3. Materials and Methods
Melting points were determined with a Kofler hot-stage apparatus and are uncorrected. Optical rotations were measured with an ATAGO AP-300 Automatic Polarimeter at 25±2 °C.
1H NMR spectra were recorded in appropriate solvents with a Bruker Avance II operating at 250.13 MHz.
13C NMR spectra were recorded with the spectrometers operating at 62.9 MHz. The assignments were made, when possible, with the aid of DEPT, COSY, HSQC experiments. The first order proton chemical shifts
δ are referenced to either residual CDCl
3 (
δ H 7.26,
δ C 77.0), residual CD
3CN (
δ H 1.94,
δ C 1.28), residual CD
3OD (
δ H 3.31,
δ C 49.0) or residual DMSO-
d6 (
δ H 2.49,
δ C 39.5) and
J-values are given in Hz. Spin multiplicity was indicated by s = singlet, d = doublet, t = triplet, bt = broad triplet, q = quartet, m = multiplet. Coupling constants
J were reported in Hertz (Hz). All reactions were followed by TLC on Kieselgel 60 F
254 with detection by UV light and/or with ethanolic 10% phosphomolybdic or sulfuric acid, and heating. Kieselgel 60 (Merck, S.p.A., Milano, Italy, 70–230 and 230–400 mesh, respectively) was used for column and flash chromatography. Some of flash chromatography were conducted by the automated system Isolera Four SV
TM (Biotage
®, Uppsala, Sweden), equipped with UV detector with variable wavelength (200–400 nm). Solvents were dried by distillation according to standard procedures, and storage over 4Å molecular sieves activated for at least 24 h at 200 °C. All reagents were purchased from Aldrich Chemical Co. and were used without further purification. Molecular sieves (4Å and AW-300) and PPh
3 were activated (2–8 h at 150 °C.) before use. All reactions involving air- or moisture-sensitive reagents were performed under an argon atmosphere using anhydrous solvents. Anhydrous dimethylformamide (DMF), dichloromethane (CH
2Cl
2), 1,2-dichloethane (DCE), and THF were purchased from Sigma-Aldrich. Other dried solvents were obtained by distillation according to standard procedure and stored over 4Å molecular sieves activated. MgSO
4 or Na
2SO
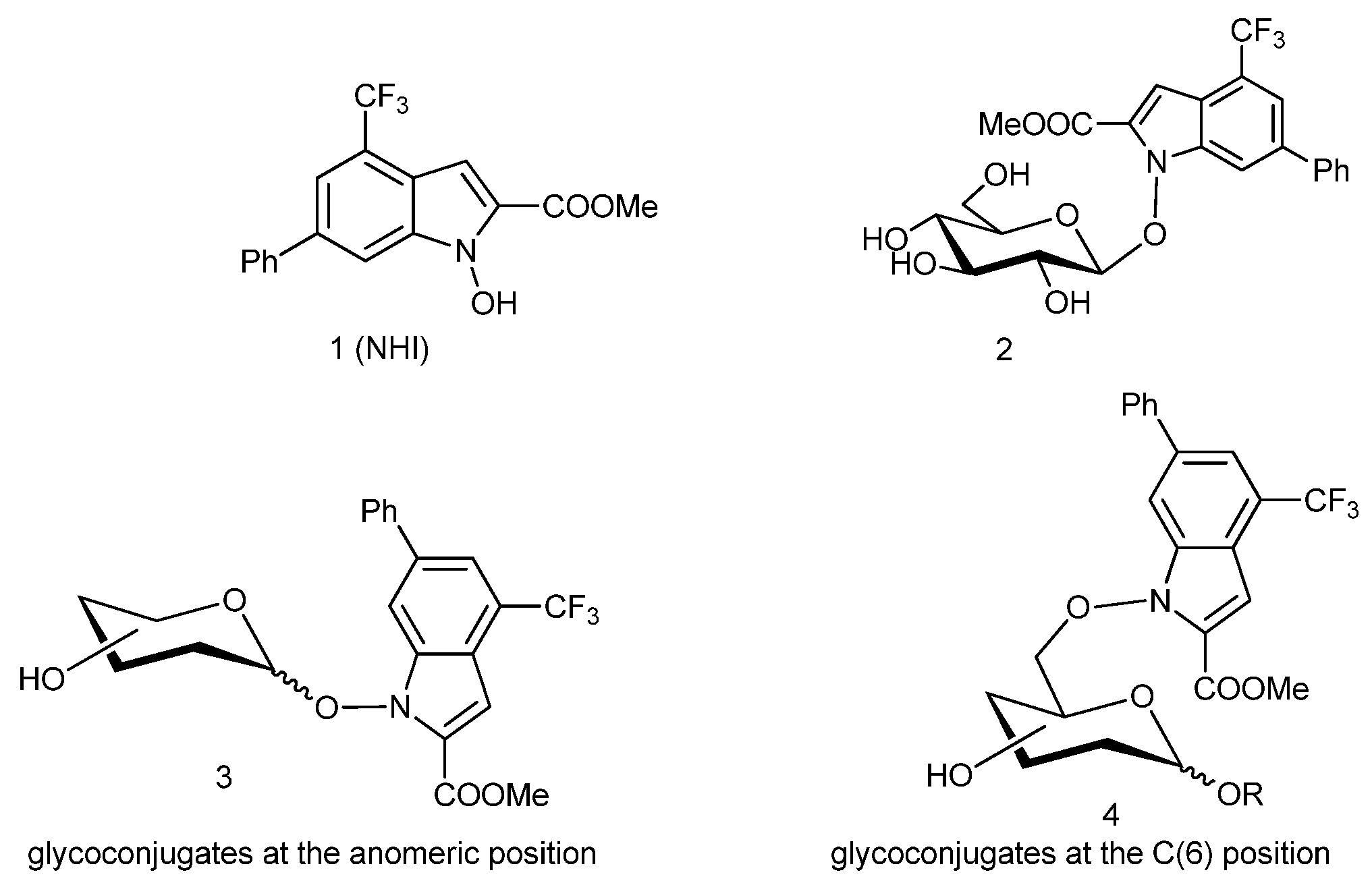
4 were used as the drying agents for solutions. 1-Hydroxy-6-phenyl-4-(trifluoromethyl)-1H-indole-2-methyl-carboxylate (
1) [
14], 2,3,4,6-tetra-
O-acetyl-α-
d-galactopyranosyl trichloroacetimidate (
5α) [
11], 4-
O-(2,3,4,6-tetra-
O-acetyl-β-
d-galactopyranosyl)-2,3,6-tri-
O-acetyl-α-
d-glucopyranosyl trichloroacetimidate (
8α) [
12,
13], methyl 2,3,4-tri-
O-acetyl-α-
d-mannopyranoside (
11) [
19], methyl 2,3,4-tri-
O-acetyl-β-
d-glucopyranoside (
14) [
20], methyl 2,3,4-tri-
O-acetyl-β-
d-galactopyranoside (
15) [
21], and 4-
O-(2-
O-acetyl-3,4-
O-isopropylidene-β-
d-galactopyranosyl)-2,3:5,6-di-
O-isopropylidene-
aldehydo-
d-glucose dimethyl acetal (
20) [
26] were prepared according to the reported procedure.
3.1. General Procedures for Glycosylation
3.1.1. Method A (with TMSOTf in CH2Cl2 or CH3CN)
A solution of the glycosyl donor
5α [
11] (1.30 mmol, 1.3 equiv) and
N-hydroxyindole derivative
1 [
14] (1.00 mmol, 1.0 equiv) in anhyd toluene (10.0 mL) was concentrated under diminished pressure (2 h) for removed any traces of water. The residue was dissolved in anhyd CH
2Cl
2 or CH
3CN (10.0 mL), molecular sieves AW 300 (1.25 g) were added and the resulting mixture was stirred under an argon atmosphere at 0 °C for 30 min. TMSOTf (36 μL, 0.20 mmol, 0.2 equiv) was added and the reaction mixture was stirred for 30 min at 0 °C and then at room temp until TLC analysis showed the complete disappearance of the
N-hydroxyindole derivative
1. The reaction mixture was diluted with CH
2Cl
2 or CH
3CN (10.0 mL), neutralized with Et
3N, filtered through a short pad of Celite
® and organic solution was concentrated under diminished pressure. The crude residue was subjected to flash chromatography on silica gel.
3.1.2. Method B (with BF3·Et2O in CH2Cl2)
The appropriate glycosyl donor 5α or 8α (1.50 mmol, 1.5 equiv) was dried under diminished pressure (1 h). Anhyd CH2Cl2 (26.0 mL), molecular sieves AW 300 (500 mg), the N-hydroxyindole derivative 1 (1.00 mmol, 1.0 equiv) was added and the resulting mixture was stirred under an argon atmosphere at room temp for 1 h. The suspension was cooled to −10 °C, treated with BF3·Et2O (160 μL, 1.30 mmol, 1.3 equiv) and slowly warmed to room temp. When TLC analysis showed the complete disappearance of the N-hydroxyindole derivative 1 the reaction mixture was diluted with CH2Cl2 (10.0 mL), neutralized with Et3N, filtered through a short pad of Celite® and organic solution was concentrated under diminished pressure. The crude residue was subjected to flash chromatography on silica gel.
