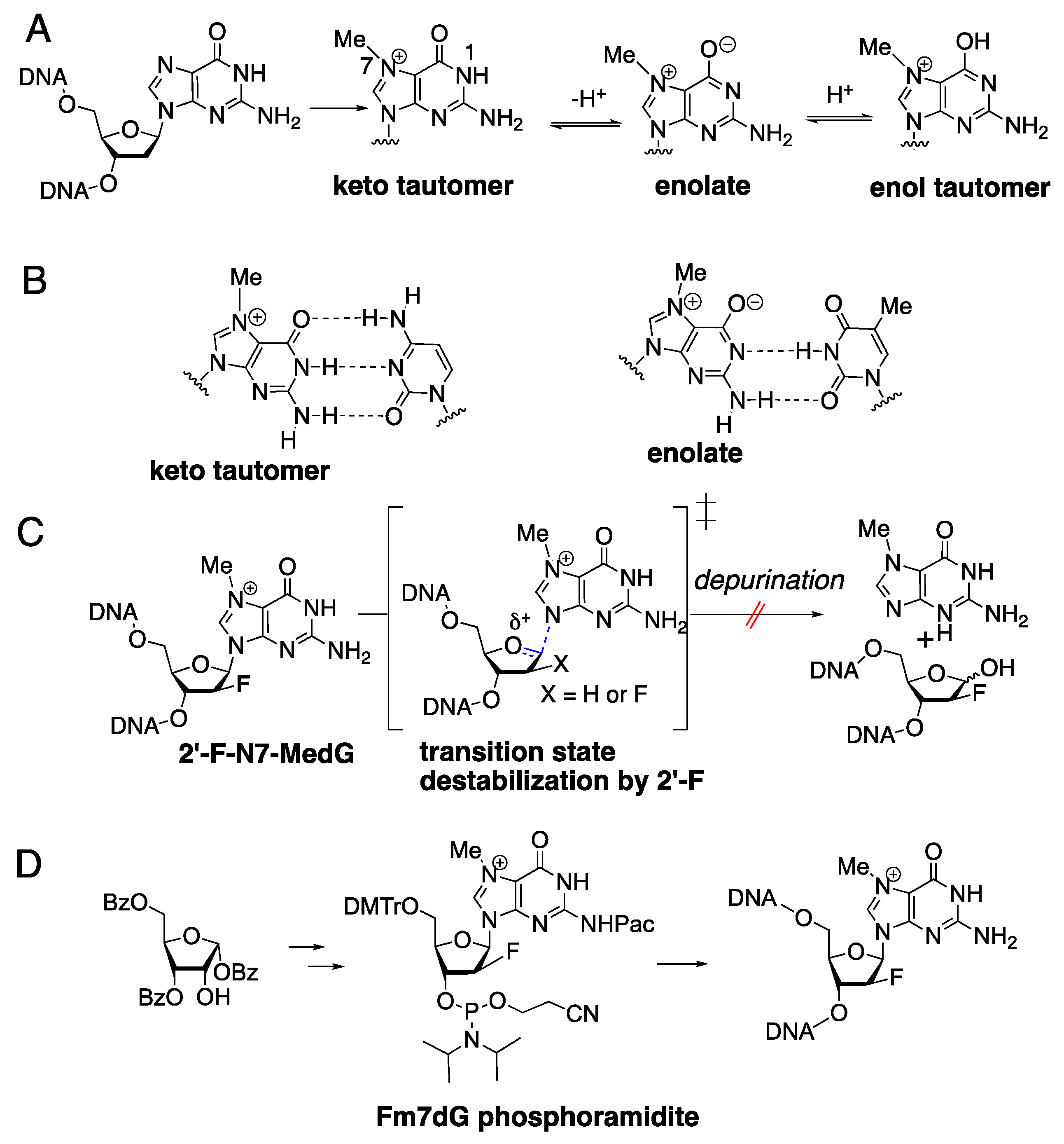
1. Introduction
2. Results and Discussion
2.1. Steady-State Kinetic Studies
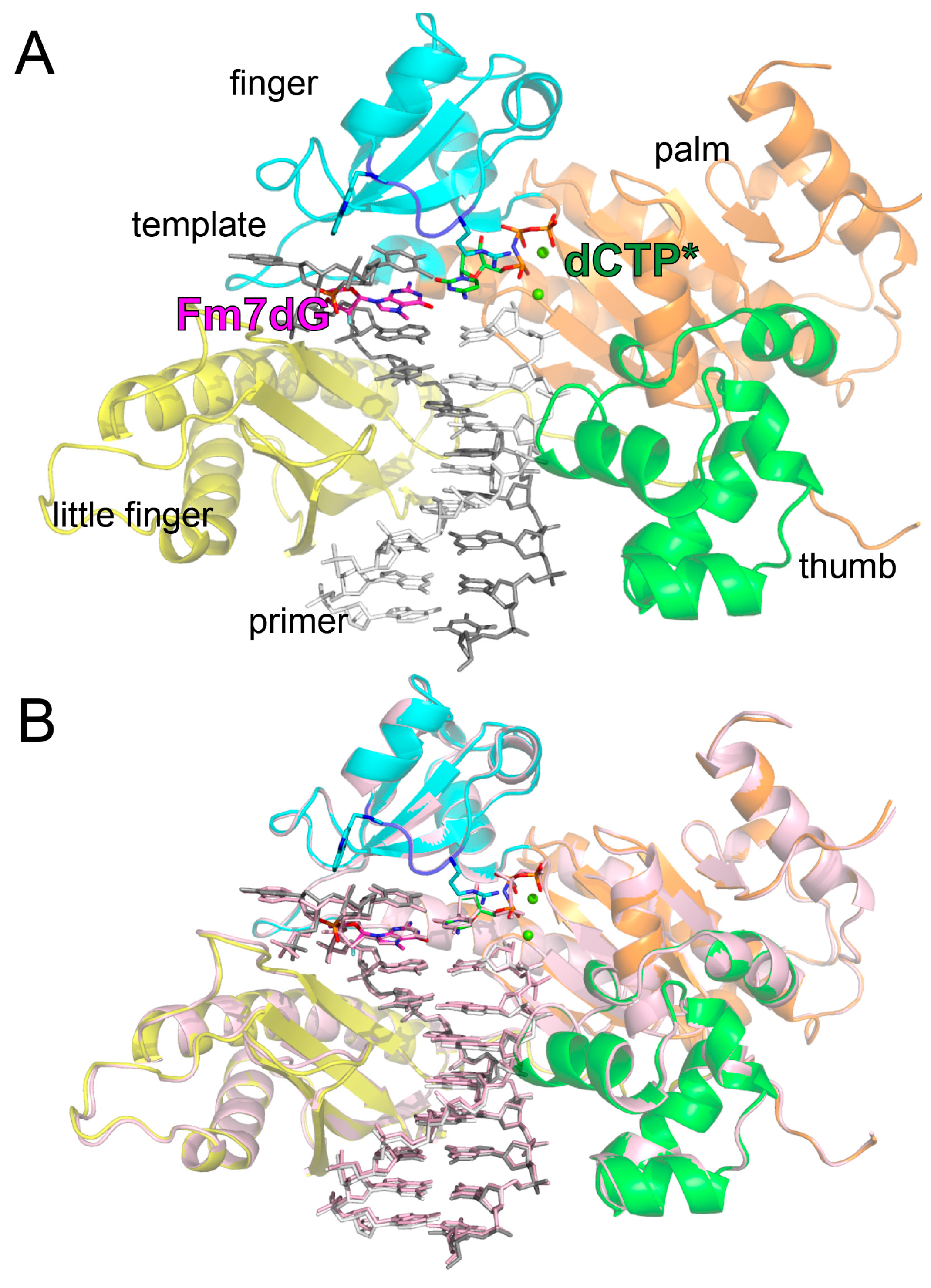
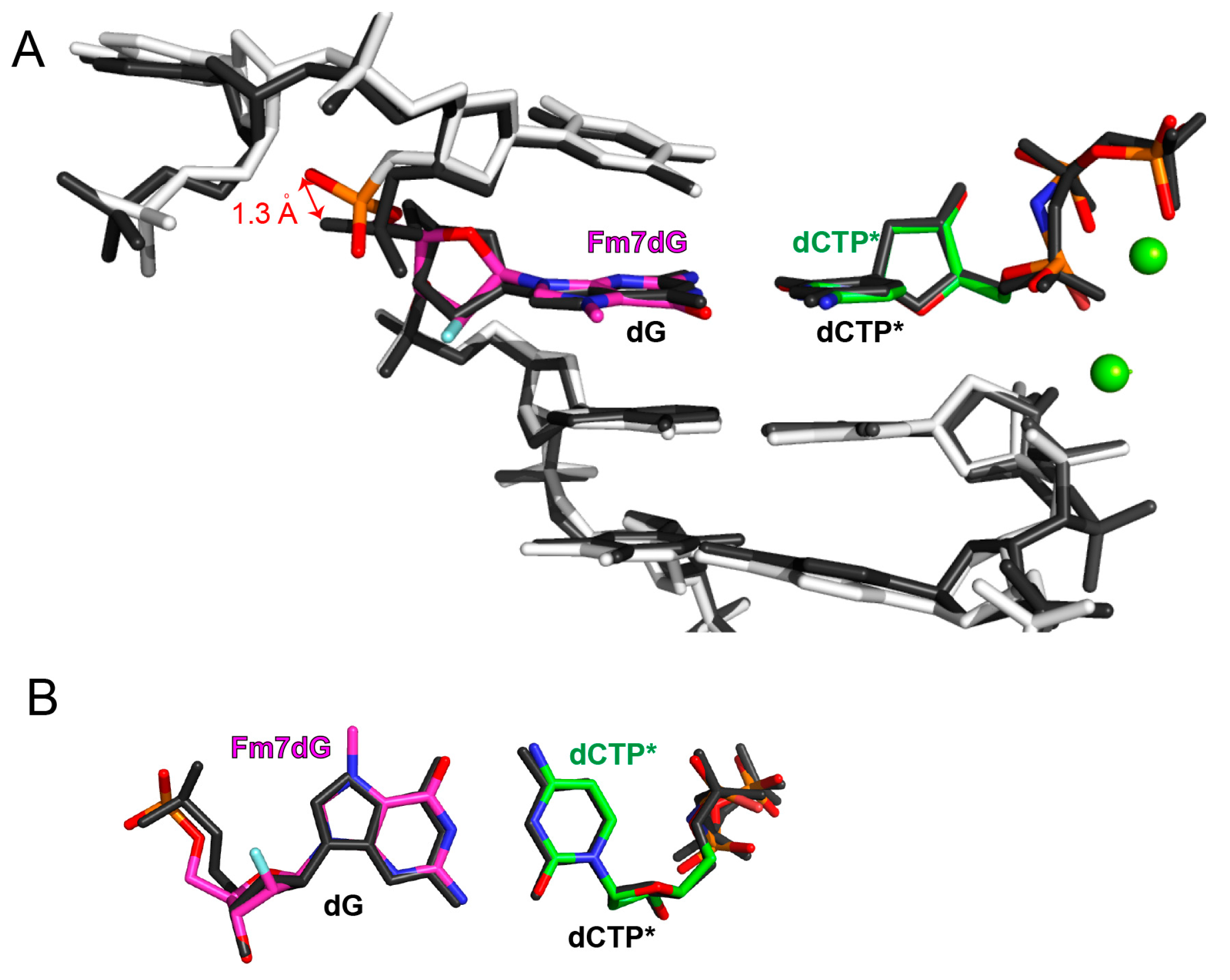
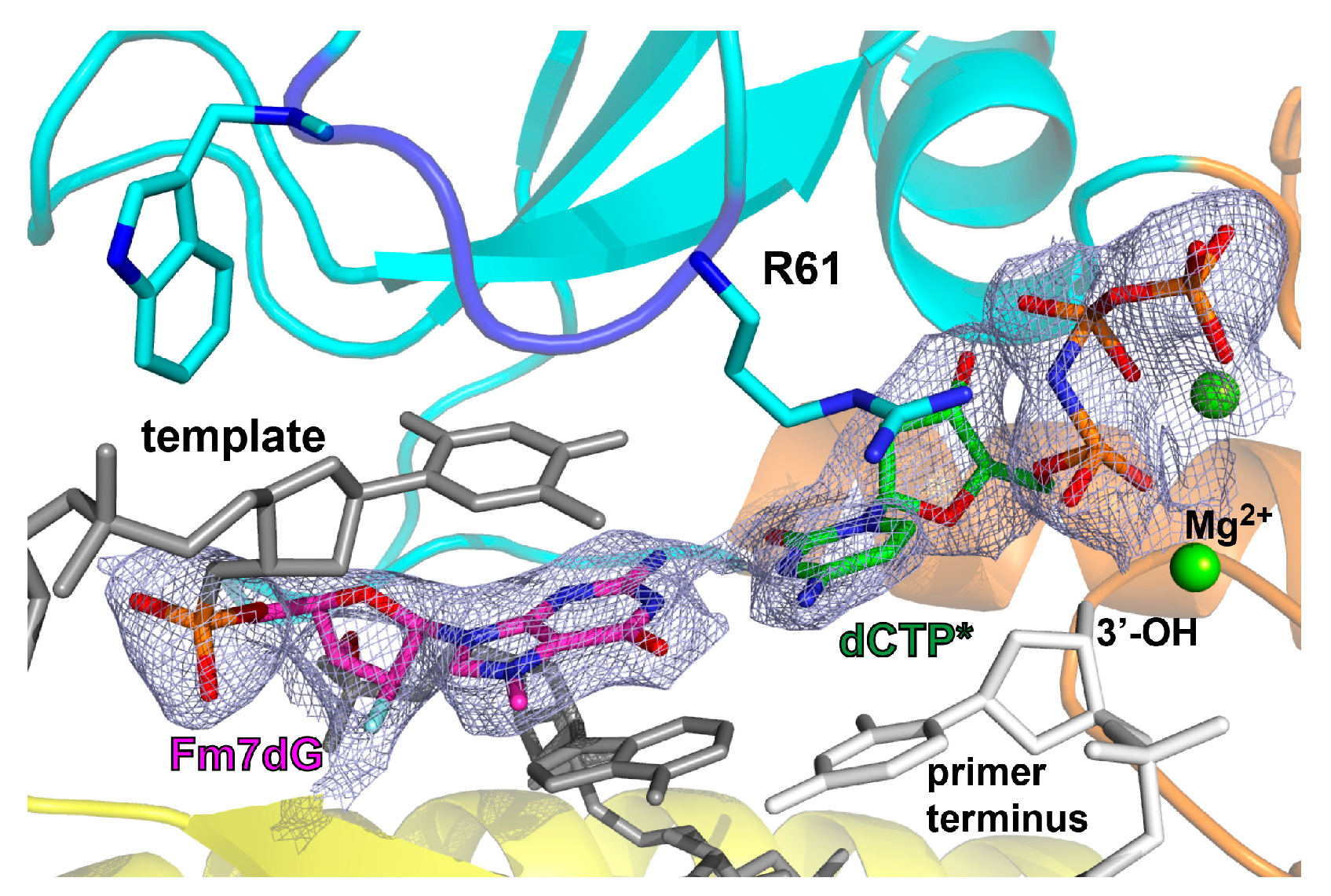
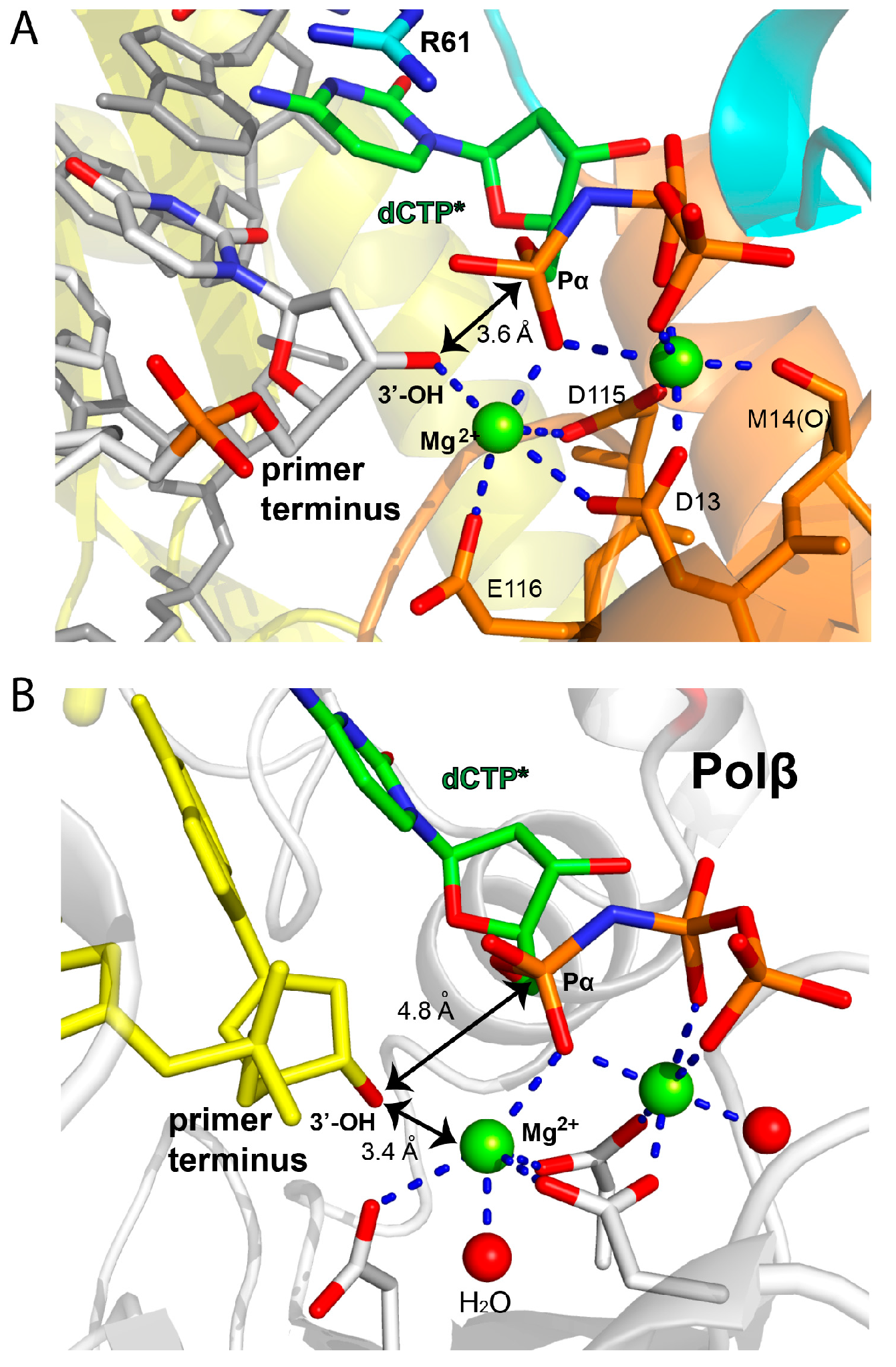
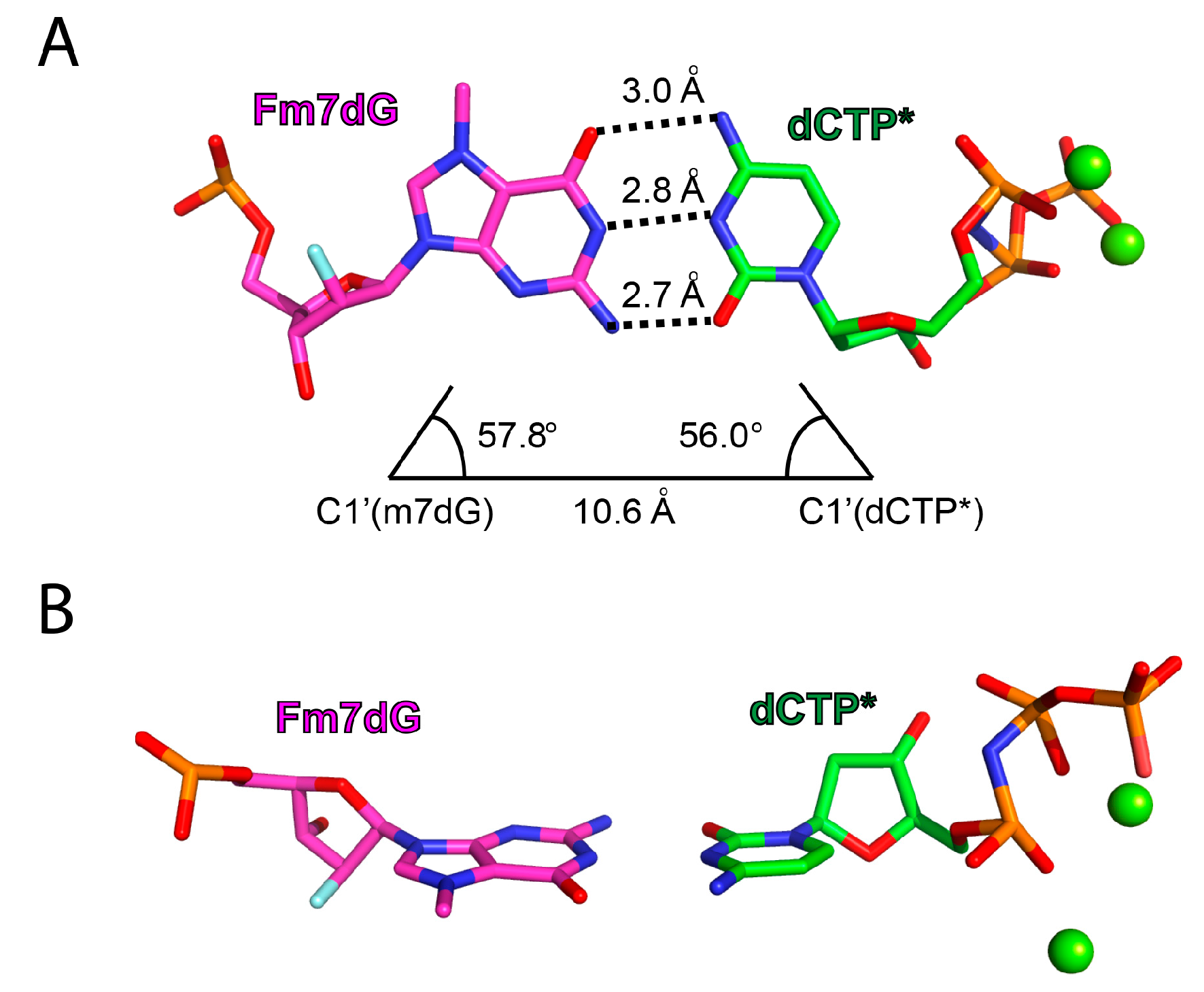
2.2. Structure of Polη Incorporating dCTP Opposite Templating Fm7dG
3. Materials and Methods
3.1. Synthesis of Fm7dG-Containing Oligonucleotide
3.2. Cloning and Protein Expression and Purification
3.3. Steady-State Kinetics of Single Nucleotide Incorporation Opposite Templating Fm7dG by Polη
3.4. Crystallization, Data Collection, and Refinement
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kumar, S.; Cheng, X.; Klimasauskas, S.; Mi, S.; Posfai, J.; Roberts, R.J.; Wilson, G.G. The DNA (cytosine-5) methyltransferases. Nucleic Acids Res. 1994, 22, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lu, Z.; Gomez, A.; Hon, G.C.; Yue, Y.; Han, D.; Fu, Y.; Parisien, M.; Dai, Q.; Jia, G.; et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 2014, 505, 117–120. [Google Scholar] [CrossRef] [PubMed]
- Rydberg, B.; Lindahl, T. Nonenzymatic methylation of DNA by the intracellular methyl group donor S-adenosyl-L-methionine is a potentially mutagenic reaction. EMBO J. 1982, 1, 211–216. [Google Scholar] [CrossRef] [PubMed]
- Barrows, L.R.; Carcinogenesis, P.M. Nonenzymatic methylation of DNA by S-adenosylmethionine in vitro. Carcinogenesis 1982, 3, 349–351. [Google Scholar] [CrossRef]
- Fu, D.; Calvo, J.A.; Samson, L.D. Balancing repair and tolerance of DNA damage caused by alkylating agents. Nat. Rev. Cancer 2012, 12, 104–120. [Google Scholar] [CrossRef]
