Expanding the Reaction Space of Linkage-Specific Sialic Acid Derivatization


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
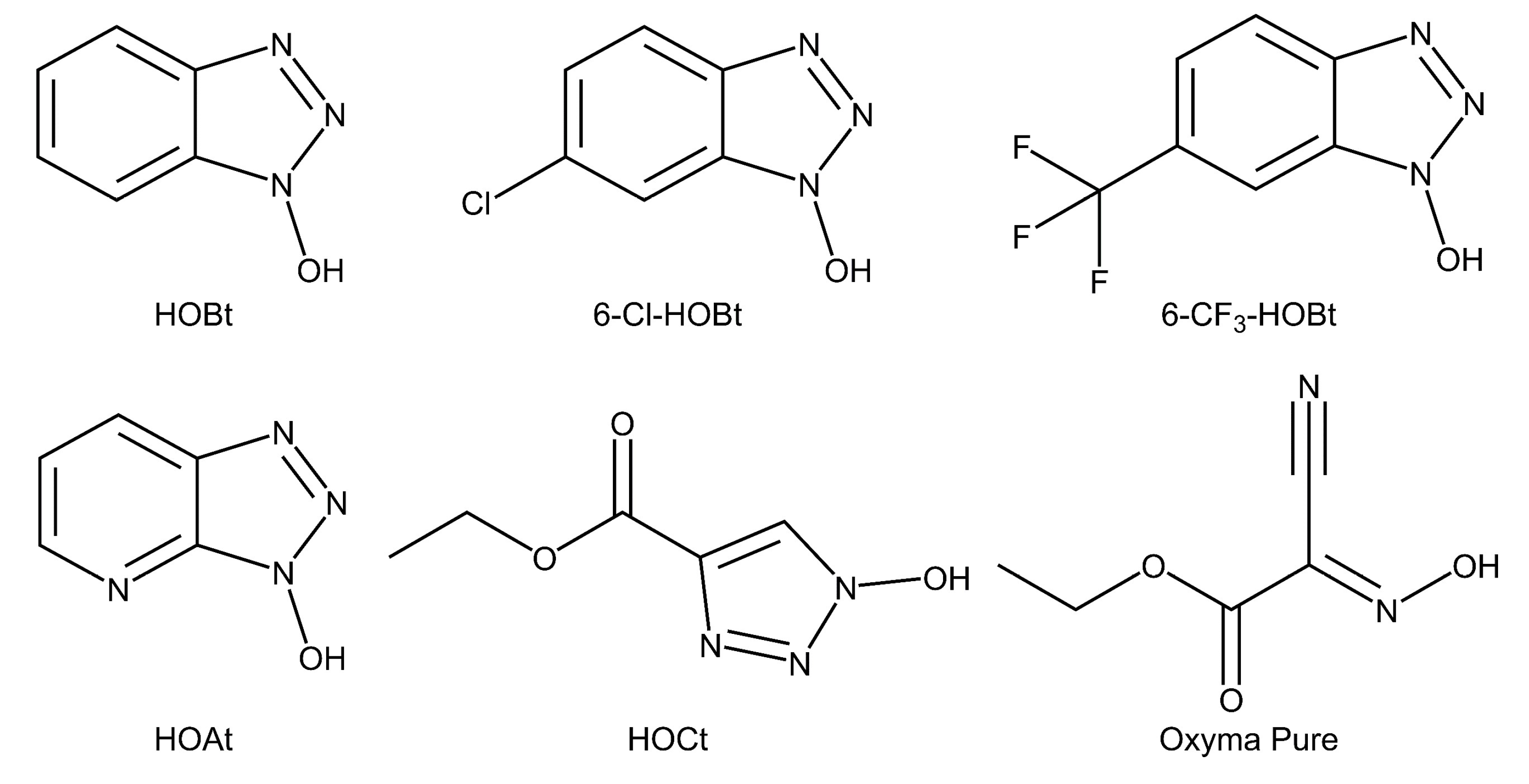
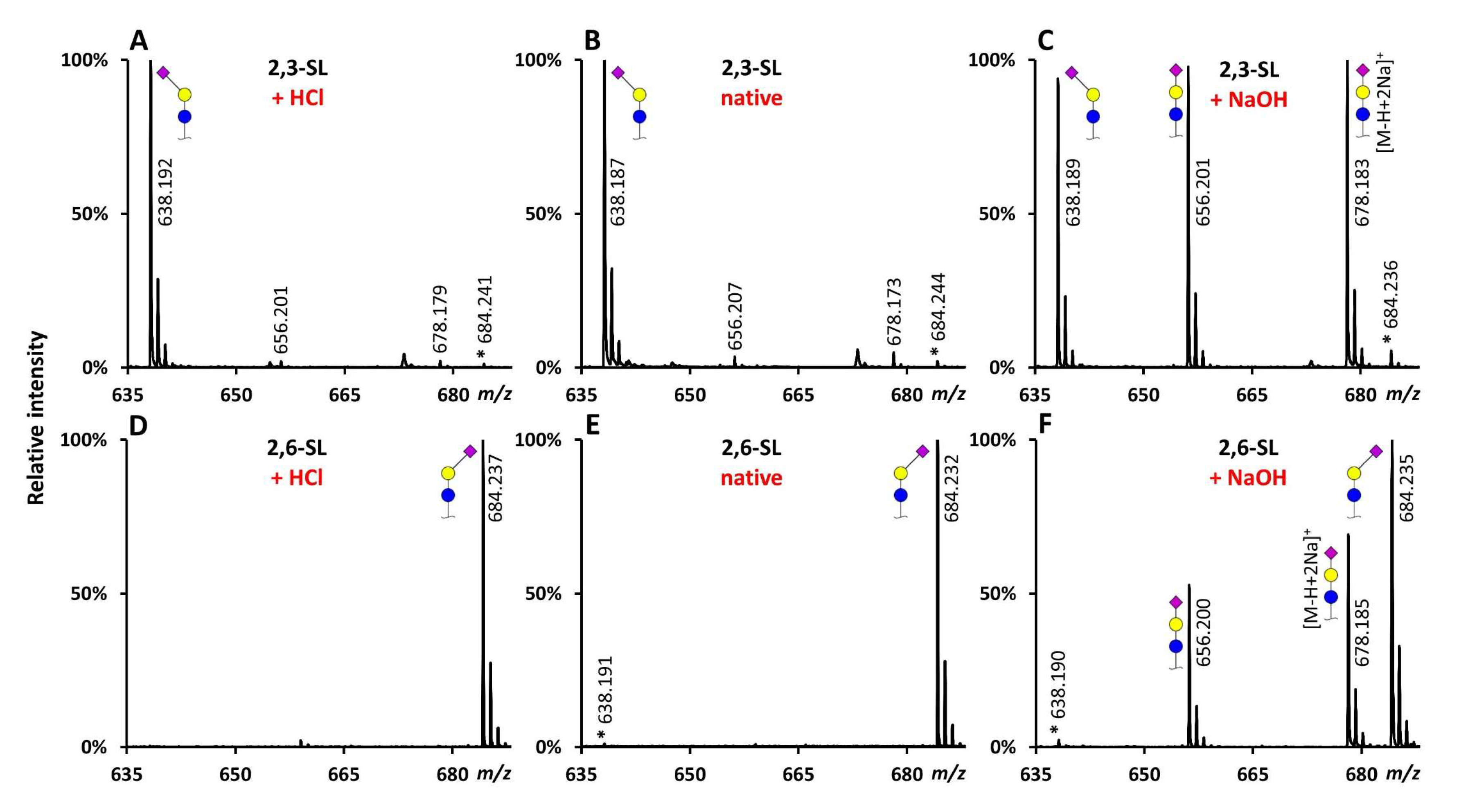
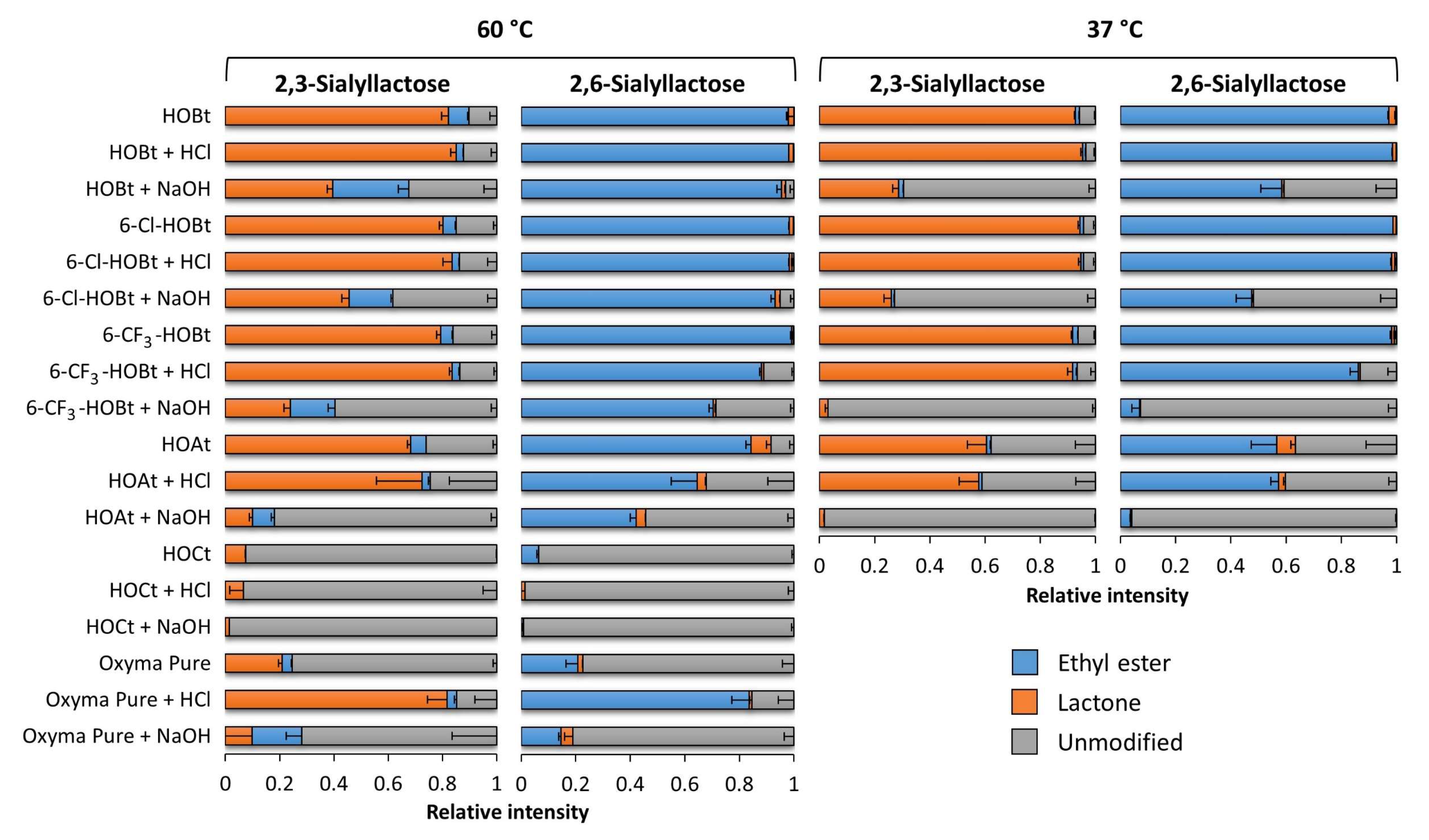
2.1. Evaluation of the Catalysts Using Sialyllactose Standards
2.2. Evaluation of the Catalysts Using Complex N-glycan Samples
3. Discussion
4. Materials and Methods
4.1. Chemicals, Reagents, and Enzymes
4.2. Samples
4.3. Preparation of Catalysts
4.4. N-glycan Release
4.5. Sialic Acid Derivatization
4.6. Cotton-HILIC SPE
4.7. MALDI-TOF-MS Analysis
4.8. Data Analysis
5. Patents
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Moremen, K.W.; Tiemeyer, M.; Nairn, A.V. Vertebrate protein glycosylation: Diversity, synthesis and function. Nat. Rev. Mol. Cell Biol. 2012, 13, 448–462. [Google Scholar] [CrossRef] [PubMed]
- Varki, A. Biological roles of glycans. Glycobiology 2017, 27, 3–49. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Varki, A. Perspectives on the significance of altered glycosylation of glycoproteins in cancer. Glycoconj. J. 1997, 14, 569–576. [Google Scholar] [CrossRef] [PubMed]
- Khoury, G.A.; Baliban, R.C.; Floudas, C.A. Proteome-wide post-translational modification statistics: Frequency analysis and curation of the swiss-prot database. Sci. Rep. 2011, 1. [Google Scholar] [CrossRef] [PubMed]
- Stowell, S.R.; Ju, T.; Cummings, R.D. Protein glycosylation in cancer. Annu. Rev. Pathol. 2015, 10, 473–510. [Google Scholar] [CrossRef] [PubMed]
- Varki, A. Sialic acids in human health and disease. Trends Mol. Med. 2008, 14, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Hedlund, M.; Ng, E.; Varki, A.; Varki, N.M. alpha 2-6-Linked sialic acids on N-glycans modulate carcinoma differentiation in vivo. Cancer Res. 2008, 68, 388–394. [Google Scholar] [CrossRef] [PubMed]