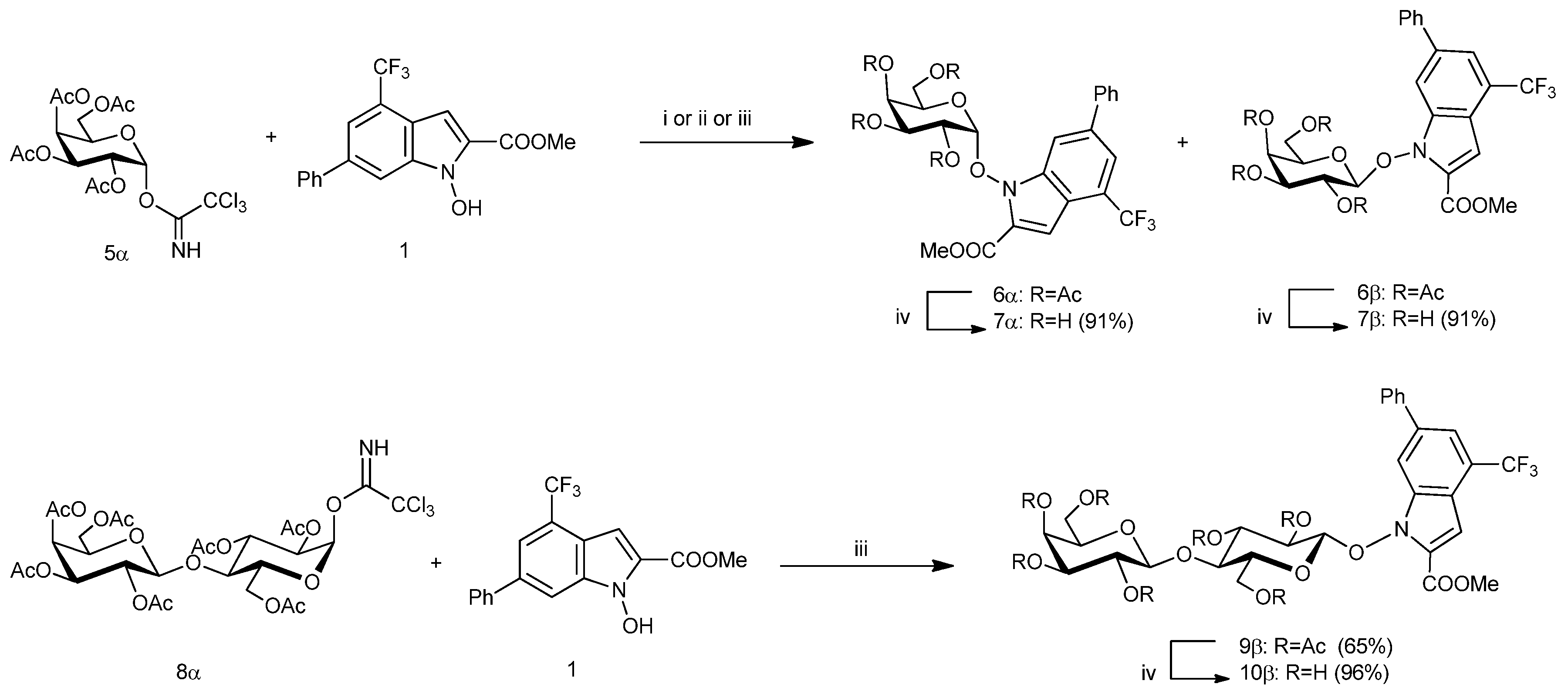
3.2. Methyl 1-(2,3,4,6-Tetra-O-Acetyl-α-d-Galactopyranosyl)-6-Phenyl-4-Trifluoromethyl-1H-Indole-2-Carboxylate (6α) and Methyl 1-(2,3,4,6-Tetra-O-Acetyl-β-d-Galactopyranosyl)-6-Phenyl-4-Trifluoromethyl-1H-Indole-2-Carboxylate (6β)
The glycosylation of
5α [
11] (100 mg, 0.20 mmol, 1.3 equiv) with
N-hydroxyindole derivative
1 [
14] (52 mg, 0.16 mmol, 1.0 equiv), was performed in dry CH
2Cl
2 (1.6 mL) in according to the general procedure (Method A). The reaction was stirred until TLC (4 h, TLC, 7:3 hexane-EtOAc, double elution) showed the complete disappearance of the acceptor
1 (
Rf 0.66) and the formation of two spots at
Rf 0.37 and 0.35. The crude product was constituted (NMR) by a mixture of the anomers
6α and
6β in ratio of 35:65, calculated on the basis of the relative H-1 signal intensities. Purification of crude product by flash chromatography on silica gel (7:3 hexane-EtOAc) afforded pure
6α (31 mg, 29%) and
6β (44 mg, 42%).
The glycosylation of
5α [
11] (100 mg, 0.20 mmol, 1.3 equiv) with
N-hydroxyindole derivative
1 [
14] (52 mg, 0.16 mmol, 1.0 equiv), was performed in dry CH
3CN (1.6 mL) in according to the general procedure (Method A). The reaction was stirred until TLC (3 h, TLC, 7:3 hexane-EtOAc, double elution) showed the complete disappearance of the acceptor
1 (
Rf 0.66) and the formation of two spots at
Rf 0.37 and 0.35. The crude product was constituted (NMR) by a mixture of the anomers
6α and
6β in ratio of 78:22, calculated on the basis of the relative H-1 signal intensities. Purification of crude product by flash chromatography on silica gel (7:3 hexane-EtOAc) afforded pure
6α (57.4 mg, 54%) and
6β (14.4 mg, 13%).
The glycosylation of
5α [
11] (200 mg, 0.40 mmol, 1.5 equiv) with
N-hydroxyindole derivative
1 [
14] (91 mg, 0.28 mmol, 1.0 equiv) was performed in dry CH
2Cl
2 (7.0 mL) according to the general procedure (Method B). The reaction was stirred until TLC (2 h, TLC, 7:3 hexane-EtOAc, double elution) showed the complete disappearance of the acceptor
1 (
Rf 0.66) and the formation of two spots at R
f 0.37 and 0.35. The crude product was constituted (NMR) by a mixture of the anomers
6α and
6β in ratio of 5:95, calculated on the basis of the relative H-1 signal intensities. Purification of crude product by flash chromatography on silica gel (7:3 hexane-EtOAc) afforded pure
6β (142 mg, 78%) and
6α (5 mg, 3%).
Compound 6α is a yellow foam; Rf 0.37 (7:3 hexane-EtOAc, double elution); mp 64–67 °C (crom); [α]D -10.3 (c 0.90 in CHCl3); 1H NMR (250.13 MHz, CD3CN) δ: 7.99 (m, 1H, Ar-H), 7.86 (m, 1H, Ar-H), 7.80–7.74 (m, 2H, Ar-H), 7.55–7.40 (m, 3H, Ar-H), 7.30 (qd, 1H, J 1.0 Hz, J 1.6 Hz, Ar-H), 5.91 (d, 1H, J1,2 4.0 Hz, H-1), 5.61 (ddd, 1H, J3,4 3.0 Hz, J4,5 1.4 Hz, J4,6b 0.6 Hz, H-4), 5.47 (dd, 1H, J2,3 11.2 Hz, H-2), 5.36 (dd, 1H, H-3), 5.02 (m, 1H, H-5), 4.05 (ddd, 1H, J5,6b 3.3 Hz, J6a,6b 12.1 Hz, H-6b), 4.00 (dd, 1H, J5,6a 4.7 Hz, H-6a), 3.91 (s, 3H, COOMe), 2.12, 2.11, 1.99, 1.67 (4s, each 3H, 4 × COMe); 13C NMR (62.9 MHz, CD3CN) δ: 171.2, 171.1, 170.8, 170.7 (4 × COMe), 160.6 (COOMe), 140.4, 139.7, 138.0, 130.4 (4 × Ar-C), 130.1, 129.2, 128.5 (Ar-CH), 124.8–123.2 (CF3 + Ar-C), 120.4 (Ar-CH), 117.2 (Ar-C), 113.3, 107.1 (2 × Ar-CH), 104.4 (C-1), 70.6 (C-5), 68.9 (C-4), 67.6 (C-2), 66.8 (C-3), 62.5 (C-6), 53.1 (COOMe), 21.8, 20.4 (4 × COMe). Anal. Calcd for C31H30F3NO12: C, 55.94; H, 4.54%; N, 2.10%; Found: C, 55.98; H, 4.56%; N, 2.14%.
