
Linear, Non-Conjugated Cyclic and Conjugated Cyclic Paraphenylene under Pressure

 , , ,
, , ,  and
and
Abstract
1. Introduction
2. Results
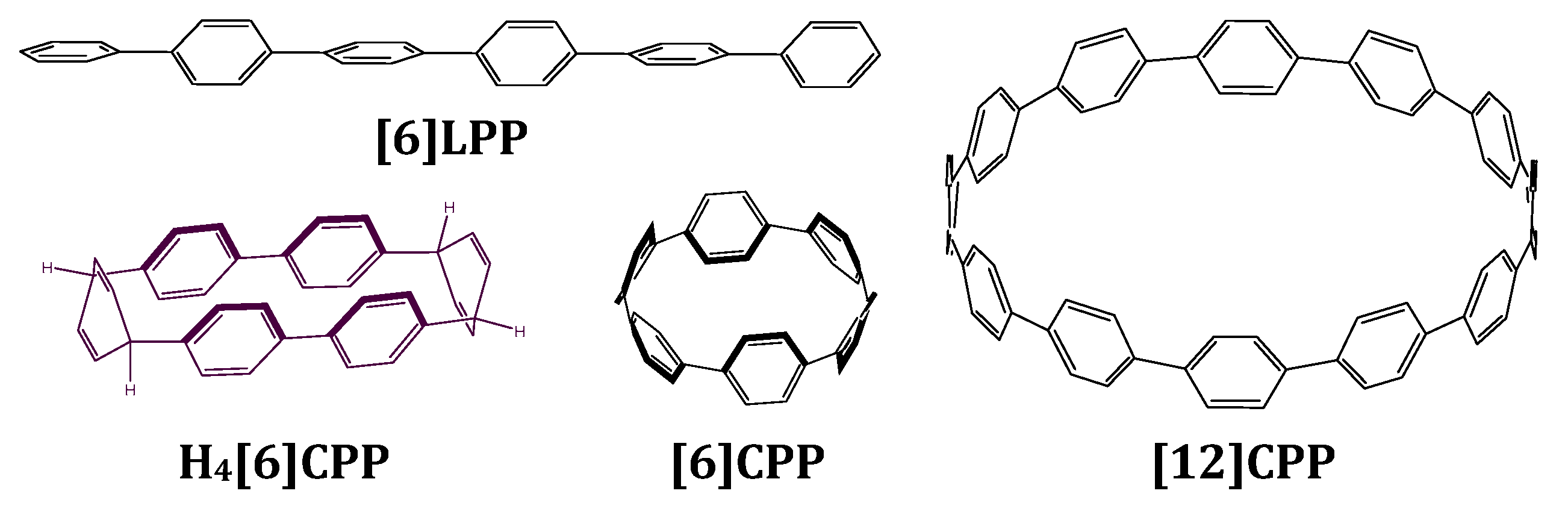
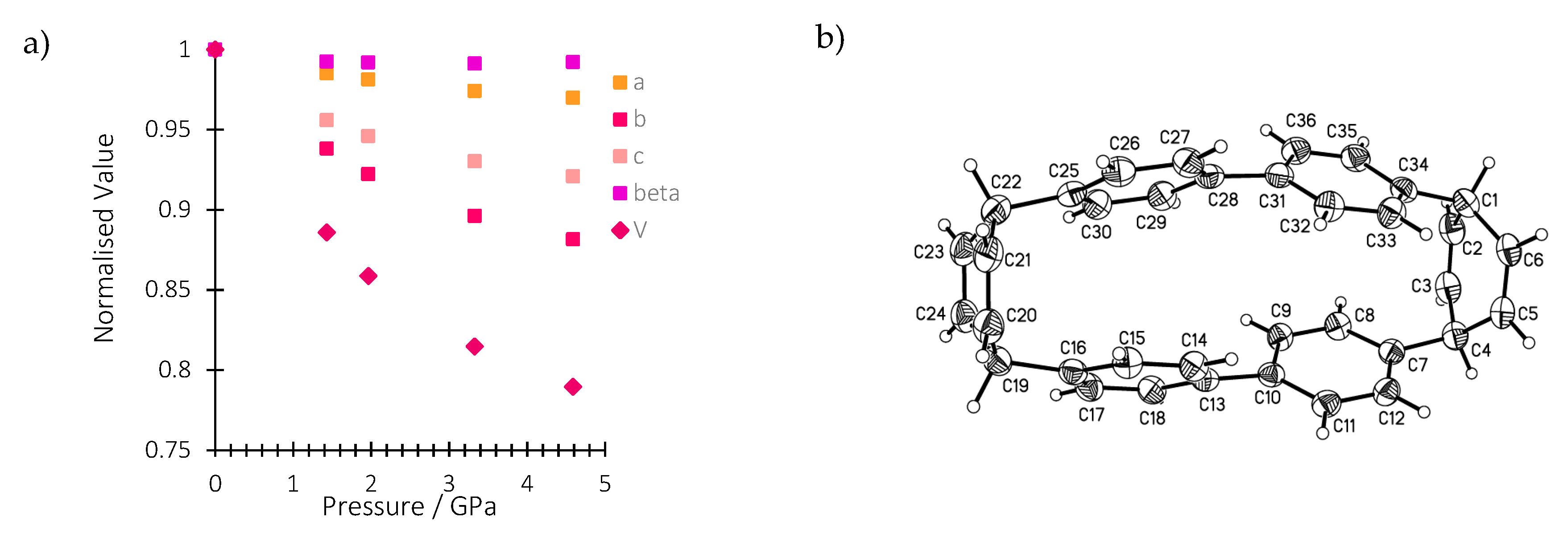
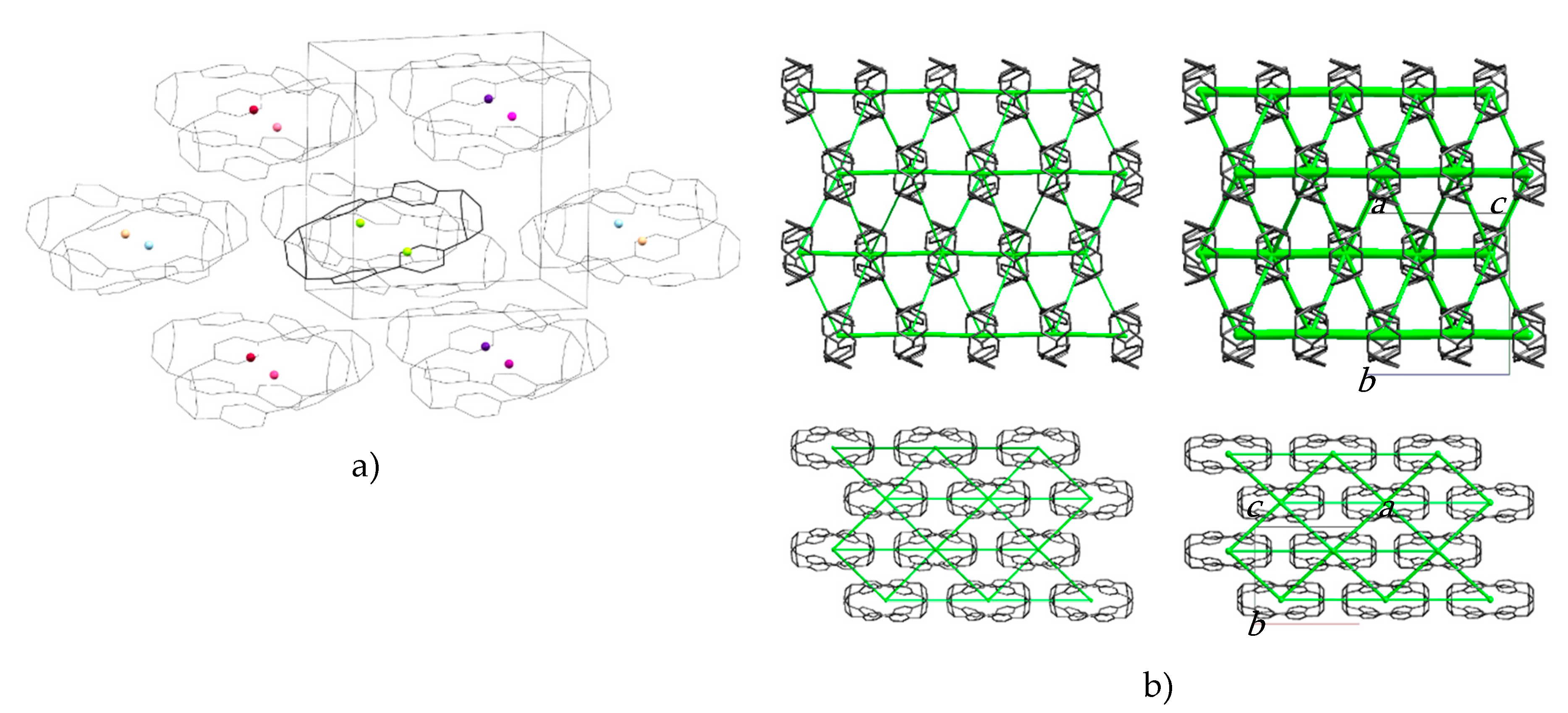
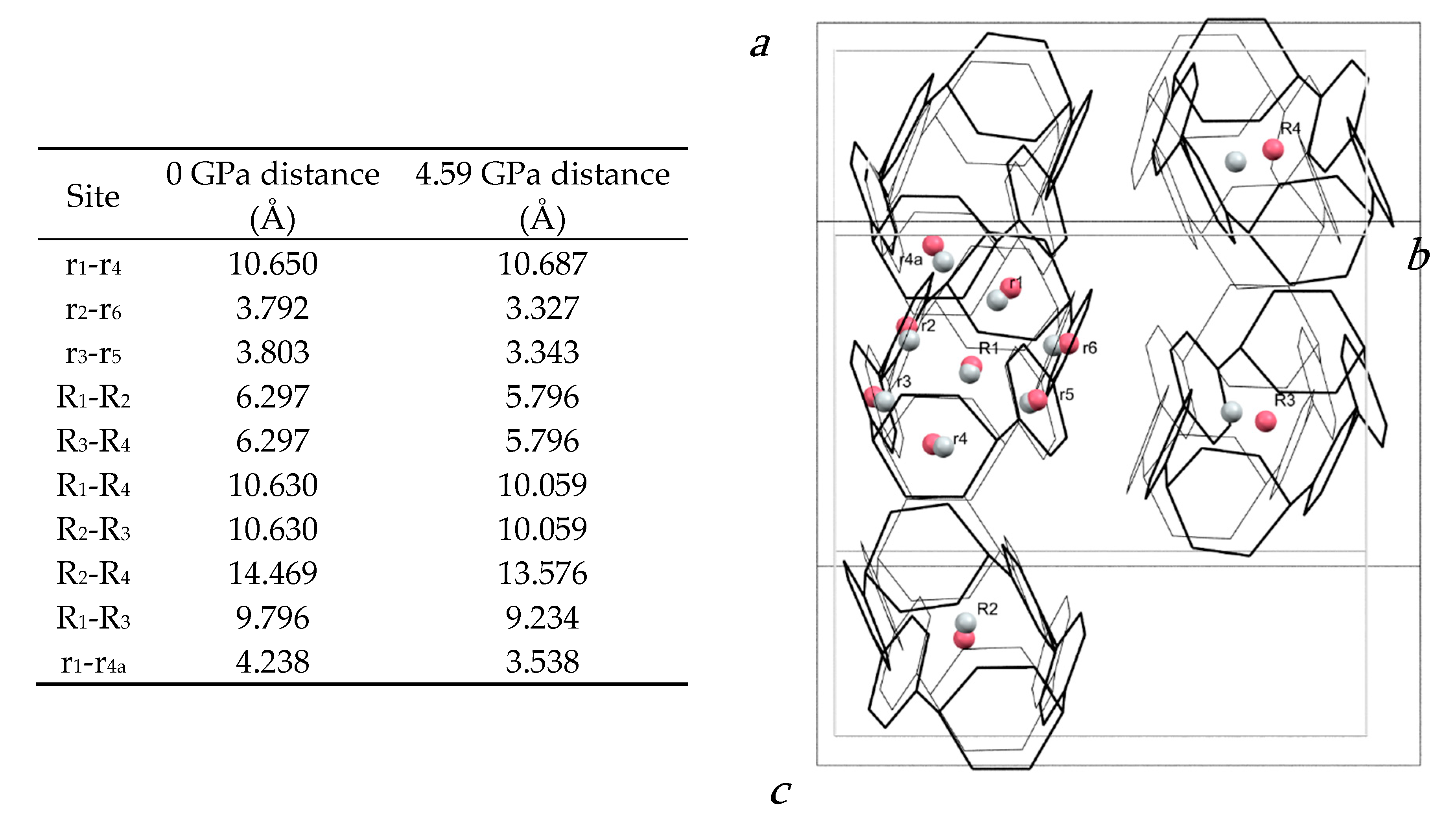
2.1. Effect of Pressure on the Crystal Structure
2.2. FTIR
2.3. Raman Spectroscopy
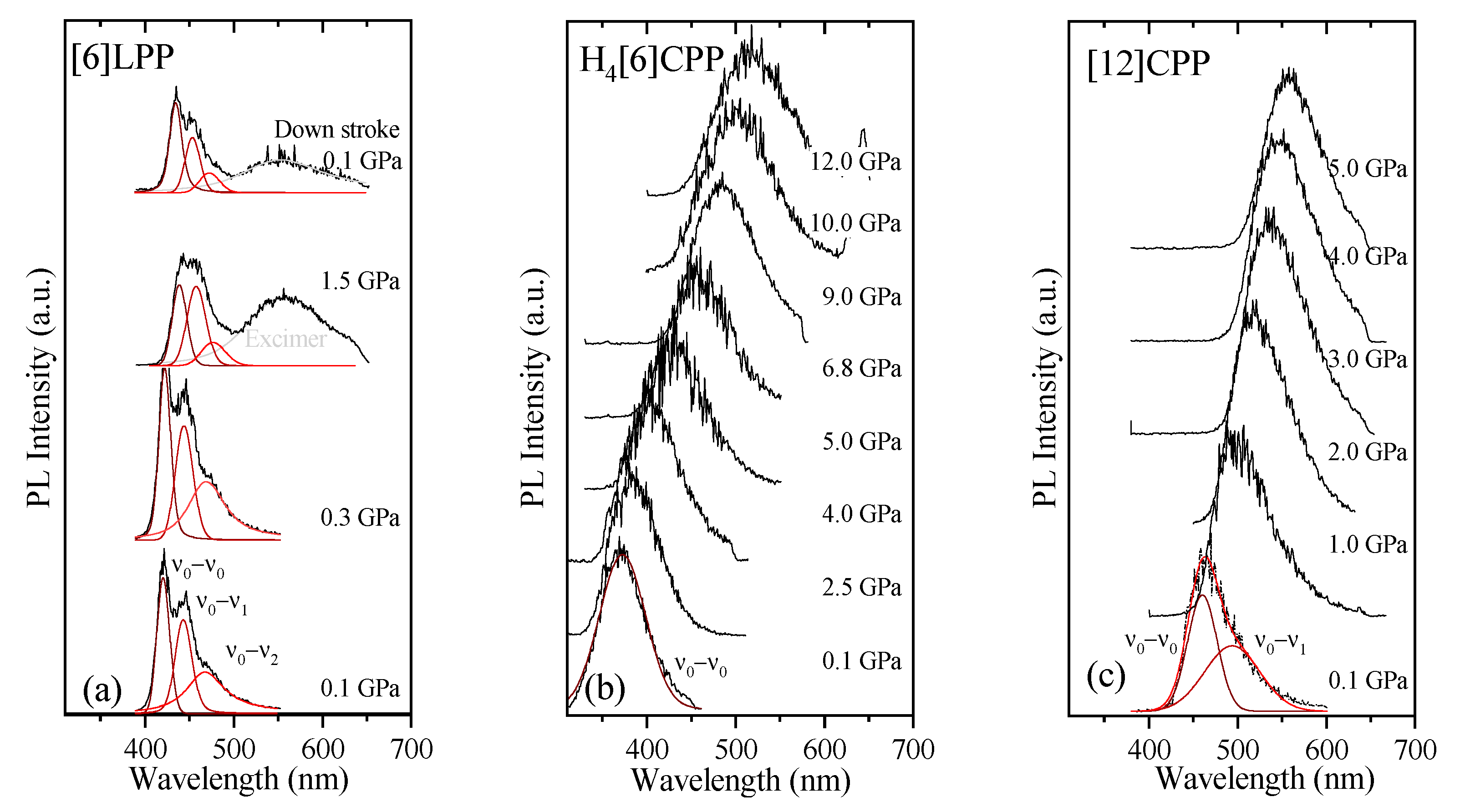
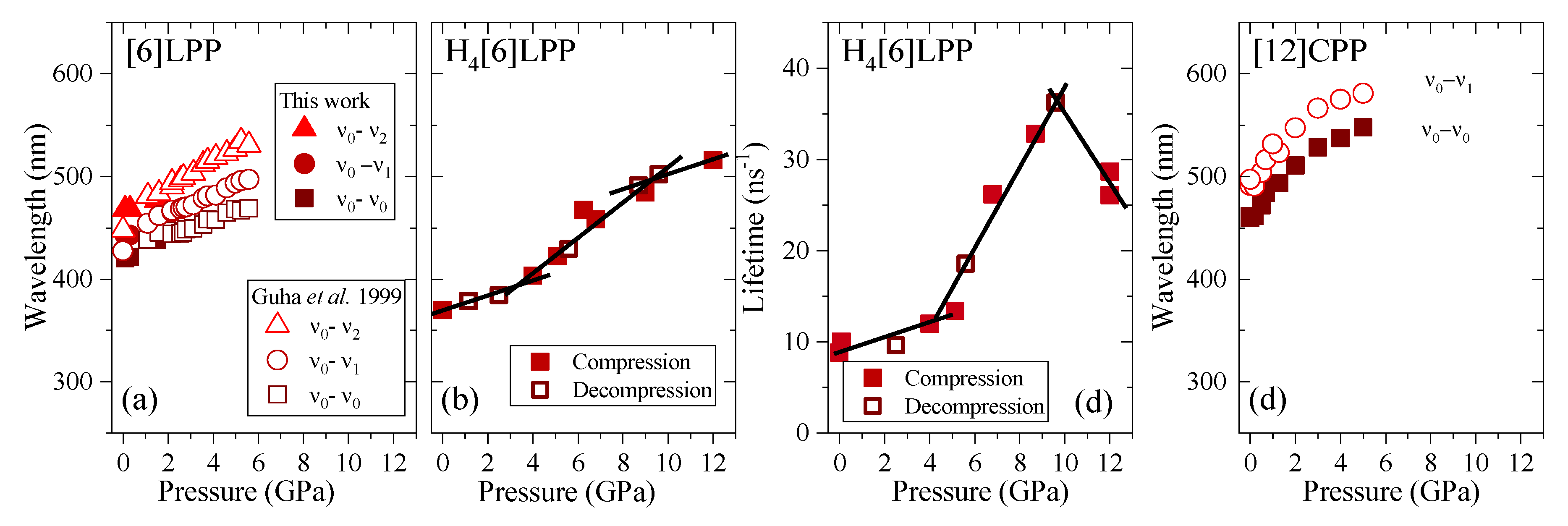
2.4. Absorption and Fluorescence at Low Pressures
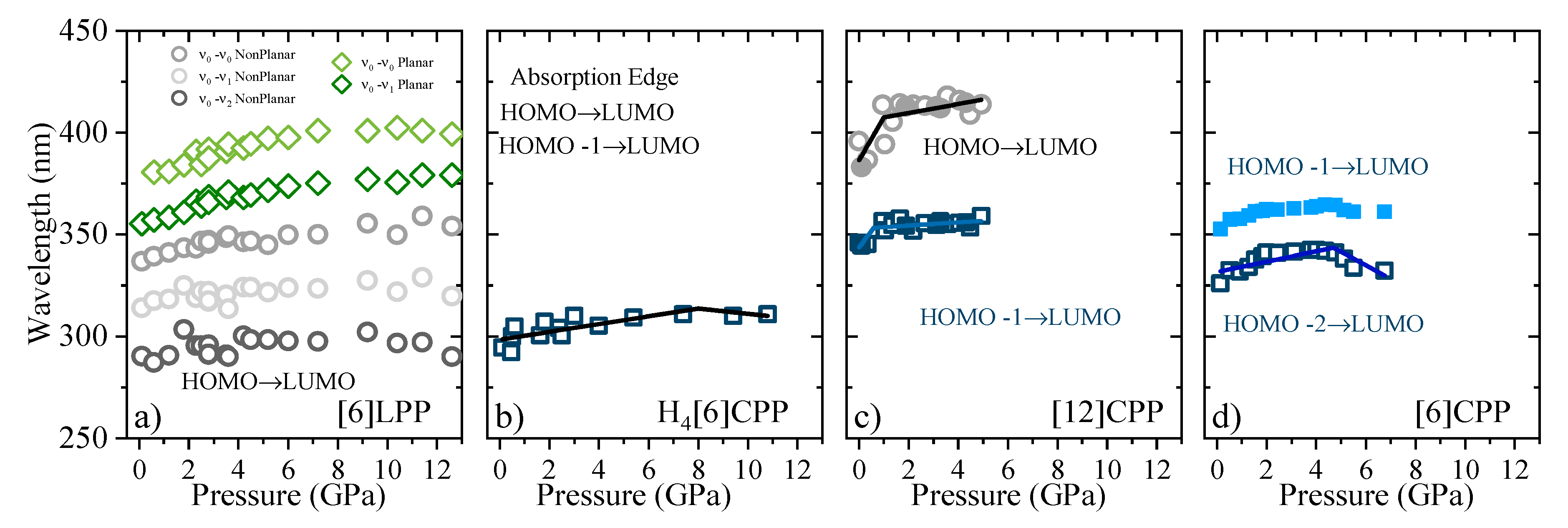
2.5. Optical Absorption in Compression
2.6. Fluorescence in Compression
3. Discussion
4. Materials and Methods
4.1. X-ray Crystallography
4.2. Structure Analysis
4.3. Interaction Energies Calculation
4.4. FTIR
4.5. Raman Spectroscopy
4.6. Near-UV−Visible Absorption
4.7. Fluorescence
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chiang, C.K.; Fincher, C.R., Jr.; Park, Y.W.; Heeger, A.J.; Shirakawa, H.; Louis, E.J.; Gau, S.C.; MacDiarmid, A.G. Electrical conductivity in doped polyacetylene. Phys. Rev. Lett. 1977, 39, 1098–1101. [Google Scholar] [CrossRef]
- Nicol, M.; Yin, G.Z. Organic chemistry at high pressure: Can unsaturated bonds survive 10 GPa? J. Phys. Colloq. 1984, 45, C8-163–C8-172. [Google Scholar] [CrossRef][Green Version]
- Aguiar, A.L.; Capaz, R.B.; Filho, A.G.S.; San-Miguel, A. Structural and phonon properties of bundled single-and double-wall carbon nanotubes under pressure. J. Phys. Chem. C 2012, 116, 22637–22655. [Google Scholar] [CrossRef][Green Version]
- Imtani, A.N.; Jindal, V.K. Structure of chiral single-walled carbon nanotubes under hydrostatic pressure. Comput. Mater. Sci. 2009, 46, 297–302. [Google Scholar] [CrossRef][Green Version]
- Fitzgibbons, C.; Guthrie, M.; Xu, E.; Crespi, V.H.S.; Davidowski, K.; Cody, G.D.; Alem, N.; Badding, J.V. Benzene-derived carbon nanothreads. Nat. Mater. 2015, 14, 43. [Google Scholar] [CrossRef]
- Hanfland, M.; Brillante, A.; Syassen, K.; Stamm, M.; Fink, J. Polyparaphenylene under pressure: Optical absorption and vibrational modes. J. Chem. Phys. 1989, 90, 1930–1934. [Google Scholar] [CrossRef]
- Alvarez, M.P.; Burrezo, P.M.; Kertesz, M.; Iwamoto, T.; Yamago, S.; Xia, J.; Jasti, R.; Navarrete, J.T.L.; Taravillo, M.; Baonza, V.G.; et al. Properties of Sizeable [n] Cycloparaphenylenes as Molecular Models of Single-Wall Carbon Nanotubes Elucidated by Raman Spectroscopy: Structural and Electron-Transfer Responses under Mechanical Stress. Ang. Chem. Int. Ed. 2014, 53, 7033–7037. [Google Scholar]
- Cuff, L.; Kertesz, M. Ab initio oligomer approach to vibrational spectra of polymers: Comparison of helical and planar poly (p-phenylene). Macromolecules 1994, 27, 762–770. [Google Scholar] [CrossRef]
- Zerbi, G.; Chierichetti, B.; Inganas, O. Vibrational spectra of oligothiophenes as model of polythiophenes. J. Chem. Phys. 1991, 94, 4637–4645. [Google Scholar] [CrossRef]