- Kang, P.; Mechref, Y.; Klouckova, I.; Novotny, M.V. Solid-phase permethylation of glycans for mass spectrometric analysis. Rapid Commun. Mass Spectrom. 2005, 19, 3421–3428. [Google Scholar] [CrossRef]
- Harvey, D.J. Analysis of carbohydrates and glycoconjugates by matrix-assisted laser desorption/ionization mass spectrometry: An update for 2003-2004. Mass Spectrom. Rev. 2009, 28, 273–361. [Google Scholar] [CrossRef]
- Powell, A.K.; Harvey, D.J. Stabilization of sialic acids in N-linked oligosaccharides and gangliosides for analysis by positive ion matrix-assisted laser desorption ionization mass spectrometry. Rapid Commun. Mass Spectrom. 1996, 10, 1027–1032. [Google Scholar] [CrossRef]
- Selman, M.H.; Hoffmann, M.; Zauner, G.; McDonnell, L.A.; Balog, C.I.; Rapp, E.; Deelder, A.M.; Wuhrer, M. MALDI-TOF-MS analysis of sialylated glycans and glycopeptides using 4-chloro-alpha-cyanocinnamic acid matrix. Proteomics 2012, 12, 1337–1348. [Google Scholar] [CrossRef] [PubMed]
- Reiding, K.R.; Hipgrave Ederveen, A.L.; Rombouts, Y.; Wuhrer, M. Murine Plasma N-Glycosylation Traits Associated with Sex and Strain. J. Proteome Res. 2016, 15, 3489–3499. [Google Scholar] [CrossRef] [PubMed]
- Reiding, K.R.; Blank, D.; Kuijper, D.M.; Deelder, A.M.; Wuhrer, M. High-throughput profiling of protein N-glycosylation by MALDI-TOF-MS employing linkage-specific sialic acid esterification. Anal. Chem. 2014, 86, 5784–5793. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, S.F.; Domann, P.; Harvey, D.J. Derivatization of sialic acids for stabilization in matrix-assisted laser desorption/ionization mass spectrometry and concomitant differentiation of alpha(2 --> 3)- and alpha(2 --> 6)-isomers. Rapid Commun. Mass Spectrom. 2009, 23, 303–312. [Google Scholar] [CrossRef] [PubMed]
- El-Faham, A.; Albericio, F. Peptide coupling reagents, more than a letter soup. Chem. Rev. 2011, 111, 6557–6602. [Google Scholar] [CrossRef] [PubMed]
- Valeur, E.; Bradley, M. Amide bond formation: Beyond the myth of coupling reagents. Chem. Soc. Rev. 2009, 38, 606–631. [Google Scholar] [CrossRef]