Compound 6β is a pale yellow solid; Rf 0.35 (hexane-AcOEt 7:3, double elution); mp 139–142 °C (crom); [α]D + 26.3 (c 1.0 in CHCl3); 1H NMR (250.13 MHz, CD3CN) δ: 8.12 (m, 1H, Ar-H), 7.84 (m, 1H, Ar-H), 7.75 (m, 1H, Ar-H), 7.71 (m, 1H, Ar-H), 7.54–7.40 (m, 3H, Ar-H), 7.23 (qd, 1H, J 1.0 Hz, J 1.7 Hz, Ar-H), 5.58 (d, 1H, J1,2 8.1 Hz, H-1), 5.40 (m, 2H, 2-H, H-4), 5.27 (dd, 1H, J2,3 9.1 Hz, J3,4 3.3 Hz, H-3), 4.10–3.98 (m, 3H, H-5, H-6a, H-6b), 3.91 (s, 3H, COOMe), 2.18, 2.16, 1.98, 1.60 (4s, each 3H, 4 × COMe); 13C NMR (62.9 MHz, CD3CN) δ: 171.2, 171.1, 170.8, 170.7 (4 × COMe), 160.5 (COOMe), 140.5, 139.8, 139.5, 130.4 (4 × Ar-C), 130.1, 129.1, 128.2 (Ph-CH), 124.1–123.2 (CF3 + Ar-C), 120.3 (Ar-CH), 117.2 (Ar-C), 114.8, 106.8 (2 × Ar-CH), 106.2 (C-1), 72.1 (C-5), 71.3 (C-3), 68.4 (C-4), 68.1 (C-2), 62.4 (C-6), 52.9 (COOMe), 21.2, 20.8, 20.7, 20.4 (4 × COMe). Anal. Calcd for C31H30F3NO12: C, 55.94; H, 4.54%; N, 2.10%; Found: C, 55.97; H, 4.58%; N, 2.13%.
3.3. Methyl 1-[4-O-(2,3,4,6-Tetra-O-Acetyl-β-d-Galactopyranosyl)-2,3,6-Tri-O-Acetyl-β-d-Glucopyranosyl)]-6-Phenyl-4-Trifluoromethyl-1H-Indole-2-Carboxylate (9β)
The glycosylation of
8α [
12,
13] (175 mg, 0.22 mmol, 1.5 equiv) with
N-hydroxyindole derivative
1 [
19] (50 mg, 0.15 mmol, 1.0 equiv), was performed in dry CH
2Cl
2 (4.0 mL) in according to the general procedure (Method B). The reaction was stirred until TLC (2 h, TLC, 1:1 hexane-EtOAc, double elution) showed the complete disappearance of the acceptor
1 (
Rf 0.76) and the formation of one spot at
Rf 0.45. Purification of crude product by flash chromatography on silica gel (1:1 hexane-EtOAc) afforded pure
9β (93 mg, 65%) as solid;
Rf 0.76 (7:3 hexane-EtOAc, double elution); mp 100–103 °C (crom); [α]
D +49.4 (
c 1.0 in CHCl
3);
1H NMR (250.13 MHz, CD
3CN-CDCl
3) δ: 8.08 (m, 1H, Ar-
H), 7.80 (m, 1H, Ar-
H), 7.71–7.66 (m, 2H, Ar-
H), 7.58–7.38 (m, 3H, Ar-
H), 7.22 (qd, 1H,
J 1.0 Hz,
J 1.8 Hz, Ar-
H), 5.57 (d, 1H,
J1,2 8.0 Hz, 1-H), 5.33 (dd, 1H,
J2,3 9.8 Hz,
J3,4 8.7 Hz, 3-H), 5.30 (dd, 1H,
J3′,4′ 3.5 Hz,
J4′,5′ 1.3 Hz, H-4′), 5.23 (dd, 1H, H-2), 5.03 (dd, 1H,
J2′,3′ 10.4 Hz, H-3′), 4.90 (dd, 1H,
J1′,2′ 7.8 Hz, H-2′), 4.59 (d, 1H, H-1′), 4.19 (dd, 1H,
J5,6b 2.3 Hz,
J6a,6b 12.1 Hz, H-6b), 4.13–4.02 (m, 4H, H-6a, H-5′, H-6′a, H-6′b), 3.99 (dd, 1H,
J4,5 9.9 Hz, H-4), 3.91 (s, 3H, COO
Me), 3.75 (ddd, 1H,
J5,6a 5.6 Hz H-5), 2.13, 2.09, 2.06, 2.02, 1.93, 1.89, 1.58 (7s, each 3H, 7 × CO
Me);
13C NMR (62.9 MHZ, CD
3CN-CDCl
3) δ: 171.1–170.2 (7 ×
COMe), 160.3 (
COOMe), 140.6, 139.8, 135.5, 130.4 (4 × Ar-
C), 129.9, 128.9, 128.2 (Ph-
CH), 123.9–122.9 (
CF
3 + Ar-
C), 120.3 (Ar-
CH), 117.3 (Ar-
C), 114.9, 106.8 (2 × Ar-
CH), 105.6 (C-1), 101.4 (C-1′), 76.9 (C-4), 73.4 (C-5), 72.7 (C-3), 71.5 (C-3′, C-5′), 70.5 (C-2), 69.8 (C-2′), 68.0 (C-4′), 62.7 (C-6′), 62.0 (C-6), 52.8 (COO
Me), 21.1–20.4 (7 × COMe). Anal. Calcd for C
43H
46F
3NO
20: C, 54.15; H, 4.86%; N, 1.47%; Found: C, 54.18; H, 4.89%; N, 1.50%.
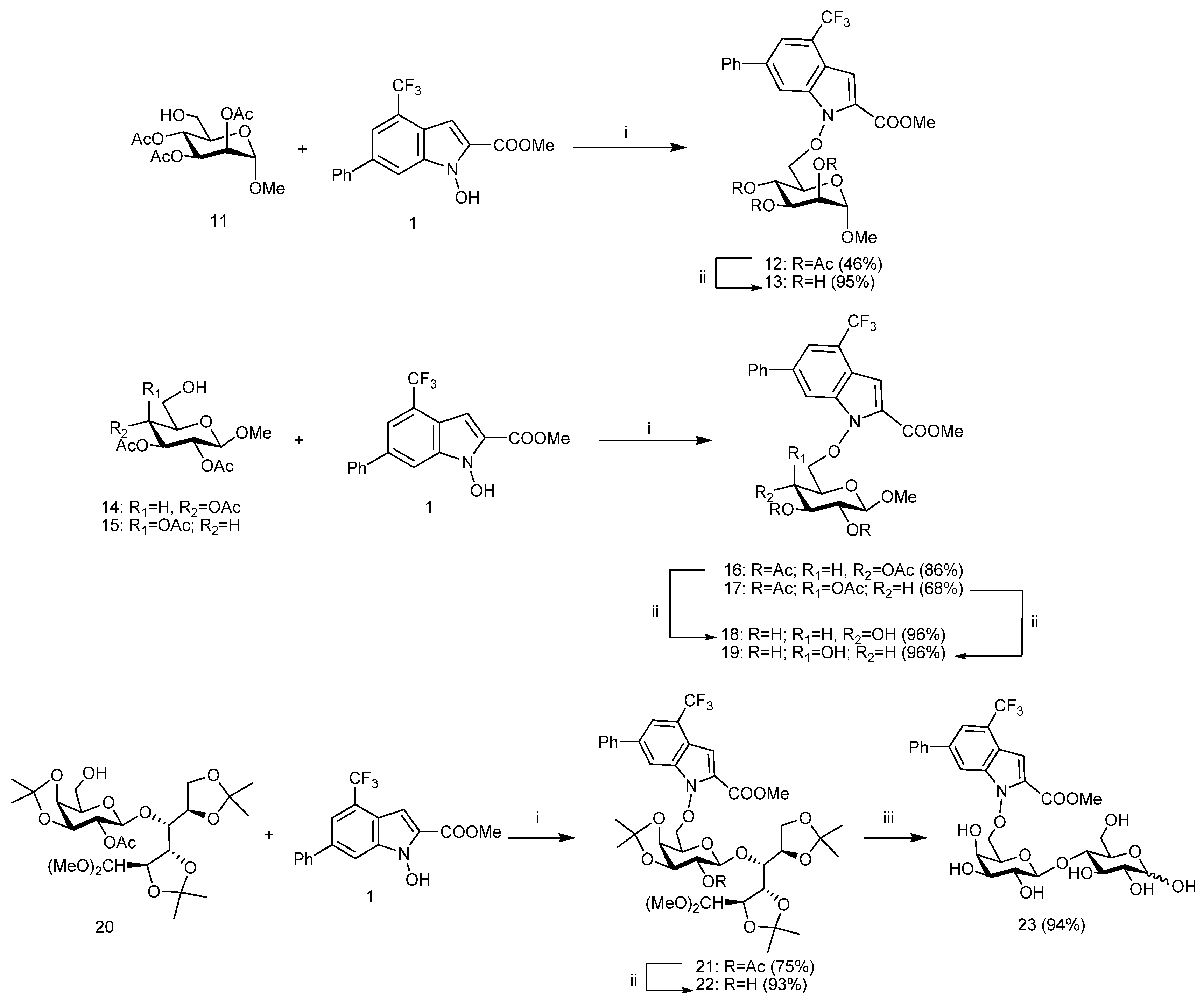
3.4. General Procedures for Mitsunobu Reaction
A solution of appropriate 6-OH-sugar
11 [
19],
14 [20],
15 [
21], or
20 [
26] (1.00 mmol, 1.0 equiv) and the
N-hydroxyindole derivative
1 (1.30 mmol, 1.3 equiv) in anhyd THF (20.0 mL) was cooled to 0 °C and treated with dry PPh
3 (3.50 mmol, 3.5 equiv) and di-isopropyl azodicarboxylate (DIAD) or di-2-methoxyethyl azodicarboxylate (DMEAD) (3.50 mmol, 3.5 equiv). The reaction mixture was stirred at room temp until TLC analysis showed the complete disappearance of the starting compound. The reaction mixture was diluted with CH
2Cl
2 (35.0 mL) and washed with satd aq Na
2HCO
3 (35 mL). The aqueous phases were extracted with CH
2Cl
2 (2 × 25 mL) and the combined organic layers were dried over MgSO
4, filtered and concentrated under diminished pressure. The crude residue was subjected to flash chromatography on silica gel.