- Lindahl, T. Instability and decay of the primary structure of DNA. Nature 1993, 362, 709–715. [Google Scholar] [CrossRef]
- Dosanjh, M.K.; Loechler, E.L.; Singer, B. Evidence from in vitro replication that O6-methylguanine can adopt multiple conformations. Proc. Natl. Acad. Sci. USA 1993, 90, 3983–3987. [Google Scholar] [CrossRef]
- Warren, J.J.; Forsberg, L.J.; Beese, L.S. The structural basis for the mutagenicity of O(6)-methyl-guanine lesions. Proc. Natl. Acad. Sci. USA 2006, 103, 19701–19706. [Google Scholar] [CrossRef]
- Choi, J.-Y.; Chowdhury, G.; Zang, H.; Angel, K.C.; Vu, C.C.; Peterson, L.A.; Guengerich, F.P. Translesion synthesis across O6-alkylguanine DNA adducts by recombinant human DNA polymerases. J. Biol. Chem. 2006, 281, 38244–38256. [Google Scholar] [CrossRef]
- Pence, M.G.; Choi, J.Y.; Egli, M.; Guengerich, F.P. Structural Basis for Proficient Incorporation of dTTP Opposite O6-Methylguanine by Human DNA Polymerase. J. Biol. Chem. 2010, 285, 40666–40672. [Google Scholar] [CrossRef]
- Boysen, G.; Pachkowski, B.F.; Nakamura, J.; Swenberg, J.A. The formation and biological significance of N7-guanine adducts. Mutat. Res. 2009, 678, 76–94. [Google Scholar] [CrossRef] [PubMed]
- Mustonen, R.; Hemminki, K. 7-Methylguanine levels in DNA of smokers“ and non-smokers” total white blood cells, granulocytes and lymphocytes. Carcinogenesis 1992, 13, 1951–1955. [Google Scholar] [CrossRef] [PubMed]