- Furumoto, H.W.; Ceccon, H.L. Ultraviolet organic liquid lasers. J. Quant. Elec. 1970, 6, 262. [Google Scholar] [CrossRef]
- Tiekink, E.R.; Zukerman-Schpector, J. (Eds.) The Importance of Pi-Interactions in Crystal Engineering: Frontiers in Crystal Engineering; John Wiley and Sons: Chichester, UK, 2012. [Google Scholar]
- Lukes, V.; Justina, A.; Aquino, A.; Lischka, H.; Kauffmann, H.F. Dependence of optical properties of oligo-para-phenylenes on torsional modes and chain length. J. Phys. Chem. B 2007, 111, 7954–7962. [Google Scholar] [CrossRef]
- Guha, S.; Graupner, W.; Resel, R.; Chandrasekhar, M.; Chandrasekhar, H.R.; Glaser, R.; Leising, G. Planarity of para hexaphenyl. Phys. Rev. Lett. 1999, 82, 3625–3628. [Google Scholar] [CrossRef]
- Zhuravlev, K.K.; McCluskey, M.D. Excitation of crystalline all–trans retinal under pressure. J. Chem. Phys. 2001, 114, 5465–5467. [Google Scholar] [CrossRef][Green Version]
- Zhuravlev, K.K.; McCluskey, M.D. Conformation of p-terphenyl under hydrostatic pressure. J. Chem. Phys. 2004, 120, 1841–1845. [Google Scholar] [CrossRef] [PubMed]
- Guha, S.; Graupner, W.; Resel, R.; Chandrasekhar, M.; Chandrasekhar, H.R.; Glaser, R.; Leising, G. Tuning intermolecular interactions: A study of the structural and vibrational properties of p-hexaphenyl under pressure. J. Phys. Chem. A 2001, 105, 6203–6211. [Google Scholar] [CrossRef]
- Heimel, G.; Puschnig, P.; Oehzelt, M.; Hummer, K.; Koppelhuber-Bitschnau, B.; Porsch, F.; Ambrosch-Draxl, C.; Resel, R. Chain-length-dependent intermolecular packing in polyphenylenes: A high pressure study. J. Phys. Condens. Matter 2003, 15, 3375. [Google Scholar] [CrossRef]
- Li, P.; Sisto, T.J.; Darzi, E.R.; Jasti, R. The effects of cyclic conjugation and bending on the optoelectronic properties of paraphenylenes. Org. Lett. 2014, 16, 182–185. [Google Scholar] [CrossRef]
- Tahara, K.; Tobe, Y. Molecular loops and belts. Chem. Rev. 2006, 106, 5274–5290. [Google Scholar] [CrossRef]
- Kayahara, E.; Kumar Patel, V.; Yamago, S. Synthesis and characterization of [5] cycloparaphenylene. J. Am. Chem. Soc. 2014, 136, 2284–2287. [Google Scholar] [CrossRef]
- Darzi, E.R.; Jasti, R. The dynamic, size-dependent properties of [5]–[12] cycloparaphenylenes. Chem. Soc. Rev. 2015, 44, 6401–6410. [Google Scholar] [CrossRef]
- Iwamoto, T.; Watanabe, Y.; Sadahiro, T.; Haino, T.; Yamago, S. Size-selective encapsulation of C60 by [10] cycloparaphenylene: Formation of the shortest fullerene-peapod. Angew. Chem. Int. Ed. 2011, 50, 8342–8344. [Google Scholar] [CrossRef]
- Bunz, U.H.F.; Menning, S.; Martín, N. para-Connected cyclophenylenes and hemispherical polyarenes: Building blocks for single-walled carbon nanotubes? Angew. Chem. Int. Ed. 2012, 51, 7094–7101. [Google Scholar] [CrossRef]
