Synthesis, In Vitro Screening and Docking Studies of New Thiosemicarbazide Derivatives as Antitubercular Agents

 , , , , and
, , , , and
Abstract
1. Introduction
2. Experimental
2.1. Synthesis
2.1.1. General Procedure for the Synthesis of 1-(pyridin-2-,3-,4-yl)carbonyl-4-substituted Thiosemicarbazide (1–19)
2.1.2. General Procedure for the Synthesis of 1-(pyridin-4-ylacetyl)-4-substituted Thiosemicarbazide (20–25)
2.2. X-ray Structure Determination
2.3. Antimycobacterial Assay
2.4. Computational Details
3. Results and Discussion
3.1. Chemistry
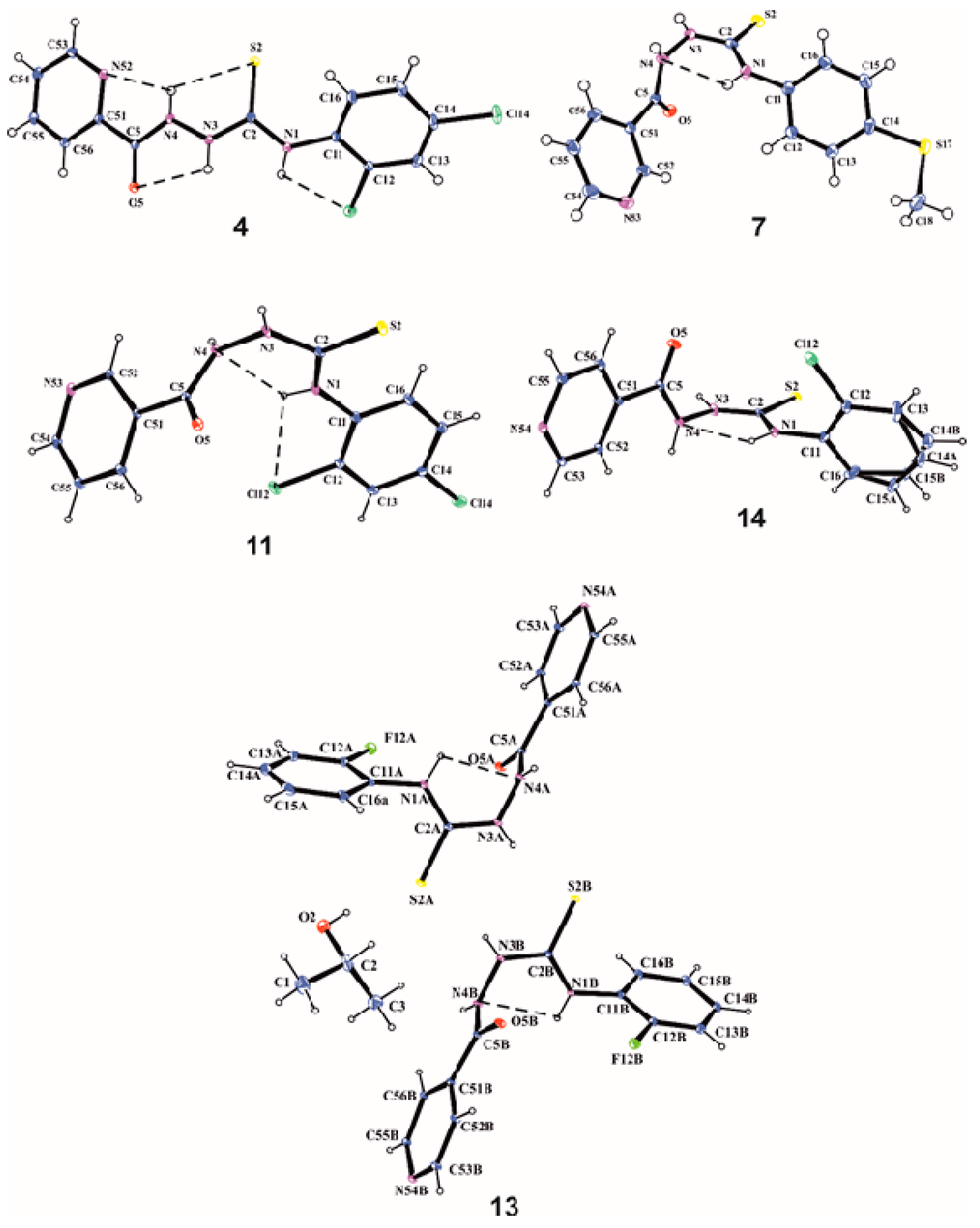
3.2. X-Ray Analysis
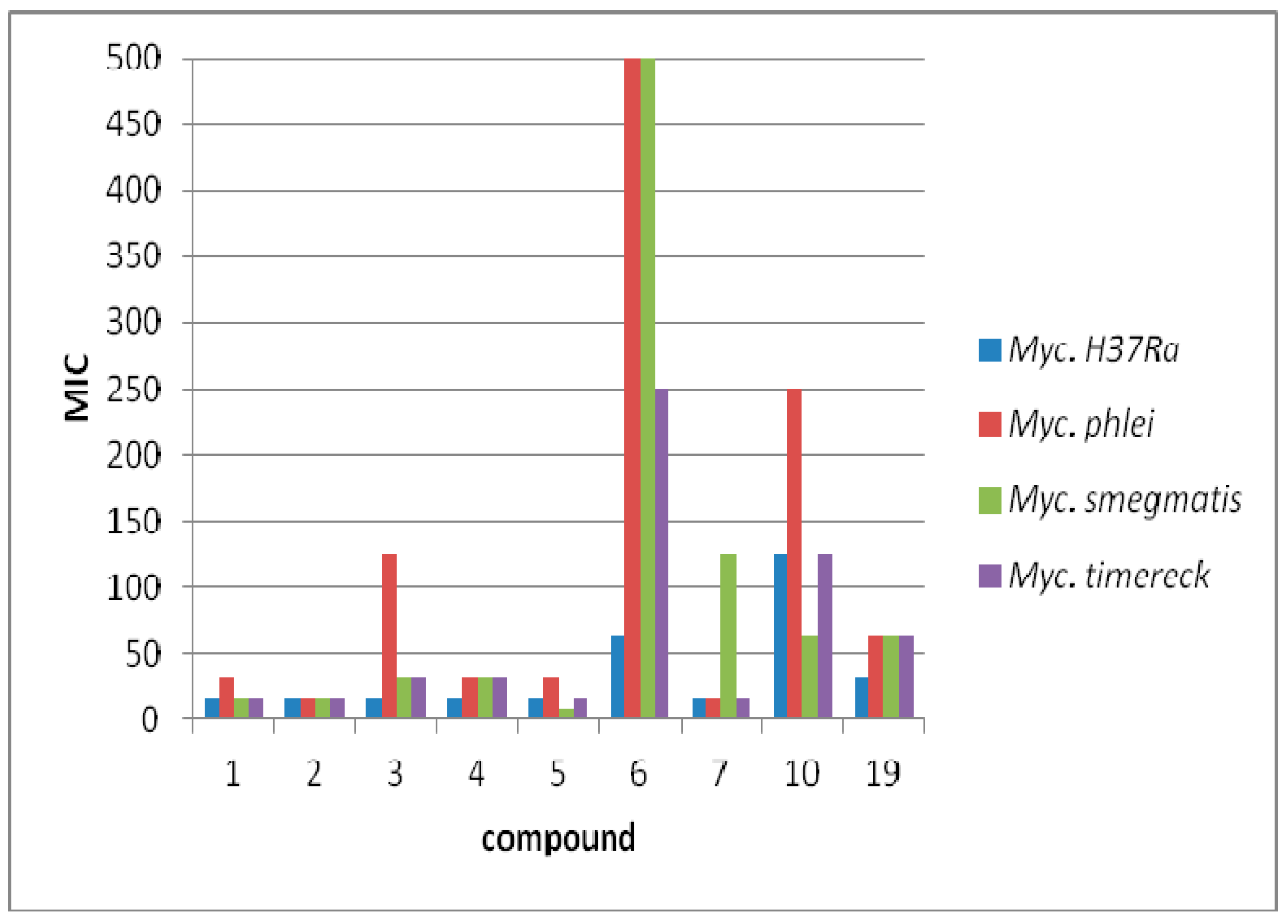
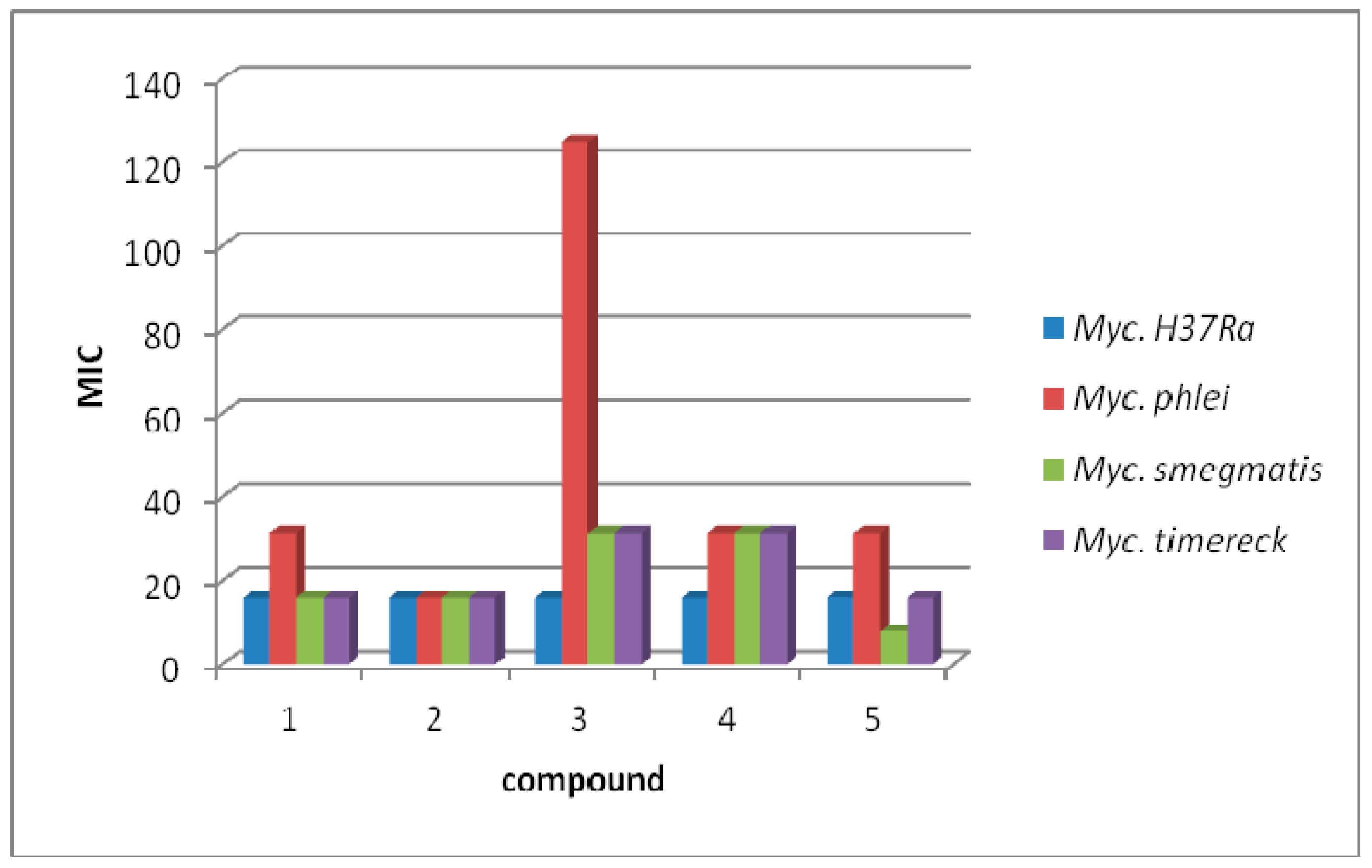