- Park, J.W.; Ames, B.N. 7-Methylguanine adducts in DNA are normally present at high levels and increase on aging: Analysis by HPLC with electrochemical detection. Proc. Natl. Acad. Sci. USA 1988, 85, 7467–7470. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, P.E.M.; Lawrence, C.W. Novel Mutagenic Properties of Abasic Sites inSaccharomyces cerevisiae. J. Mol. Biol. 1995, 251, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Boiteux, S.; Guillet, M. Abasic sites in DNA: Repair and biological consequences in Saccharomyces cerevisiae. DNA Repair 2004, 3, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Strauss, B.S. The “A rule” of mutagen specificity: A consequence of DNA polymerase bypass of non-instructional lesions? Bioessays 1991, 13, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Gates, K.S. Structural biology: FaPy lesions and DNA mutations. Nat. Chem. Biol. 2013, 9, 412–414. [Google Scholar] [CrossRef]
- Christov, P.P.; Yamanaka, K.; Choi, J.-Y.; Takata, K.-I.; Wood, R.D.; Guengerich, F.P.; Lloyd, R.S.; Rizzo, C.J. Replication of the 2,6-diamino-4-hydroxy-N(5)-(methyl)-formamidopyrimidine (MeFapy-dGuo) adduct by eukaryotic DNA polymerases. Chem. Res. Toxicol. 2012, 25, 1652–1661. [Google Scholar] [CrossRef][Green Version]
- Patra, A.; Banerjee, S.; Salyard, T.L.J.; Malik, C.K.; Christov, P.P.; Rizzo, C.J.; Stone, M.P.; Egli, M. Structural Basis for Error-Free Bypass of the 5-N-Methylformamidopyrimidine-dG Lesion by Human DNA Polymerase η and Sulfolobus solfataricus P2 Polymerase IV. J. Am. Chem. Soc. 2015, 137, 7011–7014. [Google Scholar] [CrossRef]
- Yang, K.; Park, D.; Tretyakova, N.Y.; Greenberg, M.M. Histone tails decrease N7-methyl-2′-deoxyguanosine depurination and yield DNA–protein cross-links in nucleosome core particles and cells. Proc. Natl. Acad. Sci. USA 2018, 115, E11212–E11220. [Google Scholar] [CrossRef]