- Schrettl, S.; Frauenrath, H. Elements for a rational polymer approach towards carbon nanostructures. Angew. Chem. Int. Ed. 2012, 51, 6569–6571. [Google Scholar] [CrossRef]
- Iwamoto, T.; Watanabe, Y.; Takaya, H.; Haino, T.; Yasuda, N.; Yamago, S. Size-and Orientation-Selective Encapsulation of C70 by Cycloparaphenylenes. Chem. Eur. J. 2013, 19, 14061–14068. [Google Scholar] [CrossRef]
- Gonzalez-Veloso, I.; Cabaleiro-Lago, E.M.; Rodriguez-Otero, J. Fullerene size controls the selective complexation of [11] CPP with pristine and endohedral fullerenes. J. Phys. Chem. Chem. Phys. 2018, 20, 11347–11358. [Google Scholar] [CrossRef]
- Rio, J.; Beeck, S.; Rotas, G.; Ahles, S.; Jacquemin, D.; Tagmatarchis, N.; Ewels, C.; Wegner, H.A. Electronic Communication between two [10] cycloparaphenylenes and Bis (azafullerene)(C59N) 2 Induced by Cooperative Complexation. Angew. Chem. 2018, 57, 6930–6934. [Google Scholar] [CrossRef]
- Bachrach, S.M.; Stück, D. DFT study of cycloparaphenylenes and heteroatom-substituted nanohoops. J. Org. Chem. 2010, 75, 6595–6604. [Google Scholar] [CrossRef]
- Iwamoto, T.; Watanabe, Y.; Sakamoto, Y.; Suzuki, T.; Yamago, S. Selective and random syntheses of [n] cycloparaphenylenes (n = 8–13) and size dependence of their electronic properties. J. Am. Chem. Soc. 2011, 133, 8354–8361. [Google Scholar] [CrossRef]
- Segawa, Y.; Omachi, H.; Itami, K. Theoretical studies on the structures and strain energies of cycloparaphenylenes. Org. Lett. 2010, 12, 2262–2265. [Google Scholar] [CrossRef]
- Segawa, Y.; Fukazawa, A.; Matsuura, S.; Omachi, H.; Yamaguchi, S.; Irle, S.; Itami, K. Combined experimental and theoretical studies on the photophysical properties of cycloparaphenylenes. Org. Biomol. Chem. 2012, 10, 5979–5984. [Google Scholar] [CrossRef]
- Nizhegorodov, N.I.; Downey, S.W.; Danailov, M.B. Systematic investigation of absorption, fluorescence and laser properties of some p-and m-oligophenylenes. Spectrochim. Acta A 2000, 56, 783–795. [Google Scholar] [CrossRef]
- Fujitsuka, M.; Kayahara, E.; Lu, C.; Yamago, S.; Majima, T. Significant structural relaxations of excited [n] cycloparaphenylene dications (n = 5–9). Phys. Chem. Chem. Phys. 2018, 20, 29207–29211. [Google Scholar] [CrossRef]
- Lin, J.B.; Darci, E.R.; Jasti, R.; Yavuz, I.; Houk, K.N. Solid-State Order and Charge Mobility in [5]- to [12] Cycloparaphenylenes. J. Am. Chem. Soc. 2019, 141, 952–960. [Google Scholar] [CrossRef]
- Fujitsuka, M.; Cho, D.W.; Iwamoto, T.; Yamago, S.; Majima, T. Size-dependent fluorescence properties of [n] cycloparaphenylenes (n = 8–13), hoop-shaped π-conjugated molecules. Phys. Chem. Chem. Phys. 2012, 14, 14585–14588. [Google Scholar] [CrossRef]
- Nishihara, T.; Segawa, Y.; Itami, K.; Kanemitsu, Y. Excited states in cycloparaphenylenes: Dependence of optical properties on ring length. J. Phys. Chem. Lett. 2012, 3, 3125–3128. [Google Scholar] [CrossRef]