3.3. Antimycobacterial Activity
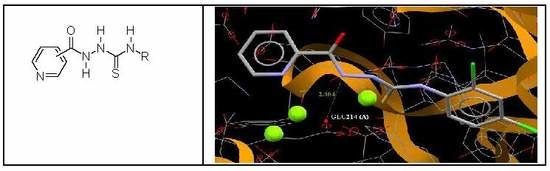
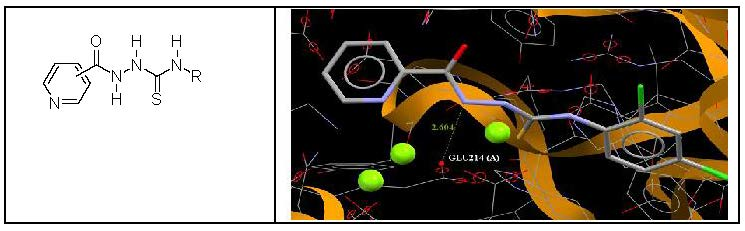
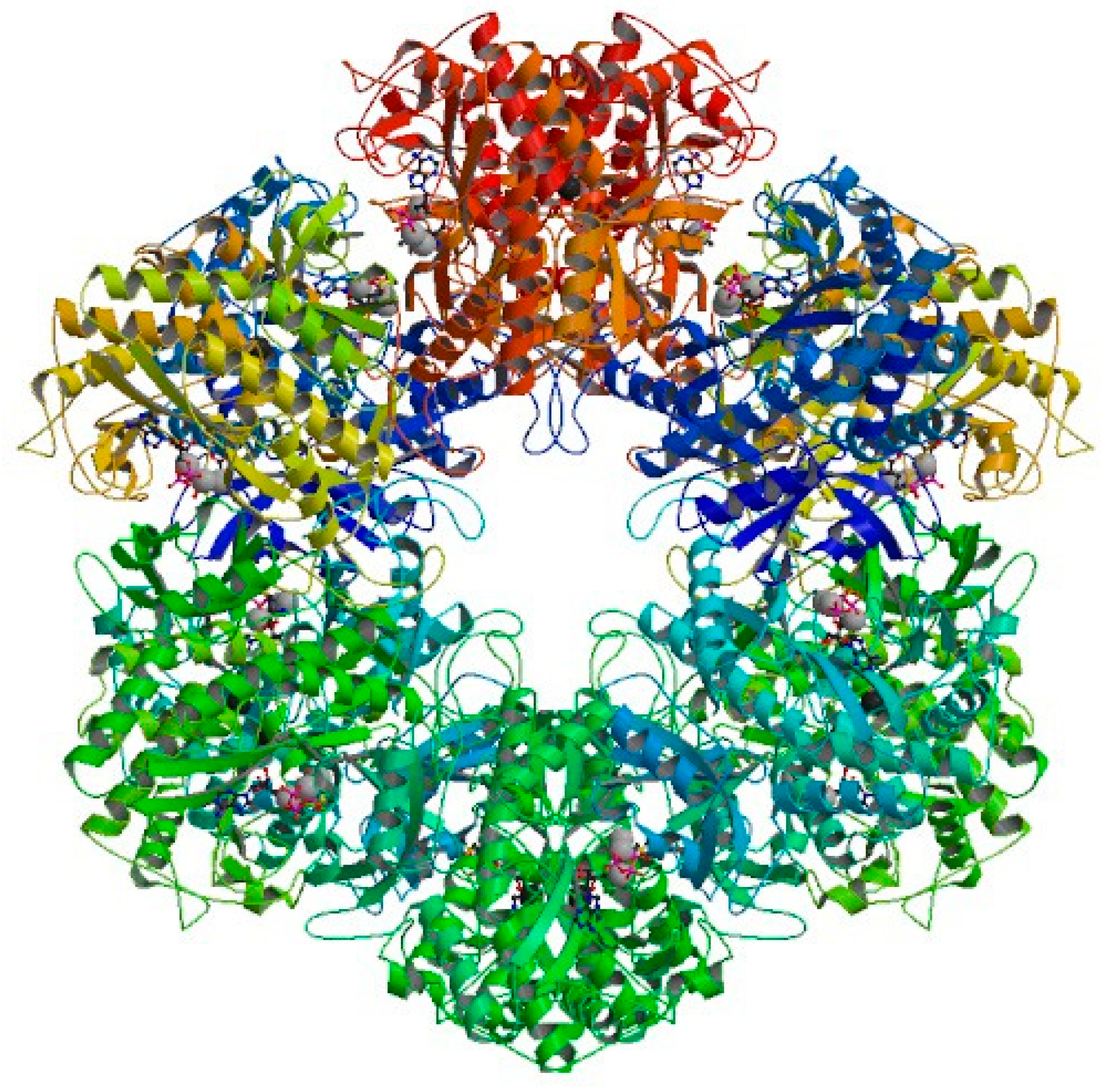
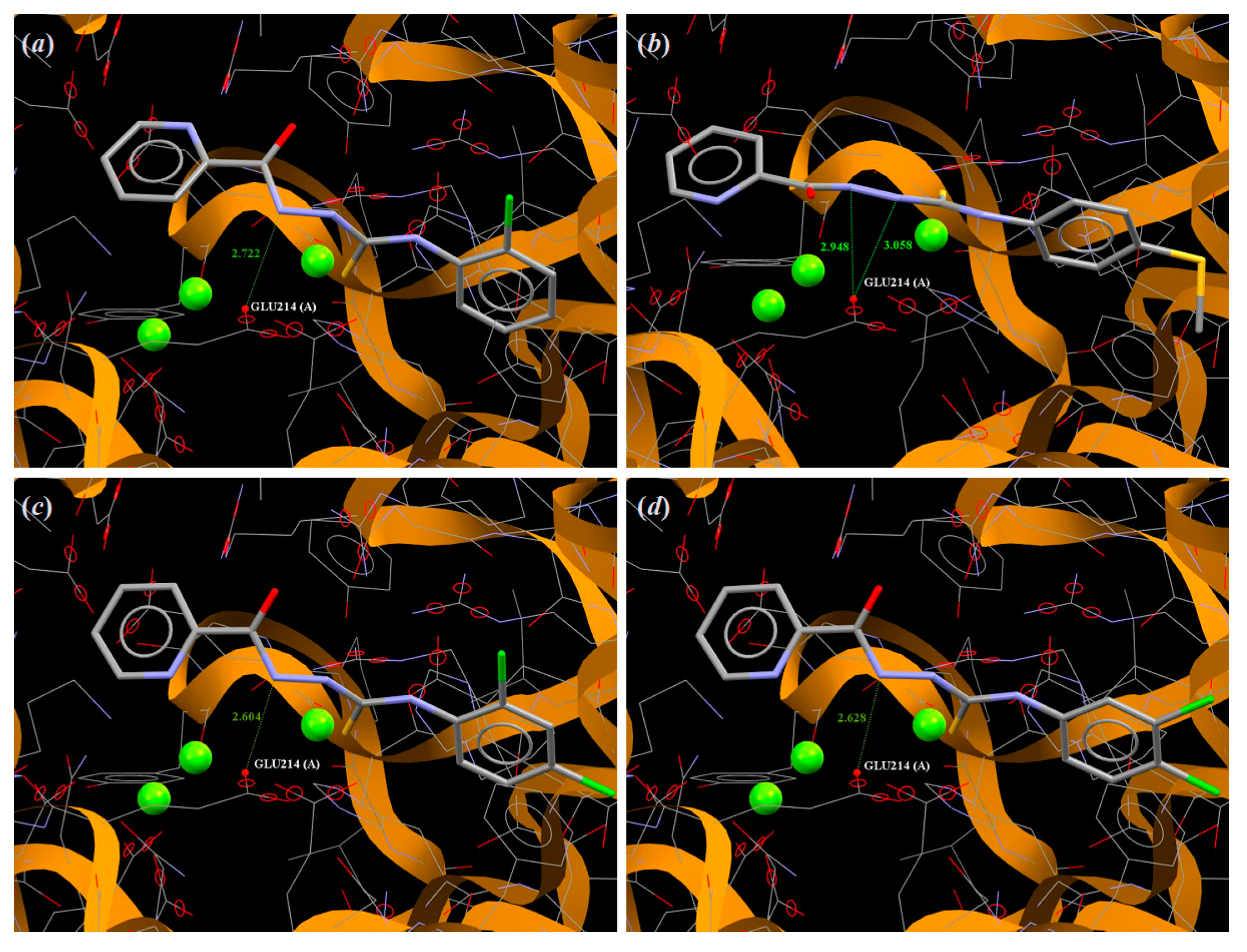
3.4. Molecular Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Global Tuberculosis Report 2016; World Health Organization: Geneva, Switzerland, 2016. [Google Scholar]
- Long, R.; Ellis, E. Introducing the sixth edition of the Canadian Tuberculosis Standards. Can. J. Infect. Dis. Med. Microbiol. 2007, 18, 283–284. [Google Scholar] [CrossRef] [PubMed]
- WHO/Europe | Tuberculosis. Available online: http://www.euro.who.int/tb (accessed on 23 August 2017).