- Mol, C.D.; Izumi, T.; Mitra, S.; Tainer, J.A. DNA-bound structures and mutants reveal abasic DNA binding by APE1 and DNA repair coordination [corrected]. Nature 2000, 403, 451–456. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, M.M. Abasic and Oxidized Abasic Site Reactivity in DNA: Enzyme Inhibition, Cross-Linking, and Nucleosome Catalyzed Reactions. Acc. Chem. Res. 2014, 47, 646–655. [Google Scholar] [CrossRef] [PubMed]
- Lawley, P.D.; Brookes, P. Acidic dissociation of 7:9-dialkylguanines and its possible relation to mutagenic properties of alkylating agents. Nature 1961, 192, 1081–1082. [Google Scholar] [CrossRef] [PubMed]
- Lawley, P.D.; Brookes, P. Further studies on the alkylation of nucleic acids and their constituent nucleotides. Biochem. J. 1963, 89, 127. [Google Scholar] [CrossRef] [PubMed]
- Sowers, L.C.; Shaw, B.R.; Veigl, M.L.; Sedwick, W.D. DNA base modification: Ionized base pairs and mutagenesis. Mutat. Res. 1987, 177, 201–218. [Google Scholar] [CrossRef]
- Lee, S.; Bowman, B.R.; Ueno, Y.; Wang, S.; Verdine, G.L. Synthesis and structure of duplex DNA containing the genotoxic nucleobase lesion N7-methylguanine. J. Am. Chem. Soc. 2008, 130, 11570–11571. [Google Scholar] [CrossRef]
- Kou, Y.; Koag, M.C.; Lee, S. N7 Methylation Alters Hydrogen-Bonding Patterns of Guanine in Duplex DNA. J. Am. Chem. Soc. 2015, 137, 14067–14070. [Google Scholar] [CrossRef]
- Koag, M.C.; Kou, Y.; Ouzon-Shubeita, H.; Lee, S. Transition-state destabilization reveals how human DNA polymerase β proceeds across the chemically unstable lesion N7-methylguanine. Nucleic Acids Res. 2014, 42, 8755–8766. [Google Scholar] [CrossRef][Green Version]