- Camacho, C.; Niehaus, T.A.; Itami, K.; Irle, S. Origin of the size-dependent fluorescence blueshift in [n] cycloparaphenylenes. Chem. Sci. 2013, 4, 187–195. [Google Scholar] [CrossRef]
- Qiu, L.; Peña-Alvarez, M.; Taravillo, M.; Evans, P.J.; Darzi, E.R.; Jasti, R.; Mayorga Burrezo, P.; López Navarrete, J.T.; Baonza, V.G.; Casado, J.; et al. High-Pressure Chemistry and the Mechanochemical Polymerization of [5]-Cyclo-p-phenylene. Chem. Eur. J. 2017, 23, 16593–16604. [Google Scholar] [CrossRef]
- Qiu, L.; Peña-Alvarez, M.; Taravillo, M.; Baonza, V.G.; Casado, J.; Kertesz, M. Mechanochemistry in [6] Cycloparaphenylene: A Combined Raman Spectroscopy and Density Functional Theory Study. Chem. Phys. Chem. 2018, 19, 1903–1916. [Google Scholar] [CrossRef]
- Peña-Alvarez, M.; Qiu, L.; Baonza, V.G.; Taravillo, M.; Kertesz, M. Casado, Molecules under Pressure: The Case of [n] Cycloparaphenylenes. J. Chem. Mat. 2019, 31, 6443–6452. [Google Scholar] [CrossRef]
- Jezowski, S.R.; Zhu, L.; Wang, Y.; Rice, A.P.; Scott, G.W.; Bardeen, C.J.; Chronister, E.L. Pressure catalyzed bond dissociation in an anthracene cyclophane photodimer. J. Am. Chem. Soc. 2012, 134, 7459–7466. [Google Scholar] [CrossRef]
- Bini, R.; Ceppatelli, M.; Citroni, M.; Schettino, V. Pressure catalyzed bond dissociation in an anthracene cyclophane photodimer. Chem. Phys. 2012, 398, 262–268. [Google Scholar] [CrossRef]
- Ciabini, L.; Gorelli, F.A.; Santoro, M.; Bini, R.; Schettino, V.; Mezouar, M. High-pressure and high-temperature equation of state and phase diagram of solid benzene. Phys. Rev. B 2005, 72, 094108. [Google Scholar] [CrossRef]
- Thiéry, M.M.; Léger, J.M. High pressure solid phases of benzene. I. Raman and X-ray studies of C6H6 at 294 K up to 25 GPa. J. Chem. Phys. 1988, 89, 4255–4271. [Google Scholar] [CrossRef]
- Baker, K.N.; Fratini, A.V.; Resch, T.; Knachel, H.C.; Adams, W.W.; Socci, E.P.; Farmer, B.L. Crystal structures, phase transitions and energy calculations of poly (p-phenylene) oligomers. Polymer 1993, 34, 1571–1587. [Google Scholar] [CrossRef]
- Fukushima, T.; Sakamoto, H.; Tanaka, K.; Hijikata, Y.; Irle, S.; Itami, K. Polymorphism of [6] Cycloparaphenylene for Packing Structure-dependent Host–Guest Interaction. Chem. Lett. 2017, 46, 855–857. [Google Scholar] [CrossRef]
- Ciabini, L.; Santoro, M.; Gorelli, F.A.; Bini, R.; Schettino, V.; Raugei, S. Triggering dynamics of the high-pressure benzene amorphization. Nat. Mater. 2007, 6, 39. [Google Scholar] [CrossRef]
- Ciabini, L.; Santoro, M.; Bini, R.; Schettino, V. High pressure reactivity of solid benzene probed by infrared spectroscopy. J. Chem. Phys. 2002, 116, 2928–2935. [Google Scholar] [CrossRef]
- Mao, H.K.; Bell, P.M.; Hemley, R. Ultrahigh pressures: Optical observations and Raman measurements of hydrogen and deuterium to 1.47 Mbar. J. Phys. Rev. Lett. 1985, 55, 99. [Google Scholar] [CrossRef]