- European Lung Foundation ELF. Available online: http://www.europeanlung.org (accessed on 20 August 2017).
- Muller, B.; Borrell, S.; Rose, G.; Gagneux, S. The heterogeneous evolution of multidrug-resistant Mycobacterium tuberculosis. Trends Genet. 2013, 29, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Zumla, A.I.; Gillespie, S.H.; Hoelscher, M.; Philips, P.P.; Cole, S.T.; Abubakar, I.; McHugh, T.D.; Schito, M.; Maeurer, M.; Nunn, A.J. New antituberculosis drugs, regimens, and adjunct therapies: Needs, advances, and future prospects. Lancet Infect. Dis. 2014, 14, 327–340. [Google Scholar] [CrossRef]
- Hu, Y.Q.; Zhang, S.; Zhao, F.; Gao, C.; Feng, L.S.; Lv, Z.S.; Xu, Z.; Wu, X. Isoniazid derivatives and their anti-tubercular activity. Eur. J. Med. Chem. 2017, 133, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Rane, R.A.; Naphade, S.S.; Bangalore, P.K.; Palkar, M.B.; Shaikh, M.S.; Karpoormath, R. Synthesis of novel 4-nitropyrrole-based semicarbazide and thiosemicarbazide hybrids with antimicrobial and anti-tubercular activity. Bioorg. Med. Chem. Lett. 2014, 24, 3079–3083. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.R.; Gangwal, R.; Sangamwar, A.T.; Jain, R. Synthesis, biological evaluation and 3D-QSAR study of hydrazide, semicarbazide and thiosemicarbazide derivatives of 4-(adamantan-1-yl)quinoline as anti-tuberculosis agents. Eur. J. Med. Chem. 2014, 85, 255–267. [Google Scholar] [CrossRef]
- Pitucha, M.; Wos, M.; Miazga-Karska, M.; Klimek, K.; Miroslaw, B.; Pachuta-Stec, A.; Gladysz, A.; Ginalska, G. Synthesis, antibacterial and antiproliferative potential of some new 1-pyridinecarbonyl-4-substituted thiosemicarbazide derivatives. Med. Chem. Res. 2016, 25, 1666–1677. [Google Scholar] [CrossRef]
- Wos, M.; Miazga-Karska, M.; Kaczor, A.A.; Klimek, K.; Karczmarzyk, Z.; Kowalczuk, D.; Wysocki, W.; Ginalska, G.; Urbanczyk-Lipkowska, Z.; Morawiak, M.; et al. Novel thiosemicarbazide derivatives with 4-nitrophenyl group as multi-target drugs: Alpha-glucosidase inhibitors with antibacterial and antiproliferative activity. Biomed. Pharmacother. 2017, 93, 1269–1276. [Google Scholar] [CrossRef]
- Pitucha, M.; Wujec, M.; Dobosz, M. Synthesis of 3-(pyridin-4-ylmethyl)-4-substituted- 1,2,4-triazoline-5-thione. J. Chin. Chem. Soc. 2013, 54, 69–73. [Google Scholar] [CrossRef]
- Moir, D.T.; Di, M.; Wong, E.; Moore, R.A.; Schweizer, H.P.; Woods, D.E.; Bowlin, T.L. Development and application of a cellular, gain-of-signal, bioluminescent reporter screen for inhibitors of type II secretion in Pseudomonas aeruginosa and Burkholderia pseudomallei. J. Biomol. Screen. 2011, 16, 694–705. [Google Scholar] [CrossRef]
- Hu, G.Q.; Li, S.; Huang, W.L.; Wang, H. Synthesis and bioactivity of arecoline derivatives containing 1,2,4-triazone. Youji Huaxue 2002, 22, 667–671. [Google Scholar]
- Baxendale, I.R.; Steven, V.; Ley, S.V.; Martinelli, M. The rapid preparation of 2-aminosulfonamide-1,3,4-oxadiazoles using polymer-supported reagents and microwave heating. Tetrahedron 2005, 61, 5323–5349. [Google Scholar] [CrossRef]
- Bhaskar, C.S.; Vidhale, N.N.; Berad, B.N. Synthesis of some γ-picolinyl-1,2,4,5-dithiadiazine and their antimicrobial activity. Asian J. Chem. 2002, 14, 162–168. [Google Scholar]
- Riccieri, F.M.; Porcelli, G.A.; Castellani, P.M. Thiourea derivatives and their antitubercular activity. Farmaco 1967, 22, 114–120. [Google Scholar]
- CrysAlisPRO Software System, Version 1.171.39.16b; Rigaku Oxford Diffraction, Rigaku Corporation: Oxford, UK, 2015.
