Efflux Inhibitor Bicalutamide Increases Oral Bioavailability of the Poorly Soluble Efflux Substrate Docetaxel in Co-Amorphous Anti-Cancer Combination Therapy

 , ,
, ,  ,
,  and
and 
Abstract
1. Introduction
2. Results and Discussion
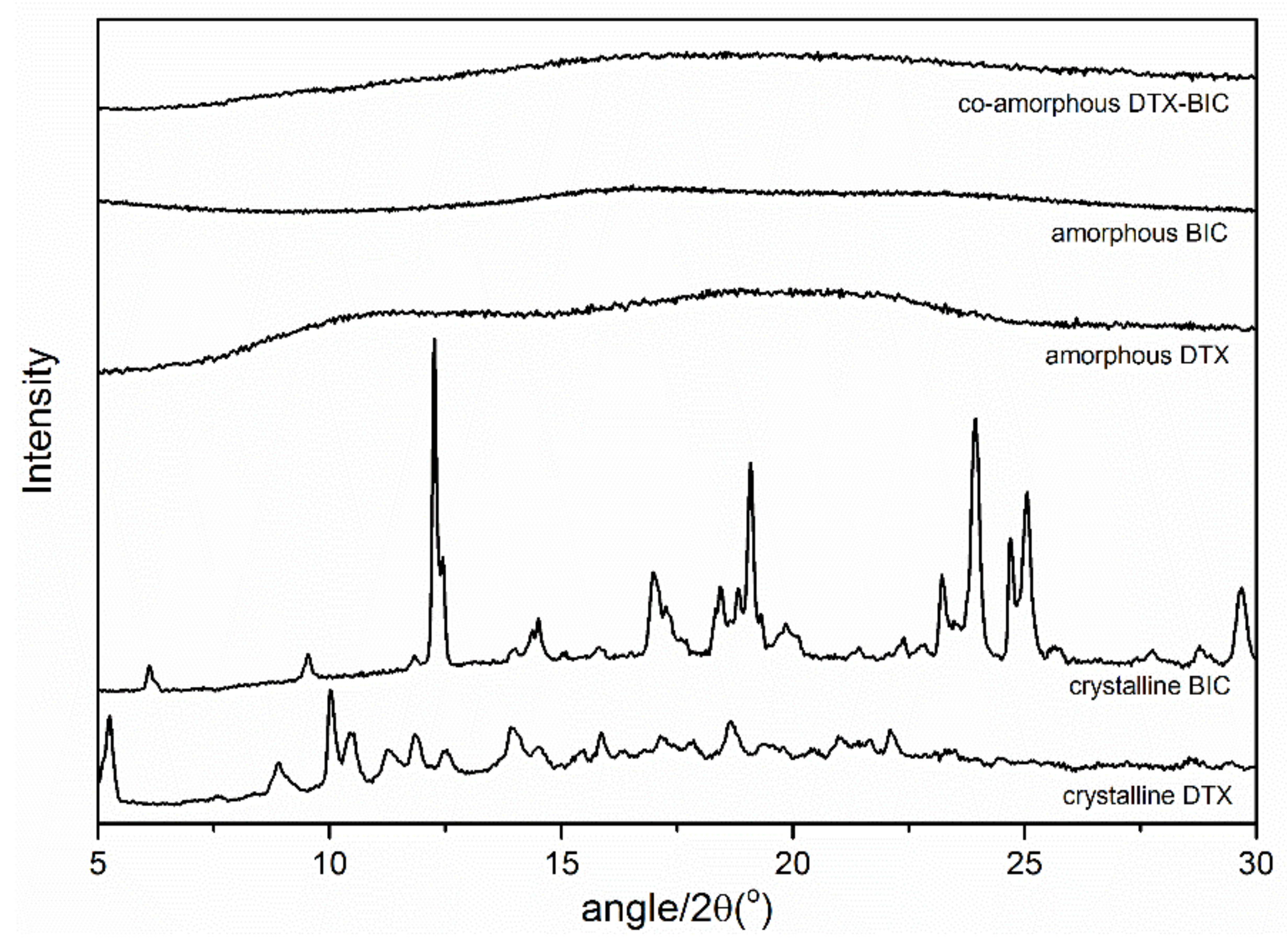
2.1. Solid State Characterization
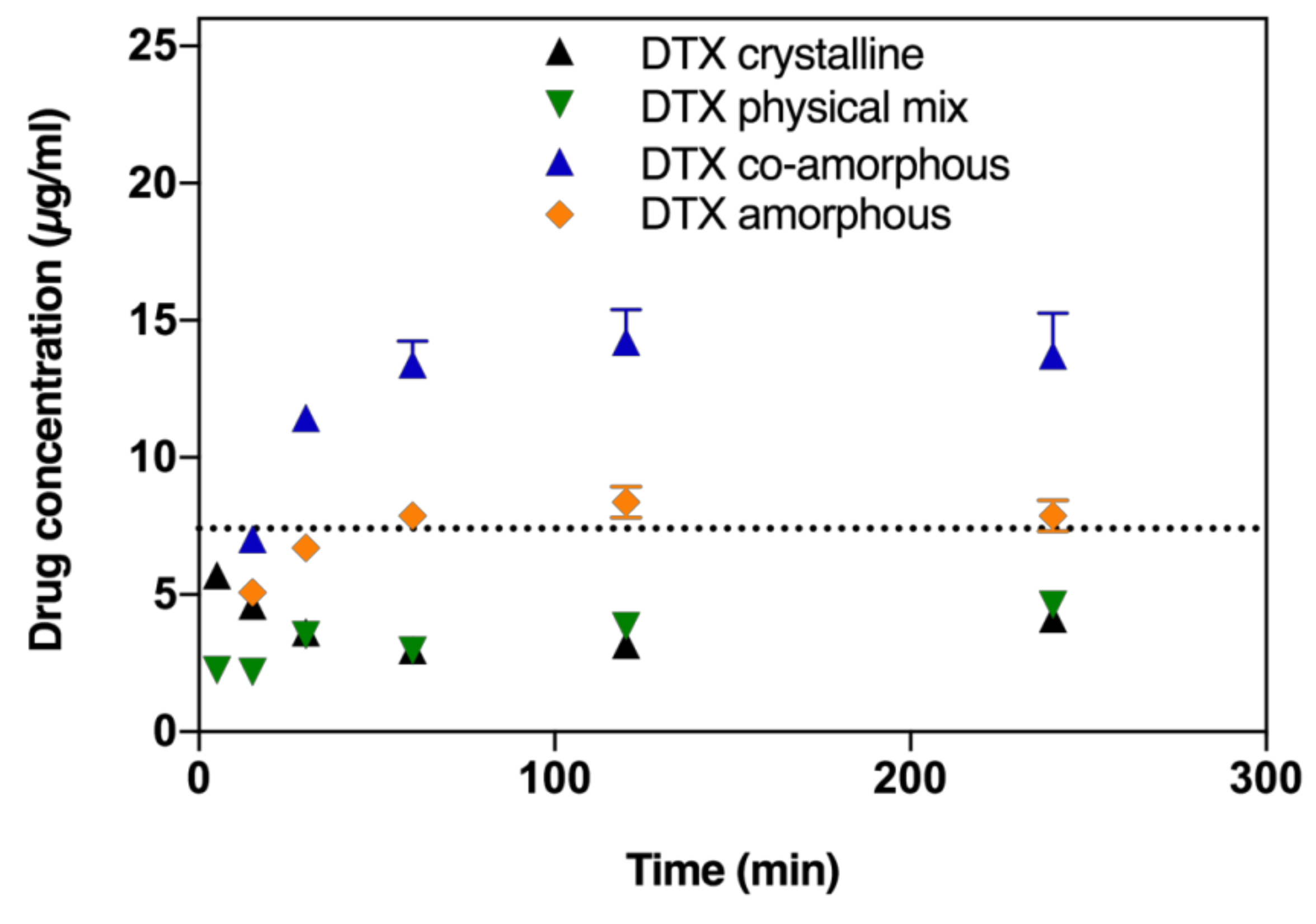
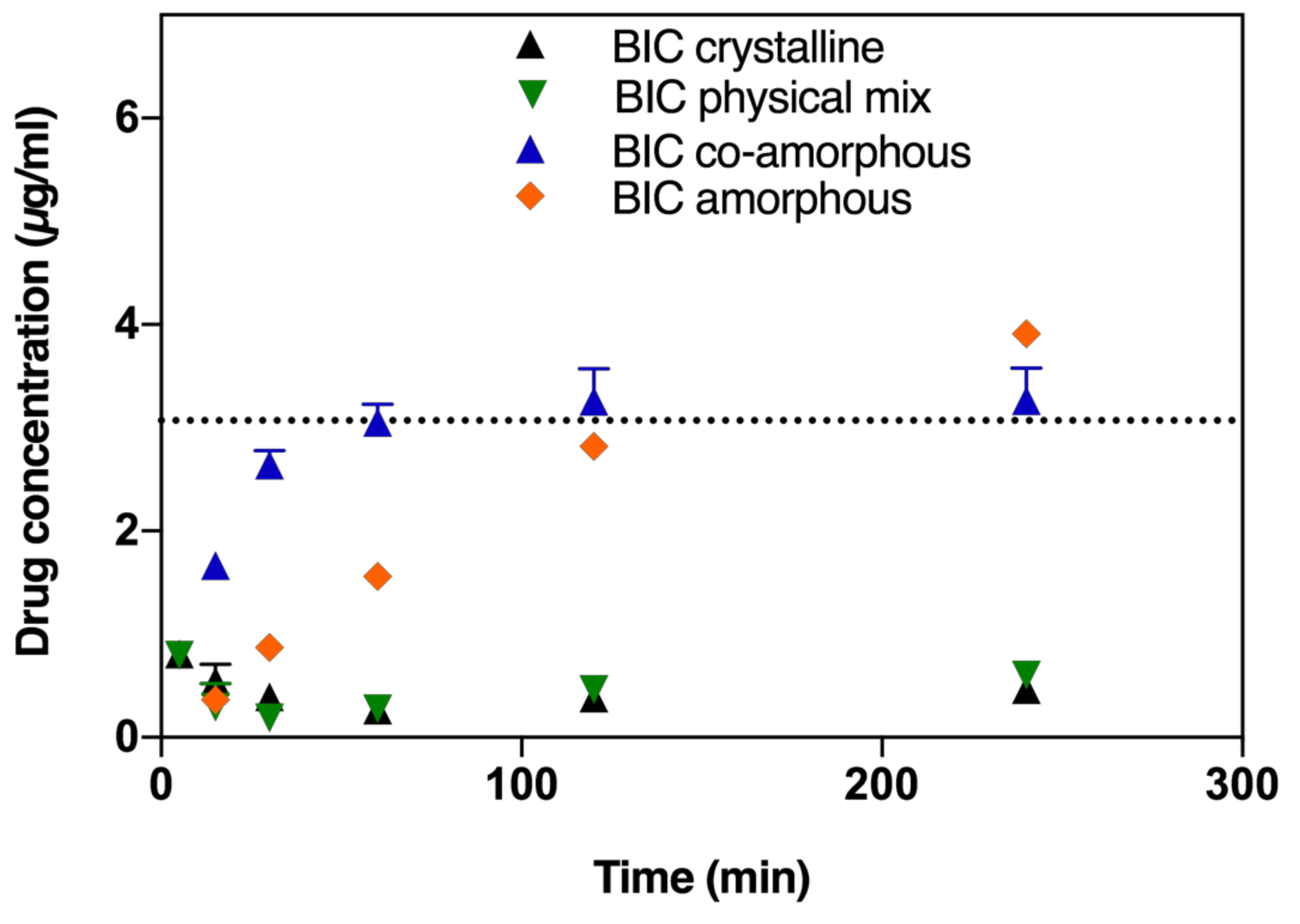
2.2. In Vitro Dissolution Testing
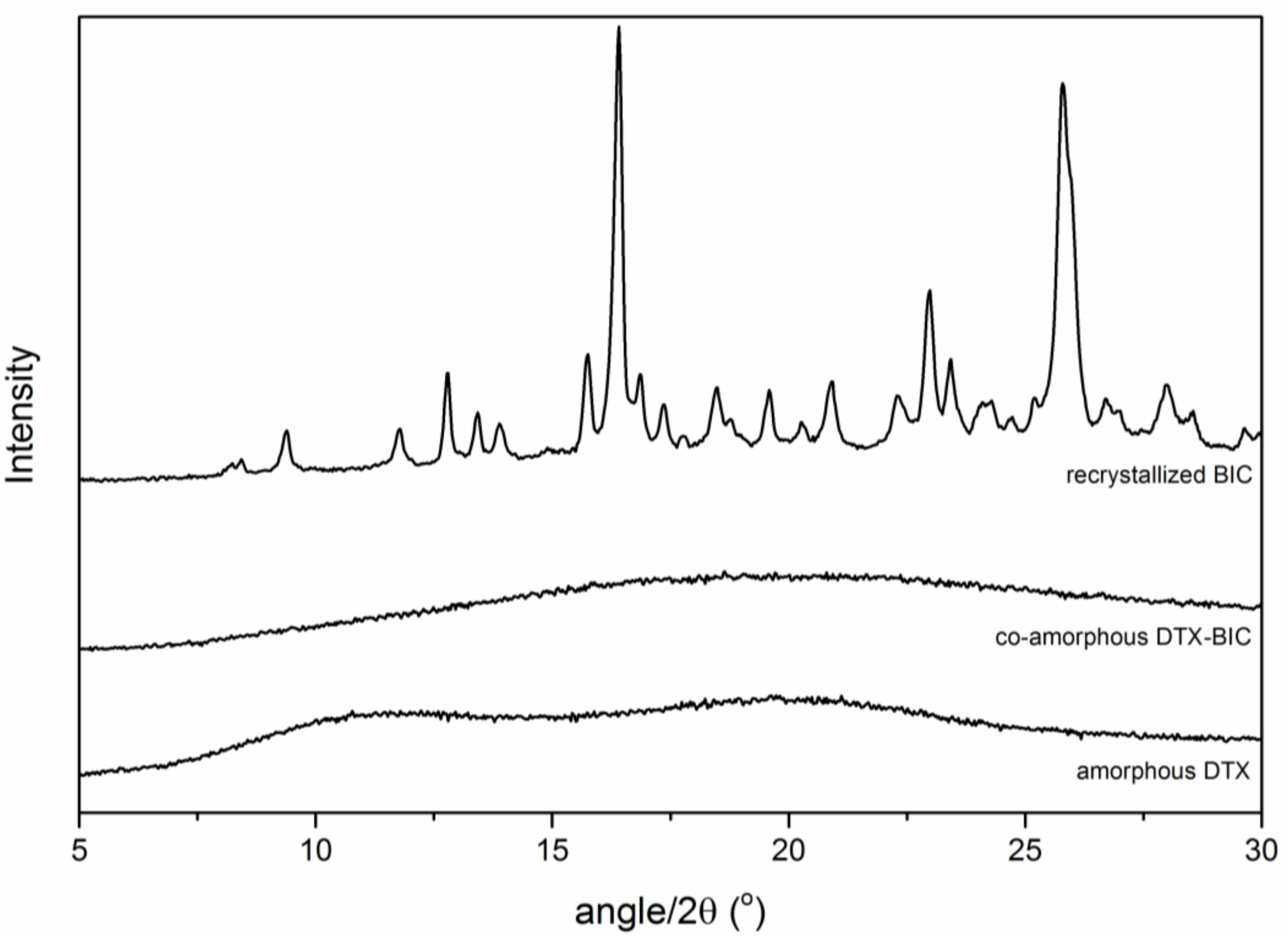
2.3. Physical Stability
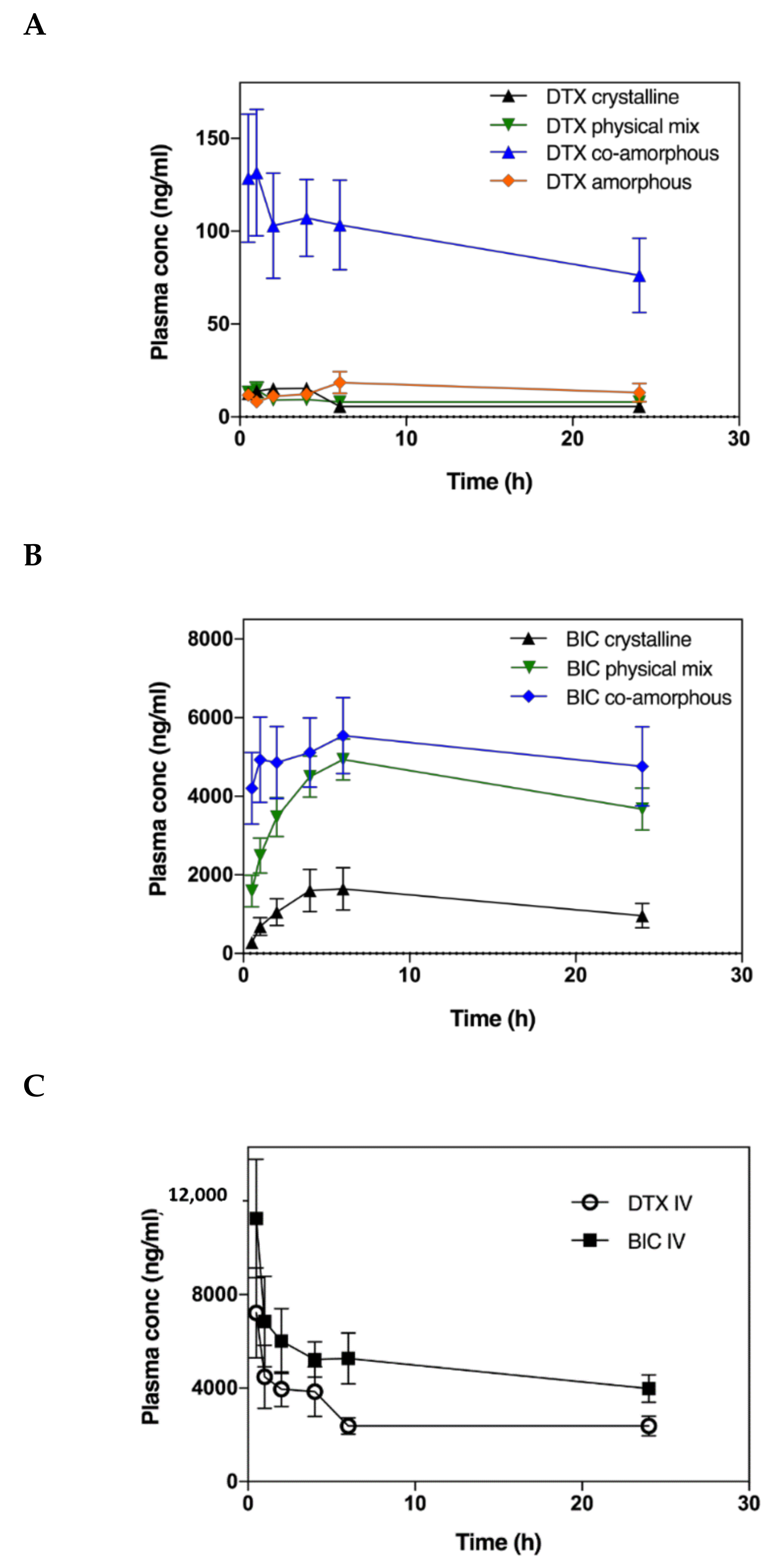
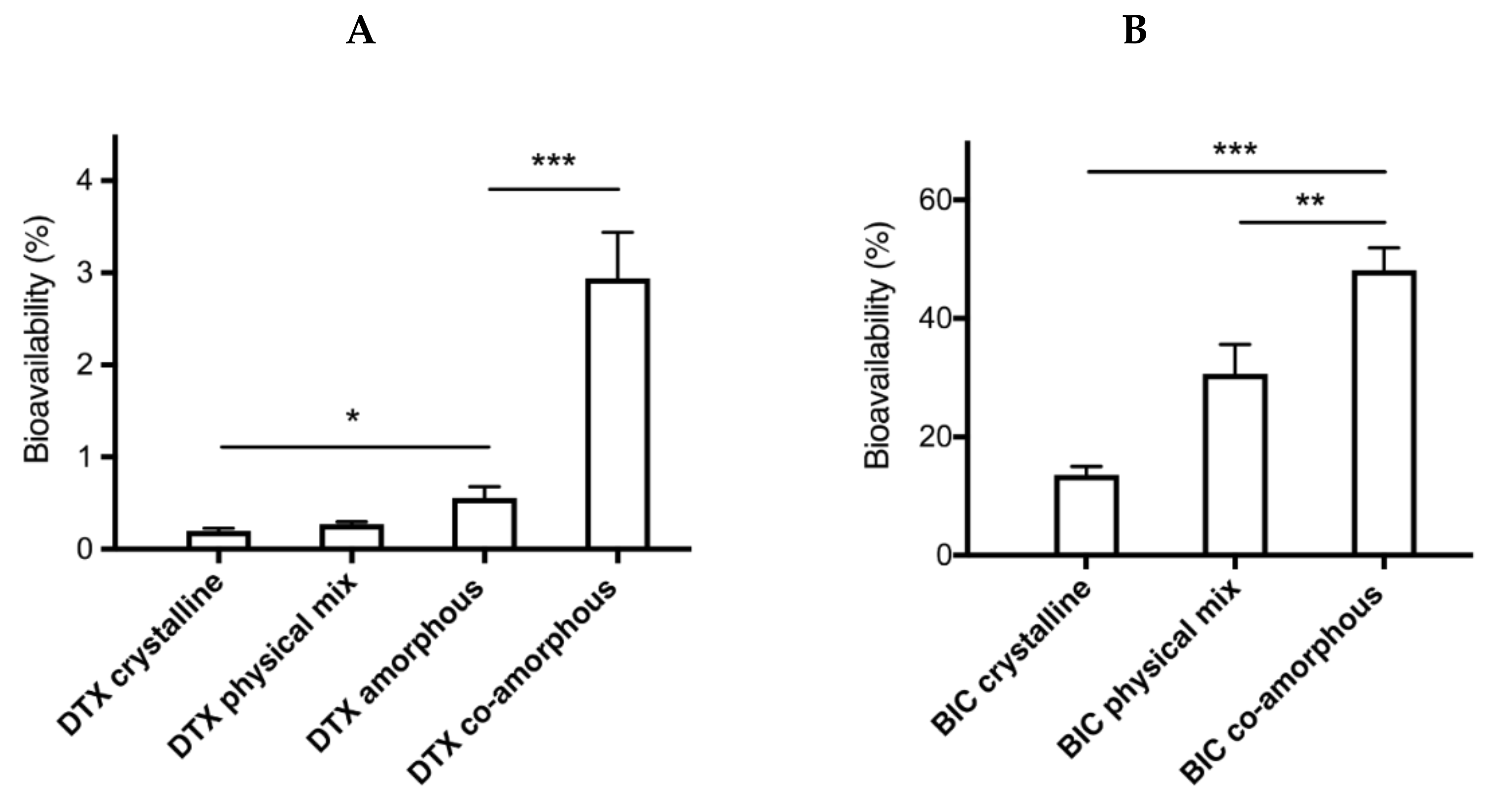
2.4. In Vivo Performance
3. Materials and Methods
3.1. Materials
3.2. Preparation of the Amorphous Materials
3.3. X-Ray Powder Diffraction (XRPD)
3.4. Differential Scanning Calorimetry (DSC)
3.5. Saturation Solubility Studies
3.6. Dissolution Studies
3.7. Stability Studies
3.8. In Vivo Pharmacokinetics Studies in Rats
3.9. Quantification of Plasma Samples
3.10. Pharmacokinetic Analysis
3.11. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sastry, S.V.; Nyshadham, J.R.; Fix, J.A. Recent technological advances in oral drug delivery–a review. Pharm. Sci. Technol. Today 2000, 3, 138–145. [Google Scholar] [CrossRef]