3.5. Methyl 2,3,4-Tri-O-Acetyl-6-O-(2-Methoxycarbonyl-6-Phenyl-4-Trifluoromethyl-1H-Indol-1-Yl)-α-d-Mannopyranoside (12)
Mitsunobu reaction applied to 11 (120 mg, 0.38 mmol, 1.0 equiv) was performed with DIAD (263 μL, 1.33 mmol, 3.5 equiv) in accordance with the general procedure. The reaction was stopped after 3 h and the crude product was purified by flash chromatography on silica gel (3:7 hexane-EtOAc) gave pure 12 (108 mg, 46%) as a colorless oil; Rf 0.58 (1:1 hexane-EtOAc); [α]D +34.2 (c 0.89 in CHCl3); 1H NMR (250.13 MHz, CD3CN) δ: 8.13 (m, 1H, Ar-H), 7.81 (m, 1H, Ar-H), 7.77–7.64 (m, 2H, Ar-H), 7.55–7.39 (m, 3H, Ar-H), 7.22 (qd, 1H, Ar-H), 5.33 (dd, 1H, J3,4 10.2 Hz, J4,5 9.8 Hz, H-4), 5.23 (dd, 1H, J2,3 6.2 Hz, H-3), 4.90 (dd, 1H, J1,2 1.4 Hz, H-2), 4.86 (d, 1H, H-1), 4.68 (dd, 1H, J5,6b 5.6 Hz, J6a,6b 10.1 Hz, H-6b), 4.52 (dd, 1H, J5,6a 2.3 Hz, H-6a), 4.28 (ddd, 1H, H-5), 3.92 (s, 3H, COOMe), 3.44 (s, 3H, OMe), 2.04, 2.02, 1.95 (3s, each 3H, 3 × COMe); 13C NMR (62.9 MHz, CD3CN) δ: 171.0–170.8 (3 × COMe), 160.5 (COOMe), 140.5, 139.4, 137.2, 130.1 (4 × Ar-C), 130.1, 129.1, 128.3 (Ph-CH), 124.3–123.3 (CF3 + Ar-C), 120.0 (Ar-CH), 117.6 (Ar-C), 112.7, 105.4 (2 × Ar-CH), 99.5 (C-1), 78.9 (C-6), 72.7 (C-2), 70.1 (C-3), 69.4 (C-5), 66.4 (C-4), 55.9 (OMe), 52.9 (COOMe), 22.1, 21.0, 20.9 (3 × COMe). Anal. Calcd for C30H30F3NO11: C, 56.52; H, 4.74%; N, 2.20%; Found: C, 56.55; H, 4.77%; N, 2.23%.
3.6. Methyl 2,3,4-Tri-O-Acetyl-6-O-(2-Methoxycarbonyl-6-Phenyl-4-Trifluoromethyl-1H-Indol-1-Yl)-β-d-Glucopyranoside (16)
Mitsunobu reaction applied to 14 (72 mg, 0.22 mmol, 1.0 equiv) was performed with DMEAD (186 mg, 0.80 mmol, 3.5 equiv) in accordance with the general procedure. The reaction was stopped after 4 h and the crude product was purified by flash chromatography on silica gel (65:35 hexane-EtOAc) gave pure 16 (104 mg, 86%) as a yellow solid; Rf 0.53 (1:1 hexane-AcOEt); mp 191–194 °C (crom); [α]D –11.5 (c 1.0 in CHCl3); 1H NMR (250.13 MHz, CD3CN) δ: 8.04 (1H, m, Ar-H), 7.80 (m, 1H, Ar-H), 7.76 (m, 1H, Ar-H), 7.73 (m, 1H, Ar-H), 7.59–7.40 (m, 3H, Ar-H), 7.18 (qd, 1H, J 0.9 Hz, J 1.7 Hz, Ar-H), 5.27 (dd, 1H, J2,3 9.8 Hz, J3,4 9.5 Hz, H-3), 5.10 (dd, 1H, J4,5 10.1 Hz, H-4), 4.93 (dd, 1H, J1,2 8.0 Hz, H-2), 4.63 (dd, 1H, J5,6b 5.8 Hz, J6a,6b 10.8 Hz, H-6b), 4.56 (d, 1H, H-1), 4.49 (dd, 1H, J5,6a 2.1 Hz, H-6a), 4.10 (ddd, 1H, H-5), 3.91 (s, 3H, COOMe), 3.33 (s, 3H, OMe), 2.02, 2.00, 1.95 (3s, each 3H, 3 × COMe); 13C NMR (62.9 MHz, CD3CN): δ 170.9–170.4 (3 × COMe), 160.5 (COOMe), 140.5, 139.5, 137.2, 130.4 (4 × Ar-C), 130.0, 129.1, 128.3 (Ph-CH), 124.3–123.3 (CF3 + Ar-C), 120.1 (Ar-CH), 117.5 (Ar-C), 112.5, 105.4 (2 × Ar-CH), 102.1 (C-1), 78.4 (C-6), 73.5 (C-3), 72.5 (C-5), 71.9 (C-2), 69.2 (C-4), 57.3 (OMe), 52.9 (COOMe), 21.0–20.8 (3 × COMe). Anal. Calcd for C30H30F3NO11: C, 56.52; H, 4.74%; N, 2.20%; Found: C, 56.54; H, 4.76%; N, 2.24%.
3.7. Methyl 2,3,4-Tri-O-Acety-6-O-(2-Methoxycarbonyl)-6-Phenyl-4-Trifluoromethyl-1H-Indol-1-Yl)-β-d-Galactopyranoside (17)
Mitsunobu reaction applied to
15 [
5] (120 mg, 0.38 mmol, 1.0 equiv) was performed with DMEAD (306 mg, 1.30 mmol, 3.5 equiv) in accordance with the general procedure. The reaction was stopped after 4 h and the crude product was purified by flash chromatography on silica gel (7:3 hexane-EtOAc) gave pure
17 (162 mg, 68%) as a yellow solid;
Rf 0.55 (1:1 hexane-EtOAc); mp 169–172 °C (crom); [α]
D + 28.5 (
c 0.96 in CHCl
3);
1H NMR (250.13 MHz, CD
3CN) δ: 7.99 (m, 1H, Ar-
H), 7.78 (m, 1H, Ar-
H), 7.77–7.72 (m, 2H, Ar-
H), 7.54–7.38 (m, 3H, Ar-
H), 7.18 (qd, 1H,
J 1.0 Hz,
J 1.7 Hz, Ar-
H), 5.46 (dd, 1H,
J3,4 3.4 Hz,
J4,5 1.1 Hz, H-4), 5.10 (dd, 1H,
J2,3 10.5 Hz, H-3), 5.03 (dd, 1H,
J1,2 7.8 Hz, H-2), 4.64 (dd, 1H,
J5,6a 7.5 Hz,
J6a,6b 9.9 Hz, H-6a), 4.62 (dd, 1H,
J5,6b 3.1 Hz, H-6b), 4.51 (d, 1H, H-1), 4.35 (ddd, 1H, H-5), 3.91 (s, 3H, COO
Me), 3.35 (s, 3H, OMe), 2.07, 2.01, 1.92 (3s, each 3H, 3 × CO
Me);
13C NMR (62.9 MHz, CD
3CN) δ: 171.4, 170.9, 170.7 (3 ×
COMe), 160.5 (
COOMe), 140.5, 139.6, 137.2 130.8 (4 × Ar-
C), 130.0, 129.5, 128.3 (Ph-
CH), 124.3–123.2 (
CF
3 + Ar-
C), 120.0 (Ar-
CH), 117.4 (Ar-
C), 112.5, 105.4 (2 × Ar-
CH), 102.5 (C-1), 78.9 (C-6), 71.7 (C-3), 71.6 (C-5), 69.7 (C-2), 69.1 (C-4), 57.5 (OMe), 52.9 (COO
Me), 20.9, 20.8, 20.7 (3 × CO
Me). Anal. Calcd for C
30H
30F
3NO
11: C, 56.52; H, 4.74%; N, 2.20%; Found: C, 56.57; H, 4.75%; N, 2.25%.