- Sheldrick, G. A short history of SHELX. Acta Crystallogr. Sect. A Found. Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Flack, H. On enantiomorph-polarity estimation. Acta Crystallogr. Sect. A Found. Crystallogr. 1983, 39, 876–881. [Google Scholar] [CrossRef]
- Farrugia, L. WinGX and ORTEP for Windows: An update. J. Appl. Cryst. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Krajewski, W.W.; Jones, T.A.; Mowbray, S.L. Structure of Mycobacterium tuberculosis glutamine synthetase in complex with a transition-state mimic provides functional insights. Proc. Natl. Acad. Sci. USA 2005, 102, 10499–10504. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef]
- Becke, A.D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5653. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision, D.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Maliszewska-Guz, A.; Wujec, M.; Pitucha, M.; Dobosz, M.; Chodkowska, A.; Jagiełło-Wójtowicz, E.; Mazur, L.; Kozioł, A.E. Cyclization of 1-{[4-methyl-4H-1,2,4-triazol-3-yl) sulfanyl]acetyl}thiosemicarbazides to 1,2,4-triazole and 1,3,4-thiadiazole derivatives and their pharmacological properties. Collect. Czech. Chem. Commun. 2005, 70, 61–62. [Google Scholar] [CrossRef]
- Bulut, N.; Kocyigit, U.M.; Gecibesler, I.H.; Dastan, T.; Karci, H.; Taslimi, P.; Durna, D.S.; Gulcin, I.; Cetin, A. Synthesis of some novel pyridine compounds containing bis-1,2,4-triazole/thiosemicarbazide moiety and investigation of their antioxidant properties, carbonic anhydrase, and acetylcholinesterase enzymes inhibition profiles. J. Biochem. Mol. Toxicol. 2018, 32, e22006. [Google Scholar] [CrossRef] [PubMed]
- Pitucha, M.; Polak, B.; Swatko-Ossor, M.; Popiołek, Ł.; Ginalska, G. Determination of the lipophilicity of some new derivatives of thiosemicarbazide and 1,2,4-triazoline-5-thione with potential antituberculosis activity. Croat. Chem. Acta 2010, 83, 299–306. [Google Scholar]
- Allen, F.H.; Kennard, O.; Watson, D.G.; Brammer, L.; Orpen, A.G.; Taylor, R. Tables of bond lengths determined by X-ray and neutron diffraction. Part 1. Bond lengths in organic compounds. J. Chem. Soc. Perkin Trans. 1987, 12, S1–S19. [Google Scholar] [CrossRef]
- Mowbray, L.S.; Kathiravan, K.M.; Pandey, A.A.; Odell, R.L. Inhibition of Glutamine Synthetase: A Potential Drug Target in Mycobacterium tuberculosis. Molecules 2014, 19, 13161–13176. [Google Scholar] [CrossRef] [PubMed]
- Harth, G.; Horwitz, M.A. Inhibition of Mycobacterium tuberculosis Glutamine Synthetase as a novel antibiotic strategy against tuberculosis: Demonstration of efficacy in vivo. Infect. Immun. 2003, 71, 456–464. [Google Scholar] [CrossRef]
- Harth, G.; Horwitz, M.A. An inhibitor of exported Mycobacterium tuberculosis glutamine synthetase selectively blocks the growth of pathogenic mycobacteria in axenic culture and in human monocytes: Extracellular proteins as potential novel drug targets. J. Exp. Med. 1999, 189, 1425–1436. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 1–25 are available from the correspondence author. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Torsion Angle | 4 | 7 | 11 | 13A | 13B | 14 |
|---|---|---|---|---|---|---|
| C11–N1–C2–N3 | 166.21(15) | 179.4(3) | −176.89(19) | 172.9(2) | −171.1(2) | −178.8(3) |
| N1–C2–N3–N4 | 162.99(14) | −11.0(5) | 3.5(2) | −10.7(3) | 9.6(3) | 5.1(4) |