- Njuma, O.J.; Su, Y.; Guengerich, F.P. The abundant DNA adduct N7-methyl deoxyguanosine contributes to miscoding during replication by human DNA polymerase η. J. Biol. Chem. 2019, 294, 10253–10265. [Google Scholar] [CrossRef]
- Zhao, L.; Christov, P.P.; Kozekov, I.D.; Pence, M.G.; Pallan, P.S.; Rizzo, C.J.; Egli, M.; Guengerich, F.P. Replication of N2,3-ethenoguanine by DNA polymerases. Angew. Chem. Int. Ed. Engl. 2012, 51, 5466–5469. [Google Scholar] [CrossRef]
- Ling, H.; Boudsocq, F.; Woodgate, R.; Yang, W. Crystal structure of a Y-family DNA polymerase in action: A mechanism for error-prone and lesion-bypass replication. Cell 2001, 107, 91–102. [Google Scholar] [CrossRef]
- Ling, H.; Boudsocq, F.; Plosky, B.S.; Woodgate, R.; Yang, W. Replication of a cis-syn thymine dimer at atomic resolution. Nature 2003, 424, 1083–1087. [Google Scholar] [CrossRef]
- Masutani, C.; Kusumoto, R.; Yamada, A.; Dohmae, N.; Yokoi, M.; Yuasa, M.; Araki, M.; Iwai, S.; Takio, K.; Hanaoka, F. The XPV (xeroderma pigmentosum variant) gene encodes human DNA polymerase eta. Nature 1999, 399, 700–704. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.E.; Kondratick, C.M.; Prakash, S.; Prakash, L. hRAD30 mutations in the variant form of xeroderma pigmentosum. Science 1999, 285, 263–265. [Google Scholar] [CrossRef] [PubMed]
- Biertümpfel, C.; Zhao, Y.; Kondo, Y.; Ramón-Maiques, S.; Gregory, M.; Lee, J.Y.; Masutani, C.; Lehmann, A.R.; Hanaoka, F.; Yang, W. Structure and mechanism of human DNA polymerase η. Nature 2010, 465, 1044–1048. [Google Scholar] [CrossRef]
- Alt, A.; Lammens, K.; Chiocchini, C.; Lammens, A.; Pieck, J.C.; Kuch, D.; Hopfner, K.-P.; Carell, T. Bypass of DNA lesions generated during anticancer treatment with cisplatin by DNA polymerase eta. Science 2007, 318, 967–970. [Google Scholar] [CrossRef]