3.8. 4-O-[2-O-Acetyl-3,4-O-Isopropylidene-6-O-(2-Methoxycarbonyl-6-Phenyl-4-Trifluoromethyl-1H-Indol-1-Yl)-β-d-Galactopyranosyl]-2,3:5,6-Di-O-Isopropylidene-Aldehydo-d-Glucose Dimethyl Acetal (21)
Mitsunobu reaction applied to
20 [
7] (170 mg, 0.30 mmol, 1.0 equiv) was performed with DMEAD (330 mg, 1.40 mmol, 3.5 equiv) in accordance with the general procedure. The reaction was stopped after 3.5 h and the crude product was purified by flash chromatography on silica gel (7:3 hexane-EtOAc) gave pure
21 (200 mg, 75%) as a yellow solid foam;
Rf 0.63 (1:1 hexane-EtOAc); mp 77–80 °C (crom); [α]
D -14.1 (
c 1.0 in CHCl
3);
1H NMR (250.13 MHz, CD
3CN) δ: 8.05 (m, 1H, Ar-
H), 7.77 (m, 1H, Ar-
H), 7.76–7.72 (m, 2H, Ar-
H), 7.56–7.40 (m, 3H, Ar-
H), 7.25 (qd, 1H,
J 0.9 Hz,
J 1.7 Hz, Ar-
H), 4.92 (dd, 1H,
J1′,2′ 8.3 Hz,
J2′,3′ 7.6 Hz, H-2′), 4.79 (d, 1H, H-1′), 4.73 (dd, 1H,
J5′,6′b 6.5 Hz,
J6′a,6′b 10.1 Hz, H-6′b), 4.68 (dd, 1H,
J5′,6′a 5.2 Hz, H-6′a), 4.39 (dd, 1H,
J1,2 5.6 Hz,
J2,3 6.7 Hz, H-2), 4.30 (d, 1H, H-1), 4.29–4.21 (m, 2H, H-4′, H-5′), 4.16 (dd, 1H,
J3′,4′ 5.2 Hz, H-3′), 4.15–3.96 (m, 2H, H-3, H-5), 3.99 (dd, 1H,
J3,4 1.6 Hz,
J4,5 3.1 Hz, H-4), 3.96 (s, 3H, COO
Me), 3.83 m, (2H, H-6a, H-6b), 3.29, 3.24 (2s, each 3H, 2 × OMe), 2.06 (s, 3H, CO
Me), 1.49, 1.38 (2s, each 3H, C
Me2), 1.28 (s, 6H, C
Me2), 1.27, 1.25 (2s, each 3H, C
Me2);
13C NMR (62.9 MHz, CD
3CN): δ 170.3 (
COMe), 160.3 (
COOMe), 140.6, 139.8, 137.6, 131.4 (4 × Ar-
C), 129.8, 128.9, 128.4 (Ph-
CH), 124.7–123.5 (
CF
3 + Ar-
C), 120.2 (Ar-
CH), 117.3 (Ar-
C), 112.3 (Ar-
CH), 111.1, 110.8, 108.5 (3 ×
CMe
2), 106.1 (C-1), 105.6 (Ar-CH), 101.0 (C-1′), 78.7 (C-6′), 78.4 (C-5), 78.2 (C-3), 77.6 (C-3′), 76.7 (C-2), 76.0 (C-4), 74.7 (C-4′), 73.3 (C-2′), 70.5 (C-5′), 65.2 (C-6), 56.2, 54.5 (2 × OMe), 52.8 (COO
Me), 28.1, 27.7, 26.8, 26.5, 26.3, 24.8 (3 × C
Me2), 21.1 (COMe). Anal. Calcd for C
42H
52F
3NO
15: C, 58.13; H, 6.04%; N, 1.61%; Found: C, 58.16; H, 6.07%; N, 1.64%.
3.9. General Procedures for Deacetylation
To a solution of appropriate acetylated NHI-glycoconjugates 6α, 6β, 9β, 12, 16, or 17 (1.00 mmol, 1.0 equiv) in a 2:3 mixture of CH2Cl2-MeOH (46.0 mL) was cooled at 0 °C and treated with a methanolic solution of MeONa (0.33 M, 0.575 mL). The reaction mixture was stirred at room temp until TLC analysis showed the complete disappearance of the starting material (2–7 h) and the formation of a lower moving product. The solution was neutralized with Amberlite® IR120 H resin and the suspension was filtered and concentrated under diminished pressure. Purification of crude product by crystallization or trituration with Et2O afforded pure NHI-glycoconjugates 7α, 7β, 10β, 13, 18, or 19.
3.10. Methyl 1-(α-d-Galactopyranosyl)-6-Phenyl-4-Trifluoromethyl-1H-Indole-2-Carboxylate (7α)
Deacetylation of 6α (74 mg, 0.11 mmol, 1.0 equiv) was performed in accordance with the general procedure. The reaction was stirred until the starting compound was completely reacted (2 h, TLC, 9:1 EtOAc-MeOH). The crude residue (52 mg, 91% yield) was constituted (NMR) exclusively by a title compound 7α. The trituration of the crude product (52 mg) with Et2O afforded a sample of pure 7α (28 mg, 49%) as a yellow solid; Rf 0.54 (9:1 EtOAc-MeOH), mp 159–162 °C (Et2O); [α]D -15.3 (c 0.63 in MeOH); 1H NMR (250.12 MHz, CD3OD-CDCl3) δ: 8.41 (m, 1H, Ar-H), 7.86–7.62 (m, 3H, Ar-H), 7.48–7.33 (m, 3H, Ar-H), 7.27 (bs, 1H, Ar-H), 5.37 (d, 1H, J1,2 3.9 Hz, H-1), 4.51 (m, 1H, H-5), 4.15 (m, 1H, H-4), 4.08 (dd, 1H, J2,3 10.6 Hz, H-2), 4.00 (dd, 1H, J3,4 2.8 Hz, H-3), 3.95 (s, 3H, COOMe), 3.91 (bs, 1H, OH), 3.80–3.65 (m, 4H, H-6a, H-6b, 2 × OH), 3.36 (bs, 1H, OH); 13C NMR (62.9 MHz, CD3OD-CDCl3) δ: 161.5 (CO), 140.6, 139.6, 137.5, 131.9 (4 × Ar-C), 129.7, 128.7, 128.0 (Ph-CH), 125.2–123.0 (CF3 + Ar-C), 119.8 (Ar-CH), 118.1 (Ar-C), 113.4 (Ar-CH), 109.4 (C-1), 106.4 (Ar-CH), 75.1 (C-5), 70.4 (C-2), 69.9 (C-3), 69.3 (C-4), 61.9 (C-6), 52.8 (OMe). Anal. Calcd for C23H22F3NO8: C, 55.54; H, 4.46%; N, 2.82%; Found: C, 55.57; H, 4.49%; N, 2.86%.