- Liu, X.; Qiu, H.; Lee, R.K.; Chen, W.; Li, J. Methylamidation for sialoglycomics by MALDI-MS: A facile derivatization strategy for both alpha2,3- and alpha2,6-linked sialic acids. Anal. Chem. 2010, 82, 8300–8306. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Gao, W.; Feng, X.; Liu, B.F.; Liu, X. MALDI-MS analysis of sialylated N-glycan linkage isomers using solid-phase two step derivatization method. Anal. Chim. Acta 2016, 924, 77–85. [Google Scholar] [CrossRef]
- Nishikaze, T.; Tsumoto, H.; Sekiya, S.; Iwamoto, S.; Miura, Y.; Tanaka, K. Differentiation of Sialyl Linkage Isomers by One-Pot Sialic Acid Derivatization for Mass Spectrometry-Based Glycan Profiling. Anal. Chem. 2017, 89, 2353–2360. [Google Scholar] [CrossRef]
- Yang, S.; Zhang, L.; Thomas, S.; Hu, Y.; Li, S.; Cipollo, J.; Zhang, H. Modification of Sialic Acids on Solid Phase: Accurate Characterization of Protein Sialylation. Anal. Chem. 2017, 89, 6330–6335. [Google Scholar] [CrossRef]
- Hanamatsu, H.; Nishikaze, T.; Miura, N.; Piao, J.; Okada, K.; Sekiya, S.; Iwamoto, S.; Sakamoto, N.; Tanaka, K.; Furukawa, J.I. Sialic Acid Linkage Specific Derivatization of Glycosphingolipid Glycans by Ring-Opening Aminolysis of Lactones. Anal. Chem. 2018, 90, 13193–13199. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, N.; Abe, T.; Natsuka, S. Quantitative LC-MS and MS/MS analysis of sialylated glycans modified by linkage-specific alkylamidation. Anal. Biochem. 2019, 567, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Wu, W.W.; Shen, R.F.; Bern, M.; Cipollo, J. Identification of Sialic Acid Linkages on Intact Glycopeptides via Differential Chemical Modification Using IntactGIG-HILIC. J. Am. Soc. Mass Spectrom. 2018, 29, 1273–1283. [Google Scholar] [CrossRef] [PubMed]