- Kalaria, D.; Sharma, G.; Beniwal, V.; Kumar, M.R. Design of biodegradable nanoparticles for oral delivery of doxorubicin: In vivo pharmacokinetics and toxicity studies in rats. Pharm. Res. 2009, 26, 492–501. [Google Scholar] [CrossRef] [PubMed]
- Hu, K.; Cao, S.; Hu, F.; Feng, J. Enhanced oral bioavailability of docetaxel by lecithin nanoparticles: Preparation, in vitro, and in vivo evaluation. Int. J. Nanomed. 2012, 7, 3537. [Google Scholar] [CrossRef] [PubMed]
- Malingré, M.M.; Beijnen, J.H.; Schellens, J.H. Oral delivery of taxanes. Invest. New Drugs 2001, 19, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Thanki, K.; Gangwal, R.P.; Sangamwar, A.T.; Jain, S. Oral delivery of anticancer drugs: Challenges and opportunities. J. Controlled Release 2013, 170, 15–40. [Google Scholar] [CrossRef] [PubMed]
- Gelderblom, H.; Verweij, J.; Nooter, K.; Sparreboom, A. Cremophor EL. Eur. J. Cancer 2001, 37, 1590–1598. [Google Scholar] [CrossRef]
- Schwander, B.; Ravera, S.; Giuliani, G.; Nuijten, M.; Walzer, S. Cost comparison of second-line treatment options for late stage non-small-cell lung cancer: Cost analysis for Italy. Clinico. Econo. Outcomes Res. 2012, 4, 237–243. [Google Scholar] [CrossRef]