| C2–N3–N4–C5 | −154.75(15) | −69.1(4) | 92.1(2) | −87.1(3) | 86.2(3) | 94.4(3) |
| N3–N4–C5–C51 | 176.16(13) | 169.8(3) | −176.47(15) | −179.79(19) | 179.15(19) | 178.3(2) |
| C11–N1–C2–S2 | −15.1(2) | 1.0(5) | 4.4(3) | −9.9(3) | 6.0(3) | 3.1(4) |
| N4–N3–C2–S2 | −15.7(2) | 167.5(3) | −177.69(12) | 171.92(16) | −174.28(17) | −176.7(2) |
| N52–C51–C5–O5 | −177.25(15) | - | - | - | - | - |
| C52–C51–C5–O5 | - | 35.0(5) | 150.14(17) | 8.7(3) | −7.0(3) | 177.2(3) |
| C56–C51–C5–O5 | 5.1(2) | −148.9(4) | −29.1(2) | −175.5(2) | 177.9(2) | −7.3(4) |
| D–H…A | D–H | H…A | D…A | D–H…A |
|---|---|---|---|---|
| 4 | ||||
| N1–H1…Cl12 | 0.79(2) | 2.69(2) | 2.9681(16) | 103.2(18) |
| N3–H3…O5 | 0.85(2) | 2.34(2) | 2.6787(19) | 104.3(17) |
| N4–H4…S2 | 0.83(2) | 2.56(2) | 2.9497(16) | 109.9(17) |
| N4–H4…N52 | 0.83(2) | 2.31(2) | 2.644(2) | 104.8(17) |
| N1–H1…O5 i | 0.79(2) | 2.12(2) | 2.849(2) | 155(2) |
| N3–H3…O5 i | 0.85(2) | 2.02(2) | 2.806(2) | 153.2(19) |
| i = −x, 2-y, −z | ||||
| 7 | ||||
| N1–H1…N4 | 0.86(2) | 2.27(5) | 2.658(4) | 107(4) |
| N1–H1…O5 i | 0.86(5) | 2.11(5) | 2.904(4) | 154(4) |
| N3–H3…N53 ii | 0.80(6) | 2.07(5) | 2.842(5) | 162(5) |
| N4–H4…S2 i | 0.94(5) | 2.44(5) | 3.337(3) | 160(4) |
| i = x, −y, ½ + z; ii = 1 + x, y, z | ||||
| 11 | ||||
| N1–H1…N4 | 0.82(2) | 2.21(2) | 2.637(2) | 113(2) |
| N1–H1…Cl12 | 0.82(2) | 2.49(2) | 2.9154(17) | 114(2) |
| N1–H1…O5 i | 0.82(2) | 2.49(2) | 3.159(2) | 140(2) |
| N3–H3…N53 ii | 0.89(2) | 1.98(2) | 2.860(2) | 172(2) |
| N4–H4…O5 i | 0.85(3) | 2.04(3) | 2.792(2) | 147(2) |
| i = −1 + x, y, z; ii = 1 + x, ½ − y, ½ + z | ||||
| 13 | ||||
| N1A–H1A…N4A | 0.89(4) | 2.31(3) | 2.697(3) | 106(3) |
| N1B–H1B…N4B | 0.81(4) | 2.38(3) | 2.701(3) | 105(3) |
| N1A–H1A…N54B | 0.89(4) | 2.05(4) | 2.883(3) | 155(3) |
| N1B–H1B…N54A | 0.81(4) | 2.12(4) | 2.896(3) | 160(3) |
| O2–H1…S2A i | 0.82 | 2.55 | 3.364(2) | 171 |
| N3A–H3A…S2B ii | 0.84(3) | 2.48(3) | 3.297(2) | 166(3) |
| N3B–H3B…S2A iii | 0.82(3) | 2.49(3) | 3.291(2) | 166(3) |
| N4A–H4A…O5A i | 0.78(3) | 2.17(3) | 2.867(2) | 149(3) |
| N4B–H4B…O5B iv | 0.84(3) | 2.09(3) | 2.849(3) | 151(3) |
| i = 3/2−x, ½ + −y, ½ − z; ii = 1 + x, y, z; iii = −1 + x, y, z; iv = ½ − x, −1/2 + y; ½ − z | ||||
| 14 | ||||
| N1–H1…N4 | 0.89(4) | 2.37(4) | 2.711(4) | 103(3) |
| N1–H1…N54 i | 0.89(4) | 2.07(4) | 2.930(3) | 163(4) |
| N3–H3…S2 ii | 1.03(4) | 2.31(4) | 3.327(3) | 170(3) |
| N4–H4…O5 iii | 0.86(5) | 2.06(5) | 2.851(4) | 151(4) |
| i = ½-x, 3/2-y, 1-z; ii = ½-x, 3/2-y, -z; ii = ½-x, ½+y, ½-z | ||||
| No | Inhibition Zone (mm) 250 μg of Compound Per Well | |||
|---|---|---|---|---|
| M. H37Ra | M. phlei | M. smegmatis | M. timereck | |
| 1 | 28.5 | 28.1 | 28.3 | 29.2 |
| 2 | 26.4 | 23.6 | 24.1 | 25.4 |
| 3 | 30.5 | 22.3 | 21.4 | 27.6 |
| 4 | 29.6 | 30.1 | 31.0 | 29.8 |
| 5 | 8.3 | 14.8 | 10.7 | 16.5 |
| 6 | 20.7 | 10.6 | 17.4 | 18.8 |
| 7 | 20.0 | 21.0 | 10.5 | 20.0 |
| 8 | 0.0 | 0.0 | 0.0 | 0.0 |
| 9 | 0.0 | 0.0 | 0.0 | 0.0 |
| 10 | 26.7 | 20.7 | 21.3 | 19.6 |
| 11 | 0.0 | 0.0 | 0.0 | 0.0 |
| 12 | 25.9 | 23.4 | 22.4 | 18.4 |
| 13 | 0.0 | 0.0 | 0.0 | 0.0 |
| 14 | 20.5 | 18.4 | 19.6 | 21.0 |
| 15 | 25.5 | 21.5 | 18.4 | 8.3 |
| 16 | 0.0 | 0.0 | 0.0 | 0.0 |
| 17 | 9.5 | 9.0 | 9.8 | 18.6 |
| 18 | 24.0 | 21.2 | 22.3 | 22.2 |
| 19 | 13.5 | 23.8 | 18.5 | 16.3 |
| 20 | 0.0 | 8.3 | 0.0 | 0.0 |
| 21 | 23.3 | 0.0 | 0.0 | 0.0 |