- Martin, C.M.; Cai, Q.; Guha, S.; Graupner, W.; Chandrasekhar, M.; Handrasekhar, H.R. Raman modes in oligophenyls under hydrostatic pressure. Phys. Stat. Sol. 2004, 241, 3339–3344. [Google Scholar] [CrossRef]
- Segawa, Y.; Miyamoto, S.; Omachi, H.; Matsuura, S.; Senel, P.; Sasamori, T.; Tokitoh, N.; Itami, K. Concise synthesis and crystal structure of [12] cycloparaphenylene. Angew. Chem. Int. Ed. 2011, 50, 3244–3248. [Google Scholar] [CrossRef]
- Xia, J.; Jasti, R. Synthesis, characterization, and crystal structure of [6] cycloparaphenylene. Angew. Chem. Int. Ed. 2012, 51, 2474–2476. [Google Scholar] [CrossRef]
- Momicchioli, F.; Bruni, M.C.; Baraldi, I. Fluorescence and absorption spectra of polyphenyls. Theoretical study on the band shape. J. Phys. Chem. 1972, 76, 3983–3990. [Google Scholar] [CrossRef]
- Martin, R.E.; Diederich, F. Linear Monodisperse π-Conjugated Oligomers: Model Compounds for Polymers and More. Angew. Chem. Int. Ed. 1999, 38, 1350–1377. [Google Scholar] [CrossRef]
- Mabbs, R.; Nijegorodov, N.; Downey, W.S. Fluorescence and laser properties of D2-, C2-and D3 symmetry series oligophenylenes. Spectrochim. Acta Part A 2003, 59, 1329–1339. [Google Scholar] [CrossRef]
- Adamska, L.; Nayyar, I.; Chen, H.; Swan, A.K.; Oldani, N.; Fernandez-Alberti, S.; Golder, M.R.; Jasti, R.; Doorn, S.K.; Tretiak, S. Self-trapping of excitons, violation of Condon approximation, and efficient fluorescence in conjugated cycloparaphenylenes. Nano Lett. 2014, 14, 6539–6546. [Google Scholar] [CrossRef]
- Darzi, E.R.; Sisto, T.J.; Jasti, R. Selective syntheses of [7]–[12] cycloparaphenylenes using orthogonal Suzuki–Miyaura cross-coupling reactions. J. Org. Chem. 2012, 77, 6624–6628. [Google Scholar] [CrossRef]
- Wong, B.M. Optoelectronic properties of carbon nanorings: Excitonic effects from time-dependent density functional theory. J. Phys. Chem. C 2009, 113, 21921–21927. [Google Scholar] [CrossRef]
- Fanetti, S.; Citroni, M.; Malavasi, L.; Artioli, G.A.; Postorino, P.; Bini, R. High-pressure optical properties and chemical stability of picene. J. Phys. Chem. C 2013, 117, 5343–5351. [Google Scholar] [CrossRef]
- Guha, S.; Graupner, W.; Yang, S.; Chrandrasekhar, M.; Chrandrasekhar, H.R.; Leising, G. Optical Properties of Poly (Para-Phenylenes) under High Pressure. Phys. Stat. Sol. 2004, 211, 177–188. [Google Scholar] [CrossRef]
- Citroni, M.; Bini, R.; Foggi, P.; Schettino, V. Role of excited electronic states in the high-pressure amorphization of benzene. Proc. Natl. Acad. Sci. USA 2008, 105, 7658–7663. [Google Scholar] [CrossRef]
- Drickamer, H.G.; Frank, C.W. Electronic Transitions and the High Pressure Chemistry and Physics of Solids; Springer: Berlin/Heidelberg, Germany, 1973. [Google Scholar]
- Fujitsuka, M.; Lu, C.; Iwamoto, T.; Kayahara, E.; Yamago, S.; Majima, T. Properties of triplet-excited [n] cycloparaphenylenes (n = 8–12): Excitation energies lower than those of linear oligomers and polymers. J. Phys. Chem. A 2014, 118, 4527–4532. [Google Scholar] [CrossRef]