- Vreeker, G.C.M.; Nicolardi, S.; Bladergroen, M.R.; van der Plas, C.J.; Mesker, W.E.; Tollenaar, R.; van der Burgt, Y.E.M.; Wuhrer, M. Automated Plasma Glycomics with Linkage-Specific Sialic Acid Esterification and Ultrahigh Resolution MS. Anal. Chem. 2018, 90, 11955–11961. [Google Scholar] [CrossRef] [PubMed]
- Klein, A. Human total serum N-glycome. Adv. Clin. Chem. 2008, 46, 51–85. [Google Scholar] [CrossRef] [PubMed]
- Clerc, F.; Reiding, K.R.; Jansen, B.C.; Kammeijer, G.S.; Bondt, A.; Wuhrer, M. Human plasma protein N-glycosylation. Glycoconj. J. 2016, 33, 309–343. [Google Scholar] [CrossRef]
- Yang, S.; Jankowska, E.; Kosikova, M.; Xie, H.; Cipollo, J. Solid-Phase Chemical Modification for Sialic Acid Linkage Analysis: Application to Glycoproteins of Host Cells Used in Influenza Virus Propagation. Anal. Chem. 2017, 89, 9508–9517. [Google Scholar] [CrossRef] [PubMed]
- Lageveen-Kammeijer, G.S.M.; de Haan, N.; Mohaupt, P.; Wagt, S.; Filius, M.; Nouta, J.; Falck, D.; Wuhrer, M. Highly sensitive CE-ESI-MS analysis of N-glycans from complex biological samples. Nat. Commun. 2019, 10, 2137. [Google Scholar] [CrossRef]
- Miura, Y.; Shinohara, Y.; Furukawa, J.; Nagahori, N.; Nishimura, S. Rapid and simple solid-phase esterification of sialic acid residues for quantitative glycomics by mass spectrometry. Chemistry 2007, 13, 4797–4804. [Google Scholar] [CrossRef]
- Schwedler, C.; Kaup, M.; Petzold, D.; Hoppe, B.; Braicu, E.I.; Sehouli, J.; Ehlers, M.; Berger, M.; Tauber, R.; Blanchard, V. Sialic acid methylation refines capillary electrophoresis laser-induced fluorescence analyses of immunoglobulin GN-glycans of ovarian cancer patients. Electrophoresis 2014, 35, 1025–1031. [Google Scholar] [CrossRef]
- Mitra, I.; Snyder, C.M.; Zhou, X.; Campos, M.I.; Alley, W.R.; Novotny, M.V.; Jacobson, S.C. Structural Characterization of Serum N-Glycans by Methylamidation, Fluorescent Labeling, and Analysis by Microchip Electrophoresis. Anal. Chem. 2016, 88, 8965–8971. [Google Scholar] [CrossRef] [PubMed]