3.11. Methyl 1-(β-d-Galactopyranosyl)-6-Phenyl-4-Trifluoromethyl-1H-Indole-2-Carboxylate (7β)
Deacetylation of 6β (60 mg, 0.089 mmol, 1.0 equiv) was performed in accordance with the general procedure. The reaction was stirred until the starting compound was completely reacted (2 h, TLC, 9:1 EtOAc-MeOH). The crude residue (43 mg, 91% yield) was constituted (NMR) exclusively by a title compound 7β. The trituration of the crude product with Et2O afforded a sample of pure 7β (30 mg, 66%) as a white solid; Rf 0.83 (9:1 EtOAc-MeOH); mp 207–210 °C (Et2O); [α]D +39.4 (c 0.505 in MeOH); 1H NMR (250.13 MHz, CD3OD-CDCl3) δ: 8.24 (bs, 1H, Ar-H), 7.71 (m, 1H, Ar-H), 7.68–7.73 (2m, each 1H, Ar-H), 7.48–7.39 (m, 2H, Ar-H), 7.38–7.32 (m, 1H, Ar-H), 7.27 (qd, 1H, Ar-H), 5.09 (d, 1H, J1,2 8.1 Hz, H-1), 3.95 (m, 1H, H-2), 3.94 (s, 3H, COOMe), 3.81 (dd, 1H, J5,6b 6.9 Hz, J6a,6b 11.0 Hz, H-6b), 3.67 (dd, 1H, J5,6a 5.5 Hz, H-6a), 3.62 (dd, 1H, J2,3 9.4 Hz, J3,4 3.4 Hz, H-3), 3.60 (m, 1H, H-4), 3.49 (ddd, 1H, J4,5 0.8 Hz, H-5); 13C NMR (62.9 MHz, CD3OD-CDCl3) δ: 161.8 (CO), 140.5, 139.5, 137.4, 132.1 (4 × Ar-C), 129.6, 128.9, 128.4 (Ph-CH), 125.4–123.6 (CF3 + Ar-C), 119.7 (Ar-CH), 118.3 (Ar-C), 113.6 (Ar-CH), 110.3 (C-1), 106.8 (Ar-CH), 76.7 (C-5), 74.3 (C-3), 72.3 (C-2), 69.9 (C-4), 62.2 (C-6), 53.4 (COOMe). Anal. Calcd for C23H22F3NO8: C, 55.54; H, 4.46%; N, 2.82%; Found: C, 55.57; H, 4.49%; N, 2.85%.
3.12. Methyl 1-[4-O-(β-d-Galactopyranosyl)-β-d-Glucopyranosyl)]-6-Phenyl-4-Trifluoromethyl-1H-Indole-2-Carboxylate (10β)
Deacetylation of 9β (53 mg, 0.055 mmol, 1.0 equiv) was performed in accordance with the general procedure. The reaction was stirred until the starting compound was completely reacted (8 h, TLC, 9:1 EtOAc-MeOH). The crude residue (37 mg, 96% yield) was constituted (NMR) exclusively by a title compound 10β. The trituration of the crude product (37 mg) with Et2O afforded a sample of pure 10β (27 mg, 74%) as a white solid: Rf 0.10 (9:1 EtOAc-MeOH); mp 199–202 °C. [α]D +63.3 (c 0.59 in MeOH); 1H NMR (250.13 MHz, DMSO-d6) δ: 8.27 (m, 1H, Ar-H), 7.83–7.80 (m, 3H, Ar-H), 7.55–7.49 (m, 3H, Ar-H), 7.10 (bs, 1H, Ar-H), 5.78 (bd, 1H, J 5.0 Hz, OH), 5.14–5.01 (m, 2H, 2 × OH), 4.87–4.78 (m, 2H, 1-H, OH), 4.69–4.61 (m, 2H, 2 × OH), 4.56–4.43 (m, 2H, H-1′, OH), 4.31–4.16 (m, 3H, H-6a, H-6b, H-5′), 3.88 (s, 3H, COOMe), 3.46–3.22 (m, 9H, H-2, H-3, H-4, H-5, H-2′, H-3′, H-4′, H-6′a, H-6′b; 13C NMR (62.9 MHz, DMSO-d6) δ: 159.7 (CO), 139.0, 139.0, 137.0, 130.0 (4 × Ar-C), 129.2, 128.2, 127.3, 127.2 (Ph-CH), 124.0–121.8 (CF3 + Ar-C), 118.8 (Ar-CH), 116.7 (Ar-C), 113.5, 113.0 (2 × Ar-CH), 107.7 (C-1), 103.7 (C-1′), 79.4 (C-4), 75.6 (C-5′), 74.8, 74.3 (C-5, C-3), 73.3, 72.0 (C-2, C-3′), 70.5 (C-2′), 68.1 (C-4′), 60.4, 60.3 (C-6′, C-6), 52.4 (COOMe). Anal. Calcd for C29H32F3NO13: C, 52.81; H, 4.89%; N, 2.12%; Found: C, 52.84; H, 4.91%; N, 2.15%.
3.13. Methyl 6-O-(2-Methoxycarbonyl-6-Phenyl-4-Trifluoromethyl-1H-Indol-1-Yl)-α-d-Mannopyra-Noside (13)
Deacetylation of 12 (54 mg, 0.085 mmol, 1.0 equiv) was performed in accordance with the general procedure. The reaction was stirred until the starting compound was completely reacted (3.5 h, TLC, 9:1 EtOAc-MeOH). The crude residue (42 mg, 95% yield) was constituted (NMR) exclusively by a title compound 13. The crystallization (Et2O-hexane) of crude product (42 mg) afforded a sample of pure 13 (32 mg, 73%) as a pale yellow solid; Rf 0.69 (9:1 EtOAc-MeOH); mp 59–62 °C (Et2O-hexane); [α]D +21.6(c 0.92 in MeOH); 1H NMR (250.13 MHz, CD3OD-CDCl3) δ: 7.95 (bs, 1H, Ar-H), 7.67–7.61 (m, 3H, Ar-H), 7.49–7.32 (m, 3H, Ar-H), 7.16 (bs, 1H, Ar-H), 4.81 (bs, 1H, H-1), 4.64 (dd, 1H, J5,6b 3.8 Hz, J6a,6b 9.4 Hz, H-6b), 4.53 (dd, 1H, J5,6a 1.8 Hz, H-6a), 4.19 (dd, 1H, J3,4 9.6 Hz, J4,5 9.4 Hz, H-4), 4.02 (bs, 1H, H-2), 3.93–3.75 (m, 5H, H-3, H-5, 3 × OH), 3.89 (s, 3H, COOMe), 3.35 (s, 3H, OMe); 13C NMR (62.9 MHz, CD3OD-CDCl3) δ: 161.2 (CO), 140.6, 139.5, 136.8, 131.9 (4 × Ar-C), 129.7, 128.6, 128.1 (3 × Ph-CH), 124.8–122.7 (CF3 + Ar-C), 120.1 (Ar-CH), 117.2 (Ar-C), 112.0, 106.4 (2 × Ar-CH), 101.9 (C-1), 78.1 (C-6), 72.1 (C-3), 71.2 (C-2), 71.0 (C-5), 67.5 (C-4), 55.9 (OMe), 51.3 (COOMe). Anal. Calcd for C24H24F3NO8: C, 56.36; H, 4.73%; N, 2.74%; Found: C, 56.39; H, 4.75%; N, 2.77%.
3.14. Methyl 6-O-(2-Methoxycarbonyl-6-Phenyl-4-Trifluoromethyl-1H-Indol-1-Yl)-β-d-Glucopyra-Noside (18)
Deacetylation of 16 (52 mg, 0.081 mmol, 1.0 equiv) was performed in accordance with the general procedure. The reaction was stirred until the starting compound was completely reacted (3.5 h, TLC, 9:1 EtOAc-MeOH). The crude residue (40 mg, 96% yield) was constituted (NMR) exclusively by a title compound 18. The trituration of the crude product (40 mg) with Et2O afforded a sample pure 18 (18 mg, 43% yield) as a white solid; Rf 0.53 (9:1 EtOAc-MeOH); mp 211–214 °C (Et2O); [α]D -73.7 (c 0.66 in MeOH); 1H NMR (250.13 MHz, CD3OD) δ: 8.10 (m, 1H, Ar-H), 7.70–7.62 (m, 3H, Ar-H), 7.50–7.38 (m, 3H, Ar-H), 7.15 (qd, 1H, Ar-H), 4.79 (dd, 1H, J5,6b 1.6 Hz, J6a,6b 10.5 Hz, H-6b), 4.58 (dd, 1H, J5,6a 6.3 Hz, H-6a), 4.18 (d, 1H, J1,2 7.7 Hz, H-1), 3.95 (s, 3H, COOMe), 3.70 (ddd, 1H, J4,5 9.8 Hz, H-5), 3.41–3.30 (m, 2H, H-3, H-4), 3.36 (s, 3H, OMe), 3.24 (dd, 1H, J2,3 9.4 Hz, H-2); 13C NMR (62.9 MHz, CD3OD) δ: 161.2 (CO), 141.0, 140.1, 137.8, 131.8 (4 × Ar-C), 130.1, 129.1, 128.4 (Ph-CH), 124.9–123.2 (CF3 + Ar-C), 119.9 (Ar-CH), 117.8 (Ar-C), 112.7 (Ar-CH), 105.8 (C-1), 105.4 (Ar-CH), 80.1 (C-6), 77.8, 71.3 (C-3, C-4), 75.5 (C-5), 74.9 (C-2), 57.5 (OMe), 52.7 (COOMe). Anal. Calcd for C24H24F3NO8: C, 56.36; H, 4.73%; N, 2.74%; Found: C, 56.40; H, 4.76%; N, 2.78%.