- Colthrup, N.B.; Daly, L.H.; Wiberley, S.E. Introduction to Infrared and Raman Spectroscopy, 3rd ed.; Academic Press: San Diego, CA, USA, 1990. [Google Scholar]
- Alvarez, M.P.; Delgado, M.C.R.; Taravillo, M.; Baonza, V.G.; Navarrete, J.T.L.; Evans, P.; Jasti, R.; Yamago, S.; Kertesz, M.; Casado, J. The Raman fingerprint of cyclic conjugation: The case of the stabilization of cations and dications in cycloparaphenylenes. J. Chem. Sci. 2016, 7, 3494–3499. [Google Scholar] [CrossRef]
- Froyer, G.; Goblot, J.Y.; Guilbert, J.L.; Maurice, F.; Pelous, Y. Poly(para phenylene): Some properties related to the synthesis method. J. Phys. Colloq. 1983, 44, C3-745–C3-748. [Google Scholar]
- Mao, H.K.; Bell, P.M.; Shaner, J.W.; Steinberg, D.J. Specific volume measurements of Cu, Mo, Pd, and Ag and calibration of the ruby R 1 fluorescence pressure gauge from 0.06 to 1 Mbar. J. App. Phys. 1978, 49, 3279–3283. [Google Scholar] [CrossRef]
- Agilent Technologies Ltd. CrysAlis PRO; Agilent Technologies Ltd.: Santa Clara, CA, USA, 2014. [Google Scholar]
- Evans, P.R.; Murshudov, G.N. How good are my data and what is the resolution? Acta Crystallogr. Sect. D Biol. Crystallogr. 2013, 69, 1204–1214. [Google Scholar] [CrossRef]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.; McCoy, A.; et al. Overview of the CCP4 suite and current developments. J. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 235–242. [Google Scholar] [CrossRef]
- Winter, G. xia2: An expert system for macromolecular crystallography data reduction. J. Appl. Cryst. 2010, 43, 186–190. [Google Scholar] [CrossRef]
- Winter, G.; Waterman, D.G.; Parkhurst, J.M.; Brewster, A.S.; Gildea, R.J.; Gerstel, M.; Fuentes-Montero, L.; Vollmar, M.; Michels-Clark, T.; Young, I.D.; et al. DIALS: Implementation and evaluation of a new integration package. Acta Crystallogr. Sect. D Biol. Crystallogr. 2018, 74, 85–97. [Google Scholar] [CrossRef]
- Evans, P. Scaling and assessment of data quality. Acta Crystallogr. Sect. D Biol. Crystallogr. 2006, 62, 72–82. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT–Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Crystallogr. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.P.; Taylor, R.; Towler, M.; Streek, J.V.D. Mercury CSD 2.0—New features for the visualization and investigation of crystal structures. J. Appl. Cryst. 2006, 39, 453–457. [Google Scholar] [CrossRef]
- Mackenzie, C.F.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer model energies and energy frameworks: Extension to metal coordination compounds, organic salts, solvates and open-shell systems. IUCrJ 2017, 4, 575–587. [Google Scholar] [CrossRef] [PubMed]