- Martinez, M.N.; Amidon, G.L. A Mechanistic Approach to Understanding the Factors Affecting Drug Absorption: A Review of Fundamentals. J. Clin. Pharmacol. 2002, 42, 620–643. [Google Scholar] [CrossRef]
- Dahan, A.; Beig, A.; Lindley, D.; Miller, J.M. The solubility–permeability interplay and oral drug formulation design: Two heads are better than one. Adv. Drug Delivery Rev. 2016, 101, 99–107. [Google Scholar] [CrossRef]
- Vasconcelos, T.; Marques, S.; das Neves, J.; Sarmento, B. Amorphous solid dispersions: Rational selection of a manufacturing process. Adv. Drug Delivery Rev. 2016, 100, 85–101. [Google Scholar] [CrossRef]
- Laitinen, R.; Löbmann, K.; Strachan, C.J.; Grohganz, H.; Rades, T. Emerging trends in the stabilization of amorphous drugs. Int. J. Pharm. 2013, 453, 65–79. [Google Scholar] [CrossRef] [PubMed]
- Hancock, B.C.; Zografi, G. Characteristics and significance of the amorphous state in pharmaceutical systems. Int. J. Pharm. 1997, 86, 1–12. [Google Scholar] [CrossRef]
- Dengale, S.J.; Grohganz, H.; Rades, T.; Löbmann, K. Recent advances in co-amorphous drug formulations. Adv. Drug Delivery Rev. 2016, 100, 116–125. [Google Scholar] [CrossRef]
- Löbmann, K.; Grohganz, H.; Laitinen, R.; Strachan, C.; Rades, T. Amino acids as co-amorphous stabilizers for poorly water soluble drugs-Part 1: Preparation, stability and dissolution enhancement. Eur. J. Pharm. Biopharm. 2013, 85, 873–881. [Google Scholar] [CrossRef] [PubMed]
- Chieng, N.; Aaltonen, J.; Saville, D.; Rades, T. Physical characterization and stability of amorphous indomethacin and ranitidine hydrochloride binary systems prepared by mechanical activation. Eur. J. Pharm. Biopharm. 2009, 71, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Löbmann, K.; Strachan, C.; Grohganz, H.; Rades, T.; Korhonen, O.; Laitinen, R. Co-amorphous simvastatin and glipizide combinations show improved physical stability without evidence of intermolecular interactions. Eur. J. Pharm. Biopharm. 2012, 81, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Breedveld, P.; Beijnen, J.H.; Schellens, J.H.M. Use of P-glycoprotein and BCRP inhibitors to improve oral bioavailability and CNS penetration of anticancer drugs. Trends Pharmacol. Sci. 2006, 27, 17–24. [Google Scholar] [CrossRef]