3.15. Methyl 6-O-(2-Methoxycarbonyl-6-Phenyl-4-Trifluoromethyl-1H-Indol-1-Yl)-β-d-Galactopyra-Noside (19)
Deacetylation of 17 (68 mg, 0.11 mmol, 1.0 equiv) was performed in accordance with the general procedure. The reaction was stirred until the starting compound was completely reacted (3 h, TLC, 9:1 EtOAc-MeOH). The crude residue (52 mg, 96% yield) was constituted (NMR) exclusively by a title compound 19. The trituration of the crude product (52 mg) with Et2O afforded a sample pure 19 (41 mg, 76%) as a white solid; Rf 0.60 (9:1 EtOAc-MeOH); mp 191–194 °C (Et2O); [α]D –21.6 (c 0.70 in MeOH); 1H NMR (250.13 MHz, CD3OD-CDCl3) δ: 7.99 (m, 1H, Ar-H), 7.68–7.62 (m, 3H, Ar-H), 7.47–7.32 (m, 3H, Ar-H), 7.24 (m, 1H, Ar-H), 4.74 (dd, 1H, J5,6b 3.7 Hz, J6a,6b 9.5 Hz, H-6b), 4.59 (dd, 1H, J5,6a 7.2 Hz, H-6a), 4.15 (d, 1H, J1,2 7.6 Hz, H-1), 4.01 (m, 1H, H-5), 3.96 (m, 1H, H-3), 3.93 (s, 3H, COOMe), 3.53 (m, 2H, H-2, H-4), 3.44 (s, 3H, OMe); 13C NMR (62.9 MHz, CD3OD-CDCl3): δ 160.6 (CO), 140.3, 139.4, 139.3, 131.7 (4 × Ar-C), 129.2, 128.3, 127.7 (Ph-CH), 124.7–123.3 (CF3 + Ar-C), 119.6 (Ar-CH), 117.2 (Ar-C), 111.6, 105.8 (2 × Ar-CH), 104.5 (C-1), 79.0 (C-6), 73.5 (C-2), 72.5 (C-5), 71.3 (C-4), 69.5 (C-3), 57.2 (OMe), 52.4 (COOMe). Anal. Calcd for C24H24F3NO8: C, 56.36; H, 4.73%; N, 2.74%; Found: C, 56.38; H, 4.76%; N, 2.78%.
3.16. 4-O-[3,4-O-Isopropylidene-6-O-(2-Methoxycarbonyl-6-Phenyl-4-Trifluoromethyl-1H-Indol-1-Yl)-β-d-Galactopyranosyl]-2,3:5,6-Di-O-Isopropylidene-Aldehydo-d-Glucose Dimethyl Acetal (22)
Deacetylation of 21 (75 mg, 0.087 mmol, 1.0 equiv) was performed in accordance with the general procedure. The reaction was stirred until the starting compound was completely reacted (8 h, TLC, 1:1 hexane-EtOAc). Purification of the crude product by flash chromatography on silica gel (1:1 hexane-EtOAc) gave pure 22 (67 mg, 93%) as a white foam; Rf 0.43 (1:1 hexane-EtOAc); mp 70–73 °C (chrom); [α]D -23.2 (c 1.1 in CHCl3); 1H NMR (250.13 MHz, CD3CN) δ: 8.03 (m, 1H, Ar-H), 7.79 (bs, 1H, Ar-H), 7.76–7.71 (m, 2H, Ar-H), 7.54–7.39 (m, 3H, Ar-H), 7.23 (qd, 1H, J 1.0 Hz, J 1.7 Hz, Ar-H), 4.70 (dd, 1H, J5′,6′b 6.9 Hz, J6′a,6′b 9.4 Hz, H-6′b), 4.63 (dd, 1H, J5′,6′a 5.0 Hz, H-6′a), 4.51 (d, 1H, J1′,2′ 8.1 Hz, H-1′), 4.46 (dd, 1H, J1,2 5.9 Hz, J2,3 7.2 Hz, H-2), 4.30 (dd, 1H, J4′,5′ 2.2 Hz, H-5′), 4.29 (d, 1H, H-1), 4.23 (dd, 1H, J3′,4′ 5.5 Hz, H-4′), 4.15 (m, 1H, H-5), 4.14–4.00 (m, 3H, H-6a, H-6b, H-3′), 4.03 (dd, 1H, J3,4 1.6 Hz, H-3), 3.94 (s, 3H, COOMe), 3.88 (dd, 1H, J4,5 4.0 Hz, H-4), 3.55 (bs, 1H, 2-OH), 3.43 (m, 1H, H-2′), 3.27, 3.20 (2s, each 3H, 2 × OMe), 1.46, 1.37, 1.33, 1.29, 1.27, 1.26 (6s, each 3H, 3 × CMe2); 13C NMR (62.9 MHz, CD3CN) δ: 160.4 (CO), 140.7, 139.8, 137.5, 131.4 (4 × Ar-C), 130.0, 129.1, 128.6 (Ph-CH), 124.7–123.3 (CF3 + Ar-C), 120.3 (Ar-CH), 117.5 (Ar-C), 112.6 (Ar-CH), 110.7, 110.5, 109.0 (3 × CMe2), 106.2 (C-1), 105.5 (Ar-CH), 104.4 (C-1′), 80.0 (C-3), 78.6 (C-6′), 78.5 (C-3′), 78.3 (C-4), 77.8 (C-5), 74.5 (C-4′), 74.4 (C-2′), 72.6 (C-2), 71.0 (C-5′), 65.9 (C-6), 56.3, 54.2 (2 × OMe), 52.9 (COOMe), 28.4, 27.5, 27.0, 26.5, 26.3, 25.0 (3 × CMe2). Anal. Calcd for C40H50F3NO14: C, 58.18; H, 6.10%; N, 1.70%; Found: C, 58.21; H, 6.13%; N, 1.74%.
3.17. 4-O-[6-O-(2-Methoxycarbonyl-6-Phenyl-4-Trifluoromethyl-1H-Indol-1-Yl)-β-d-Galactopyra-Nosyl]-α,β-d-Glucopyranose (23)
A solution of 22 (59 mg, 0.072 mmol, 1.0 equiv) in 80% aq AcOH (1.3 mL) was stirred at 80 °C. After 4 h, the TLC analysis (9:1 EtOAc-MeOH) showed the complete disappearance of the starting material and the formation of a lower moving product (Rf 0.10). The reaction mixture was concentrated under diminished pressure and repeatedly coevaporated with toluene (4 × 20 mL). Trituration of the residue (45 mg) with Et2O afforded an amorphous white solid (23 mg, 48% yield) composed (13C NMR, DMSO-d6) by a α-anomer 23 (>95%). 1H NMR (250.13 MHz, DMSO-d6) δ: 8.15 (m, 1H, Ar-H), 7.92–7.83 (m, 3H, Ar-H), 7.57–7.41 (m, 3H, Ar-H), 7.15 (m, 1H, Ar-H), 6.40 (d, 1H, J1,OH 4.6 Hz, OH-1), 5.23 (d, 1H, J 7.2 Hz, OH), 4.92 (m, 2H, 2 × OH), 4.83 (d, 1H, J1,2 5.1 Hz, H-1), 4.72–4.67 (m, 2H, 2 × OH), 4.58–4.47 (m, 3H, H-6′a, H-6′b, OH), 4.34 (d, 1H, J1′,2′ 7.2 Hz, H-1′), 4.12 (bt, 1H, H-5′), 3.91 (s, 3H, COOMe), 3.78–3.48 (m, 5H, H-2, H-3, H-6a, H-6b, H-3′), 3.40–3.20 (m, 4H, H-4, H-5, H-4′, H-2′); 13C NMR (62.9 MHz, DMSO-d6) δ: 159.0 (CO), 138.8, 138.2, 135.8, 129.2 (4 × Ar-C), 129.3, 128.1, 127.6 (Ph-CH), 123.5–122.3 (CF3 + Ar-C), 118.8 (Ar-CH), 115.9 (Ar-C), 111.8, 104.1 (2 × Ar-CH), 104.0 (C-1′), 92.1 (C-1), 82.2 (C-4), 79.4 (C-6′), 72.7 (C-2′), 72.3 (C-5′), 72.0 (C-5), 71.7 (C-3), 70.3 (C-4′), 69.8 (C-2), 68.9 (C-3′), 60.7 (C-6), 52.4 (COOMe). Anal. Calcd for C29H32F3NO13: C, 52.81; H, 4.89%; N, 2.12%; Found: C, 52.84; H, 4.92%; N, 2.15%.