- Bini, R.; Ballerini, R.; Pratesi, G.; Jodl, H.J. Experimental setup for Fourier transform infrared spectroscopy studies in condensed matter at high pressure and low temperatures. Rev. Sci. Instrum. 1997, 68, 3154–3160. [Google Scholar] [CrossRef]
- Peña-Alvarez, M.; Qiu, L.; Taravillo, M.; Baonza, V.G.; Ruiz Delgado, M.C.; Yamago, S.; Jasti, R.; López Navarrete, J.T.; Casado, J.; Kertesz, M. From linear to cyclic oligoparaphenylenes: Electronic and molecular changes traced in the vibrational Raman spectra and reformulation of the bond length alternation pattern. Phys. Chem. Chem. Phys. 2016, 18, 11683–11692. [Google Scholar] [CrossRef] [PubMed]
- Baonza, V.G.; Taravillo, M.; Arencibia, A.; Cáceres, M.; Núñez, J. Diamond as pressure sensor in high-pressure Raman spectroscopy using sapphire and other gem anvil cells. J. Raman Spectrosc. 2003, 34, 264–270. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Electrostatic | Polarisation | Dispersion | Repulsion | Total | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Pressure/GPa | 0 | 4.59 | 0 | 4.59 | 0 | 4.59 | 0 | 4.59 | 0 | 4.59 | |
| 2 | −x, y + 1/2, −z + 1/2 | −6.9 | −25.1 | −1.4 | −2.9 | −35.9 | −66.2 | 20.4 | 90.5 | −27.1 | −30.4 |
| 2 | −x, y + 1/2, −z + 1/2 | −6.5 | −22.0 | −1.4 | −2.5 | −30.8 | −54.1 | 15.8 | 70.0 | −24.9 | −29.0 |
| 2 | x, −y + 1/2, z + 1/2 | −0.1 | −3.3 | −0.1 | −0.3 | −6.7 | −14.3 | 1.8 | 17.3 | −4.8 | −5.6 |
| 2 | x, −y + 1/2, z + 1/2 | −8.3 | −51.5 | −0.6 | −1.7 | −71.8 | −143.5 | 36.4 | 197.2 | −49.3 | −58.9 |
| 2 | x, −y + 1/2, z + 1/2 | −4.6 | −15.0 | −0.8 | −2.2 | −25.9 | −45.5 | 16.8 | 74.7 | −10.7 | −11.0 |
| 1 | −x, −y, −z | −2.5 | −29.1 | −0.4 | −3.1 | −13.1 | −66.2 | 3.8 | 91.1 | −12.0 | −34.8 |
| 1 | −x, −y, −z | −6.8 | −9.5 | −1.3 | −1.2 | −31.5 | −33.3 | 17.2 | 30.5 | −25.0 | −21.1 |
| 1 | −x, −y, −z | −3.6 | −10.0 | −0.5 | −1.5 | −16.1 | −35.4 | 5.9 | 34.1 | −14.6 | −21.5 |
| 1 | −x, −y, −z | −8.9 | −33.0 | −2.1 | −4.1 | −47.6 | −83.0 | 29.1 | 119.9 | −34.4 | −36.1 |
| SYSTEM | ω0, (cm−1) | A, (cm−1GPa−1) |
|---|---|---|
| Benzene [44] | 2990 | 4.7 ± 0.2 |
| [6]LPP | 3030/3090 | 5.5 ± 0.4 |
| H4[6]CPP | 3030 | 8.0 ± 0.8 |
| [12]CPP | 3030/3080 | 9.0 ± 0.8 |
| [6]CPP | 3010/3070 | 6.8 ± 0.5 |
| GA1g | GE2g | |||||||
|---|---|---|---|---|---|---|---|---|
| SYSTEM | Diameter (nm) | Poval (GPa) | ω0 (cm−1) | A (cm−1GPa−1) | B (cm−1GPa−1) | ω0 (cm−1) | A (cm−1GPa−1) | B (cm−1GPa−1) |
| Benzene [45] | -- | -- | 1567 | 3.6 ± 0.1 | -- | -- | -- | -- |
| [6]LPP | -- | -- | 1592 | 3.9 ± 0.3 | -- | -- | -- | -- |
| H4[6]CPP | 0.45 | 9 | 1610 | 3.1 ± 0.1 | −1.0 ± 0.1 | 1658 | 3.2 ± 0.1 | -- |
| [12]CPP | 1.65 [51] | 1 | 1595 | 7.0 ± 2.0 | 3.4 ± 0.1 | 1602 | 9.0 ± 2.0 | 3.9 ± 0.1 |
| [6]CPP | 0.81 [52] | 5 | 1567 | 5.1 ± 0.2 | 1.0 ± 0.6 | 1584 | 4.7 ± 0.2 | 1.4 ± 0.7 |
| S0 → S1 | S0 → S2 | S0 → S3-S4 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Poval (GPa) | λ0 (nm) | A (nm/GPa) | B (nm/GPa) | λ0 (nm) | A (nm/GPa) | B (nm/GPa) | λ0 (nm) | A (nm/GPa) | B (nm/GPa) | |