- Varma, M.V.S.; Panchagnula, R. Enhanced oral paclitaxel absorption with vitamin E-TPGS: Effect on solubility and permeability in vitro, in situ and in vivo. Eur. J. Pharm. Sci. 2005, 25, 445–453. [Google Scholar] [CrossRef]
- Valicherla, G.R.; Dave, K.M.; Syed, A.A.; Riyazuddin, M.; Gupta, A.P.; Singh, A.; Mitra, K.; Datta, D.; Gayen, J.R. Formulation optimization of Docetaxel loaded self-emulsifying drug delivery system to enhance bioavailability and anti-tumor activity. Sci. Rep. 2016, 6, 26895. [Google Scholar] [CrossRef]
- Zhang, L.; Shen, Y.; Qiu, L. Loading docetaxel in β-cyclodextrin-based micelles for enhanced oral chemotherapy through inhibition of P-glycoprotein mediated efflux transport. RSC Adv. 2017, 7, 26161–26169. [Google Scholar] [CrossRef]
- Beyer, U.; Hofheinz, R.-D. Novel conjugation and combination strategies of mitomycin C with special focus on the current patent literature. Expert Opin. Ther. Patents 2005, 15, 1157–1168. [Google Scholar] [CrossRef]
- Cockshott, I.D. Bicalutamide. Clin. Pharmacokinet. 2004, 43, 855–878. [Google Scholar] [CrossRef] [PubMed]
- Baumhäkel, M.; Kasel, D.; Rao-Schymanski, R.; Böcker, R.; Beckurts, K.; Zaigler, M.; Barthold, D.; Fuhr, U. Screening for inhibitory effects of antineoplastic agents on CYP3A4 in human liver microsomes. Int. J. Clin. Pharmacol. Ther. 2001, 39, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Ghosal, A.; Chowdhury, S.K.; Tong, W.; Hapangama, N.; Yuan, Y.; Su, A.-D.; Zbaida, S. Identification of human liver cytochrome P450 enzymes responsible for the metabolism of lonafarnib (SarasarTM). Drug Metab. Dispos. 2006. [Google Scholar] [CrossRef] [PubMed]
- Yurdaydin, C.; Idilman, R.; Kalkan, C.; Karakaya, F.; Caliskan, A.; Keskin, O.; Karatayli, E.; Karatayli, S.; Bozdayi, A.M.; Koh, C. Exploring optimal dosing of Lonafarnib with Ritonavir for the treatment of chronic Delta hepatitis-Interim results from the lowr HDV-2 study. Hepatology 2016, 64, 910A. [Google Scholar]
- Chiou, W.L.; Riegelman, S. Pharmaceutical applications of solid dispersion systems. J. Pharm. Sci. 1971, 60, 1281–1302. [Google Scholar] [CrossRef]