3.18. Enzymatic Assays
The LDH inhibition activities of the compounds were evaluated against purified human lactate dehydrogenase isoform 5 (Lee Biosolution, Inc.). The ‘forward’ direction (pyruvate→lactate) of the lactate dehydrogenase reaction was conducted, and the IC50 measurements were performed by fluorescence (emission wavelength at 460 nm, excitation wavelength at 340 nm) to monitor the amount of consumed NADH. Assays were carried out in wells containing 200 μL of a reagents solution, dissolved in 100 mM phosphate buffer (pH = 7.4), in the presence of 200 μM pyruvate and 40 μM NADH. For the IC50 calculations of the compounds, seven different concentrations (in duplicate for each concentration) were used to produce the concentration–response curve. As well as the sample test wells, maximum and minimum controls were also included in each plate. After 15 min of incubation, the final measurements were carried out by using a Victor X3 Microplates Reader (PerkinElmer®). IC50 values were produced using GraphPad Prism software (GraphPad, San Diego, CA, USA).
3.19. Molecular Modeling Studies
The seven compounds were built using Maestro 9.0 and were minimized using the conjugate gradient method until a convergence value of 0.05 kcal (mol Å)
−1 was reached. The minimization was carried out in a water environment model (generalized-Born/surface-area model) using the MMFF force field and a distance-dependent dielectric constant of 1.0. The
hLDH5 chain was extracted from the minimized average structure previously obtained by us [
14]. AUTODOCK 4.2 software [
32] was employed for molecular docking. The identification of the torsion angles in the ligands, the addition of the solvent model and the determination of protein and ligand atomic charges was carried out using Autodock Tools. Kollmann charges were assigned to the protein and Gasteiger charges to the ligand. A grid spacing of 0.375 Å and a distance-dependent function of the dielectric constant were used for the energetic map calculations. The compounds were subjected to a robust docking procedure by applying 200 runs of Autodock search, using the Lamarckian Genetic Algorithm with 10,000,000 steps of energy evaluations [
33]. The number of individuals in the initial population was set to 500 and a maximum of 10,000,000 generations were simulated during each docking run.
3.20. Cancer Cell Antiproliferative Potency Assays
Determination of cellular IC
50 values was performed under normoxic conditions as described previously [
10]. Briefly, HeLa and A549 cells, grown in RPMI 1640 medium supplemented with 10% FBS and 1% Penicillin/Streptomycin, were added at a density of 5000 cells per well to 96 well plates to which 31.6 nM–200 µM compound in DMSO was already added (1% final concentration DMSO in all wells; triplicate wells at the same concentration per repetition). Plates were incubated at 37 °C in a 95% air/5% CO
2 atmosphere for 72 h. Medium was removed and cells were fixed by the addition of 50 µL 10% trichloroacetic acid in water, 4 °C, to each well. Plates were incubated at 4 °C for at least one hour, and the sulforhodamine B colorimetric assay was performed to assess remaining biomass in each well. Cells treated with 1% DMSO were used as the 100% live control for biomass, and wells incubated with medium alone were used as the baseline zero biomass control. IC
50 values were calculated using SoftMax Pro software (Molecular Devices, Sunnyvale, CA).
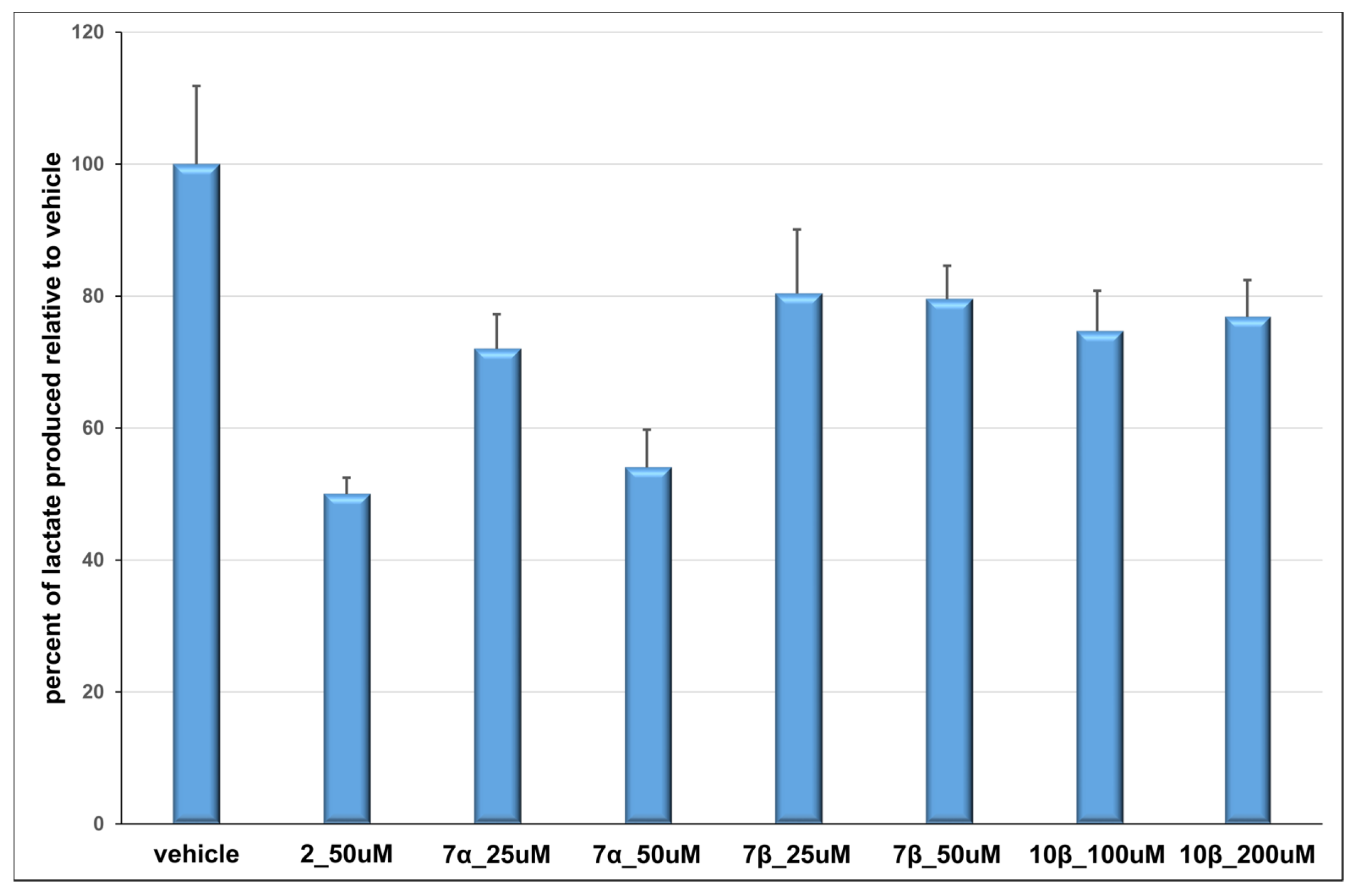
3.21. Cellular Lactate Production Inhibition Assays
This assay was performed under normoxic conditions as previously described [
10]. Briefly, confluent HeLa cervical carcinoma cells (ATCC, Manassas, VA, USA) in a 96-well plate were treated with compound or vehicle control (1% DMSO final concentration in all samples) in DMEM minus phenol red + 10% dialyzed FBS + 1% Penstrep, supplemented with 10 mM glucose, 1 mM sodium pyruvate and 4 mM glutamine, in a final volume of 125 µL per well. Immediately following compound addition, plates were incubated for 8 h at 37 °C in a 95% air/5% CO
2 atmosphere. Duplicate wells were prepared for each treatment. Following treatment, medium was collected, and 100 µL were added to 2 µL 50 mM chlorophenylalanine (CPA; internal standard for GC-MS analysis). Samples were concentrated, derivatized by a four-hour incubation with MTBSTFA + 1% TBDMCS (Thermo Scientific, Walthman, MA, USA) in acetonitrile at 85 °C, and immediately analyzed using GC-MS (Agilent 6890N GC/5973 MS, equipped with an Agilent DB-5 capillary column, 30 M × 320 µM × 0.25 µM, model number J&W 123–5032, Agilent Technologies, Santa Clara, CA, USA) and an electron impact ionization source. One microliter of each sample was injected using an automated injector, and a solvent delay of 8.20 min was implemented. The initial oven temperature was 120 °C, held for 5 min; then the temperature was increased at a rate of 10 °C min
−1 until a temperature of 250 °C was reached. Temperature was then increased by 40 °C min
−1 until a final temperature of 310 °C was reached. Total run time per sample was 22.5 min. Compounds were identified using AMDIS Chromatogram software (Amdis, freeware available from amdis.net) and programmed WIST and Niley commercial libraries. The integration area of lactate in each sample was divided by the integration area of CPA in the same sample to achieve a lactate/internal standard ratio. The ratios were averaged for duplicates, and percent lactate production over the vehicle was calculated for each independent experiment. The mean lactate production/vehicle was then averaged between three or more independent experiments.

 ,
, 










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}