- Haracska, L.; Yu, S.L.; Johnson, R.E.; Prakash, L.; Prakash, S. Efficient and accurate replication in the presence of 7,8-dihydro-8-oxoguanine by DNA polymerase eta. Nat. Genet. 2000, 25, 458–461. [Google Scholar] [CrossRef]
- Zhao, Y.; Biertümpfel, C.; Gregory, M.T.; Hua, Y.-J.; Hanaoka, F.; Yang, W. Structural basis of human DNA polymerase η-mediated chemoresistance to cisplatin. Proc. Natl. Acad. Sci. USA 2012, 109, 7269–7274. [Google Scholar] [CrossRef]
- Batra, V.K.; Beard, W.A.; Shock, D.D.; Pedersen, L.C.; Wilson, S.H. Structures of DNA polymerase beta with active-site mismatches suggest a transient abasic site intermediate during misincorporation. Mol. Cell 2008, 30, 315–324. [Google Scholar] [CrossRef]
- Clausen, A.R.; Murray, M.S.; Passer, A.R.; Pedersen, L.C.; Kunkel, T.A. Structure-function analysis of ribonucleotide bypass by B family DNA replicases. Proc. Natl. Acad. Sci. USA 2013, 110, 16802–16807. [Google Scholar] [CrossRef]
- Patra, A.; Nagy, L.D.; Zhang, Q.; Su, Y.; Müller, L.; Guengerich, F.P.; Egli, M. Kinetics, structure, and mechanism of 8-Oxo-7,8-dihydro-2’-deoxyguanosine bypass by human DNA polymerase η. J. Biol. Chem. 2014, 289, 16867–16882. [Google Scholar] [CrossRef] [PubMed]
- Ouzon-Shubeita, H.; Baker, M.; Koag, M.C.; Lee, S. Structural basis for the bypass of the major oxaliplatin-DNA adducts by human DNA polymerase η. Biochem. J. 2019, 476, 747–758. [Google Scholar] [CrossRef] [PubMed]