- Löbmann, K.; Laitinen, R.; Grohganz, H.; Gordon, K.C.; Strachan, C.; Rades, T. Coamorphous Drug Systems: Enhanced Physical Stability and Dissolution Rate of Indomethacin and Naproxen. Mol. Pharmaceutics 2011, 8, 1919–1928. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DTX | BIC | |||||||
|---|---|---|---|---|---|---|---|---|
| AUC (ng/mL × 24h) | Fa (%) | Cmax (ng/mL) | Tmax (h) | AUC (µg/mL × 24h) | Fa (%) | Cmax (ng/mL) | Tmax (h) | |
| Crystalline | 185 ± 29 | 0.20 ± 0.03 | 15 | 4 | 43.06 ± 4.79 | 14 ± 1 | 1664 | 6 |
| Physical mix | 253 ± 29 | 0.27 ± 0.02 | 15 | 1 | 97.21 ± 15.80 | 31 ± 5 | 4937 | 6 |
| Co-amorphous | 2188 ± 264 | 2.94 ± 0.50 | 132 | 1 | 138.72 ± 11.12 | 48 ± 4 | 5542 | 6 |
| Amorphous | 518 ± 113 | 0.56 ± 0.12 | 19 | 6 | * N/A | N/A | N/A | N/A |
| Intravenous | 93,196 ± 12,626 | N/A | 7212 | 0.5 | 317,208 ± 20,888 | N/A | 11,253 | 0.5 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bohr, A.; Nascimento, T.L.; Harmankaya, N.; Weisser, J.J.; Wang, Y.; Grohganz, H.; Rades, T.; Löbmann, K. Efflux Inhibitor Bicalutamide Increases Oral Bioavailability of the Poorly Soluble Efflux Substrate Docetaxel in Co-Amorphous Anti-Cancer Combination Therapy. Molecules 2019, 24, 266. https://doi.org/10.3390/molecules24020266