- Snyder, C.M.; Zhou, X.; Karty, J.A.; Fonslow, B.R.; Novotny, M.V.; Jacobson, S.C. Capillary electrophoresis-mass spectrometry for direct structural identification of serum N-glycans. J. Chromatogr. A 2017, 1523, 127–139. [Google Scholar] [CrossRef] [PubMed]
- Dedova, T.; Braicu, E.I.; Sehouli, J.; Blanchard, V. Sialic Acid Linkage Analysis Refines the Diagnosis of Ovarian Cancer. Front. Oncol. 2019, 9, 261. [Google Scholar] [CrossRef] [PubMed]
- De Haan, N.; Reiding, K.R.; Haberger, M.; Reusch, D.; Falck, D.; Wuhrer, M. Linkage-specific sialic acid derivatization for MALDI-TOF-MS profiling of IgG glycopeptides. Anal. Chem. 2015, 87, 8284–8291. [Google Scholar] [CrossRef] [PubMed]
- Shubhakar, A.; Reiding, K.R.; Gardner, R.A.; Spencer, D.I.R.; Fernandes, D.L.; Wuhrer, M. High-Throughput Analysis and Automation for Glycomics Studies. Chromatographia 2014, 78, 321–333. [Google Scholar] [CrossRef]
- Carpino, L.A. 1-Hydroxy-7-azabenzotriazole. An efficient peptide coupling additive. J. Am. Chem. Soc. 1993, 115, 4397–4398. [Google Scholar] [CrossRef]
- El-Faham, A.; Albericio, F. Synthesis and Application ofN-Hydroxylamine Derivatives as Potential Replacements for HOBt. Eur. J. Org. Chem. 2009, 2009, 1499–1501. [Google Scholar] [CrossRef]
- Jiang, L.; Davison, A.; Tennant, G.; Ramage, R. Synthesis and application of a novel coupling reagent, ethyl 1-hydroxy-1H -1,2,3-triazole-4-carboxylate. Tetrahedron 1998, 54, 14233–14254. [Google Scholar] [CrossRef]
- Subiros-Funosas, R.; Prohens, R.; Barbas, R.; El-Faham, A.; Albericio, F. Oxyma: An efficient additive for peptide synthesis to replace the benzotriazole-based HOBt and HOAt with a lower risk of explosion. Chemistry 2009, 15, 9394–9403. [Google Scholar] [CrossRef]
- Wehrstedt, K.D.; Wandrey, P.A.; Heitkamp, D. Explosive properties of 1-hydroxybenzotriazoles. J. Hazard Mater. 2005, 126, 1–7. [Google Scholar] [CrossRef]
- Schauer, R. Characterization of sialic acids. In Methods Enzymol; Academic Press: Cambridge, MA, USA, 1978; Volume 50, pp. 64–89. [Google Scholar] [CrossRef]