- Gregory, M.T.; Park, G.Y.; Johnstone, T.C.; Lee, Y.-S.; Yang, W.; Lippard, S.J. Structural and mechanistic studies of polymerase η bypass of phenanthriplatin DNA damage. Proc. Natl. Acad. Sci. USA 2014, 111, 9133–9138. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.; Batra, V.K.; Pedersen, L.C.; Beard, W.A.; Wilson, S.H.; Pedersen, L.G. Incorrect nucleotide insertion at the active site of a G:A mismatch catalyzed by DNA polymerase. Proc. Natl. Acad. Sci. USA 2008, 105, 5670–5674. [Google Scholar] [CrossRef]
- Abashkin, Y.G.; Erickson, J.W.; Burt, S.K. Quantum Chemical Investigation of Enzymatic Activity in DNA Polymerase β. A Mechanistic Study. J. Phys. Chem. B 2001, 105, 287–292. [Google Scholar] [CrossRef]
- Zhao, Y.; Gregory, M.T.; Biertümpfel, C.; Hua, Y.-J.; Hanaoka, F.; Yang, W. Mechanism of somatic hypermutation at the WA motif by human DNA polymerase η. Proc. Natl. Acad. Sci. USA 2013, 110, 8146–8151. [Google Scholar] [CrossRef]
- Otwinowski, Z.; Minor, W. Processing of X-ray diffraction data collected in oscillation mode. Macromol. Crystallogr. A 1997, 276, 307–326. [Google Scholar]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef]
- Adams, P.D.; Afonine, P.V.; Bunkóczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.-W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 213–221. [Google Scholar] [CrossRef]
- Davis, I.W.; Leaver-Fay, A.; Chen, V.B.; Block, J.N.; Kapral, G.J.; Wang, X.; Murray, L.W.; Arendall, W.B.; Snoeyink, J.; Richardson, J.S.; et al. MolProbity: All-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007, 35, W375–W383. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Template:dNTP | Km (μM) | kcat (10−3 s−1) | kcat/Km (10−3 s−1μM−1) | fa | Replication Fidelity |
|---|---|---|---|---|---|