Bohr A, Nascimento TL, Harmankaya N, Weisser JJ, Wang Y, Grohganz H, Rades T, Löbmann K. Efflux Inhibitor Bicalutamide Increases Oral Bioavailability of the Poorly Soluble Efflux Substrate Docetaxel in Co-Amorphous Anti-Cancer Combination Therapy. Molecules. 2019; 24(2):266. https://doi.org/10.3390/molecules24020266
Chicago/Turabian StyleBohr, Adam, Thais Leite Nascimento, Necati Harmankaya, Johan Juhl Weisser, Yingya Wang, Holger Grohganz, Thomas Rades, and Korbinian Löbmann. 2019. "Efflux Inhibitor Bicalutamide Increases Oral Bioavailability of the Poorly Soluble Efflux Substrate Docetaxel in Co-Amorphous Anti-Cancer Combination Therapy" Molecules 24, no. 2: 266. https://doi.org/10.3390/molecules24020266
APA StyleBohr, A., Nascimento, T. L., Harmankaya, N., Weisser, J. J., Wang, Y., Grohganz, H., Rades, T., & Löbmann, K. (2019). Efflux Inhibitor Bicalutamide Increases Oral Bioavailability of the Poorly Soluble Efflux Substrate Docetaxel in Co-Amorphous Anti-Cancer Combination Therapy. Molecules, 24(2), 266. https://doi.org/10.3390/molecules24020266