| 22 | 20.1 | 11.1 | 21.0 | 22.9 |
| 23 | 0.0 | 9.9 | 10.2 | 23.3 |
| 24 | 0.0 | 0.0 | 0.0 | 0.0 |
| 25 | 8.9 | 10.8 | 8.8 | 0.0 |
| Rifampicin | 35.4 | 36.1 | 33.2 | 28.0 |
| No | M. H37Ra | M. phlei | M. smegmatis | M. timereck | ||||
|---|---|---|---|---|---|---|---|---|
| MIC | MIC | MIC | MIC | |||||
| (µg/mL) | (µM/L) | (µg/mL) | (µM/L) | (µg/mL) | (µM/L) | (µg/mL) | (µM/L) | |
| 1 | 15.625 | 53.82 | 31.25 | 107.64 | 15.625 | 53.82 | 15.625 | 53.82 |
| 2 | 15.625 | 50.93 | 15.625 | 50.93 | 15.625 | 50.93 | 15.625 | 50.93 |
| 3 | 15.625 | 49.07 | 125 | 392.56 | 31.25 | 98.14 | 31.25 | 98.14 |
| 4 | 15.625 | 45.79 | 31.25 | 91.58 | 31.25 | 91.58 | 31.25 | 91.58 |
| 5 | 15.625 | 45.79 | 31.25 | 91.58 | 7.81 | 22.89 | 15.625 | 45.78 |
| 6 | 62.5 | 203.73 | 500 | 1629.88 | 500 | 1629.88 | 250 | 814.94 |
| 7 | 15.625 | 49.07 | 15.625 | 49.07 | 125 | 392.56 | 15.625 | 49.07 |
| 10 | 125 | 465.91 | 250 | 931.82 | 62.5 | 232.95 | 125 | 465.91 |
| 12 | 62.5 | 183.17 | 31.25 | 91.58 | 62.5 | 183.17 | 62.5 | 183.17 |
| 19 | 31.25 | 91.58 | 62.5 | 183.17 | 62.5 | 183.17 | 62.5 | 183.17 |
| Rifampicin | 0.976 | 1.18 | 15.625 | 18.99 | 15.625 | 18.99 | 3.9 | 4.74 |
| Compound | ChemPLP Value | Amino Acid Involved in Hydrogen Bonds |
|---|---|---|
| 1 | 57.60 | Glu214, Arg364 |
| 2 | 56.89 | Glu214 |
| 3 | 59.84 | Glu214*2 |
| 4 | 63.25 | Glu214 |
| 5 | 64.43 | Glu214 |
| 6 | 58.49 | Glu214 |
| 7 | 58.71 | Ser280 |
| 8 | 57.73 | Glu133, Arg352 |
| 9 | 64.38 | Glu214 |
| 10 | 66.12 | Glu214, Lys215 |
| 11 | 56.13 | Ser280 |
| 12 | 67.24 | Glu214 |
| 13 | 56.94 | Glu214 |
| 14 | 56.74 | Glu214 |
| 15 | 64.50 | Glu214, Ser280 |
| 16 | 63.33 | Glu214 |
| 17 | 65.92 | Glu214, Ser280 |
| 18 | 64.68 | Glu214 |
| 19 | 65.80 | Glu214 |
| 20 | 62.09 | Glu214, Arg364 |
| 21 | 67.00 | Glu214, Arg364 |
| 22 | 65.40 | Glu214*2 |
| 23 | 67.89 | Lys215*2, Ser280 |
| 24 | 65.72 | Glu214 |
| 25 | 70.97 | Glu214*2 |
| MSO-P | 127.47 | Lys215, Glu219*2, Glu227*2, Glu133*2, Arg352*2 |
| ADP | 137.10 | Lys215, Glu133*2, Arg364, Arg347*2, Glu219.Glu227*2 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pitucha, M.; Karczmarzyk, Z.; Swatko-Ossor, M.; Wysocki, W.; Wos, M.; Chudzik, K.; Ginalska, G.; Fruzinski, A. Synthesis, In Vitro Screening and Docking Studies of New Thiosemicarbazide Derivatives as Antitubercular Agents. Molecules 2019, 24, 251. https://doi.org/10.3390/molecules24020251
Pitucha M, Karczmarzyk Z, Swatko-Ossor M, Wysocki W, Wos M, Chudzik K, Ginalska G, Fruzinski A. Synthesis, In Vitro Screening and Docking Studies of New Thiosemicarbazide Derivatives as Antitubercular Agents. Molecules. 2019; 24(2):251. https://doi.org/10.3390/molecules24020251
Chicago/Turabian StylePitucha, Monika, Zbigniew Karczmarzyk, Marta Swatko-Ossor, Waldemar Wysocki, Maciej Wos, Kamil Chudzik, Grazyna Ginalska, and Andrzej Fruzinski. 2019. "Synthesis, In Vitro Screening and Docking Studies of New Thiosemicarbazide Derivatives as Antitubercular Agents" Molecules 24, no. 2: 251. https://doi.org/10.3390/molecules24020251
APA StylePitucha, M., Karczmarzyk, Z., Swatko-Ossor, M., Wysocki, W., Wos, M., Chudzik, K., Ginalska, G., & Fruzinski, A. (2019). Synthesis, In Vitro Screening and Docking Studies of New Thiosemicarbazide Derivatives as Antitubercular Agents. Molecules, 24(2), 251. https://doi.org/10.3390/molecules24020251