| dG:dCTP | 2.66 ± 0.29 | 120.6 ±6.1 | 45.6 | 1 | |
| dG:dTTP | 159.3 ± 2.7 | 74.8 ± 0.9 | 0.47 | 0.01 | 97 |
| Fm7dG:dCTP | 4.27 ± 0.42 | 56.4 ± 2.7 | 13.2 | 1 | |
| Fm7dG:dTTP | 52.5 ± 1.7 | 49.3 ± 0.05 | 0.94 | 0.07 | 14.3 |
| PDB CODE | Fm7dG:dCTP* (6UI2) |
| Data Collection | |
| space group | P61 |
| Cell Constants | |
| a (Å) b c α (°) β γ | 98.684 98.684 81.852 90 90 120 |
| resolution (Å)a | 20–2.34 (2.39–2.34) |
| Rmergeb (%) | 0.113 (0.441) |
| <I/σ> | 20.4 (5.28) |
| completeness (%) | 100.0 (100.0) |
| redundancy | 11.3 (11.3) |
| Refinement | |
| Rworkc/Rfreed (%) | 17.1/23.3 |
| unique reflections | 19,191 |
| Mean B Factor (Å2) | |
| protein | 24.94 |
| ligand | 23.51 |
| solvent | 26.49 |
| Ramachandran Plot | |
| most favored (%) | 96.2 |
| add. allowed (%) | 3.3 |
| RMSD bond lengths (Å) bond angles (degree) | 0.009 1.55 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koag, M.-C.; Jung, H.; Kou, Y.; Lee, S. Bypass of the Major Alkylative DNA Lesion by Human DNA Polymerase η. Molecules 2019, 24, 3928. https://doi.org/10.3390/molecules24213928
Koag M-C, Jung H, Kou Y, Lee S. Bypass of the Major Alkylative DNA Lesion by Human DNA Polymerase η. Molecules. 2019; 24(21):3928. https://doi.org/10.3390/molecules24213928
Chicago/Turabian StyleKoag, Myong-Chul, Hunmin Jung, Yi Kou, and Seongmin Lee. 2019. "Bypass of the Major Alkylative DNA Lesion by Human DNA Polymerase η" Molecules 24, no. 21: 3928. https://doi.org/10.3390/molecules24213928
APA StyleKoag, M.-C., Jung, H., Kou, Y., & Lee, S. (2019). Bypass of the Major Alkylative DNA Lesion by Human DNA Polymerase η. Molecules, 24(21), 3928. https://doi.org/10.3390/molecules24213928