- Holst, S.; Deuss, A.J.M.; van Pelt, G.W.; van Vliet, S.J.; Garcia-Vallejo, J.J.; Koeleman, C.A.M.; Deelder, A.M.; Mesker, W.E.; Tollenaar, R.A.; Rombouts, Y.; et al. N-glycosylation Profiling of Colorectal Cancer Cell Lines Reveals Association of Fucosylation with Differentiation and Caudal Type Homebox 1 (CDX1)/Villin mRNA Expression. Mol. Cell. Proteom. 2016, 15, 124. [Google Scholar] [CrossRef] [PubMed]
- Selman, M.H.J.; Hemayatkar, M.; Deelder, A.M.; Wuhrer, M. Cotton HILIC SPE Microtips for Microscale Purification and Enrichment of Glycans and Glycopeptides. Anal. Chem. 2011, 83, 2492–2499. [Google Scholar] [CrossRef] [PubMed]
- Jansen, B.C.; Reiding, K.R.; Bondt, A.; Hipgrave Ederveen, A.L.; Palmblad, M.; Falck, D.; Wuhrer, M. MassyTools: A High-Throughput Targeted Data Processing Tool for Relative Quantitation and Quality Control Developed for Glycomic and Glycoproteomic MALDI-MS. J. Proteom. Res. 2015, 14, 5088–5098. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds HOBt, 6-Cl-HOBt, 6-CF3-HOBt, HOAt, HOCt and Oxyma Pure are commercially available (Table S1). |





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pongracz, T.; Wuhrer, M.; de Haan, N. Expanding the Reaction Space of Linkage-Specific Sialic Acid Derivatization. Molecules 2019, 24, 3617. https://doi.org/10.3390/molecules24193617
Pongracz T, Wuhrer M, de Haan N. Expanding the Reaction Space of Linkage-Specific Sialic Acid Derivatization. Molecules. 2019; 24(19):3617. https://doi.org/10.3390/molecules24193617
Chicago/Turabian StylePongracz, Tamas, Manfred Wuhrer, and Noortje de Haan. 2019. "Expanding the Reaction Space of Linkage-Specific Sialic Acid Derivatization" Molecules 24, no. 19: 3617. https://doi.org/10.3390/molecules24193617
APA StylePongracz, T., Wuhrer, M., & de Haan, N. (2019). Expanding the Reaction Space of Linkage-Specific Sialic Acid Derivatization. Molecules, 24(19), 3617. https://doi.org/10.3390/molecules24193617