| Picene [55] | -- | 475 | ~8 ± 0.1 | -- | -- | -- | -- | |||
| [6]LPP | -- | 325 | ~1 ± 0.1 | -- | -- | -- | -- | |||
| H4[6]CPP | 9 | 295 | ~2 ± 0.1 | (−1) ± 1 | -- | |||||
| [12]CPP | 1 | 396 | ~30 ± 0.1 | ~2 ± 1 | 344 | ~15 ± 2 | ~1 ± 1 | |||
| [6]CPP | 5 | -- | -- | -- | 352 | ~4 ± 1 | ~(−5) ± 1 | 326 | ~8 ± 1 | ~(−9) ± 1 |
| S1 → S0 | |||||
|---|---|---|---|---|---|
| PT (GPa) | λ0 (nm) | A (nm/GPa) | B (nm/GPa) | C (nm/GPa) | |
| Picene [55] | 475 | ~6.0 | -- | ||
| [6]LPP | > 1 | 440 | ~2 ± 0.2 | -- | -- |
| H4[6]CPP | 4, 9-10 | 375 | ~5 ± 1 | ~20 ± 2 | ~8 ± 1 |
| [12]CPP | 1 | 462 | ~50 ± 4 | ~10 ± 2 | -- |
| System | Comments | FTIR | Raman | Absorption | Fluorescence |
|---|---|---|---|---|---|
| [6]LPP | 1 to 2 GPa: planarisation | Spectral changes, new bands [66] | Change in slope of the GA1g mode | Change in the absorption profile | -- |
| 1–23 GPa | Steep upshift C-H stretching modes | -- | Steep upshift, measurements conducted up to 12 GPa | Steep upshift measurements conducted up to 12 GPa | |
| P > 23 GPa: polymerisation | C-H stretching from sp3 carbon | -- | -- | -- | |
| H4[6]CPP | 1–4.59 GPa, intramolecular distance decrease, XRD | No significant changes | Change in slope at 5 GPa of the emission bands | ||
| 4.6 to 10 GPa: pressure induces π-π interactions | C-H stretching from sp3 carbon decrease in intensity | Change in slope of the GA1g mode | Change in slope at 10 GPa of the absorption bands | Change in slope 10 GPa of the emission bands | |
| P > 16 GPa:polymerisation | C-H stretching from sp3 carbon | -- | -- | -- | |
| [12]CPP | ~0.6 GPa: deformation of the cycle, ovalisation: favoured intramolecular conjugation | -- | Change in slope of the GA1g mode | Change in slope of absorption bands; HOMO-LUMO absorption becomes more intense | Change in slope of emission band |
| Up to 10 GPa | No significant changes | ||||
| [6]CPP | 5 GPa: Deformation of the cycle and intermolecular polymerisation | C-H stretching from sp3 carbon appears in decompression | Change in slope of the GA1g mode and irreversibility on decompression | Change in slope of absorption bands; HOMO-LUMO absorption becomes more intense | None observed |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peña-Álvarez, M.; Fanetti, S.; Falsini, N.; Novelli, G.; Casado, J.; G. Baonza, V.; Taravillo, M.; Parsons, S.; Bini, R.; Citroni, M. Linear, Non-Conjugated Cyclic and Conjugated Cyclic Paraphenylene under Pressure. Molecules 2019, 24, 3496. https://doi.org/10.3390/molecules24193496
Peña-Álvarez M, Fanetti S, Falsini N, Novelli G, Casado J, G. Baonza V, Taravillo M, Parsons S, Bini R, Citroni M. Linear, Non-Conjugated Cyclic and Conjugated Cyclic Paraphenylene under Pressure. Molecules. 2019; 24(19):3496. https://doi.org/10.3390/molecules24193496
Chicago/Turabian StylePeña-Álvarez, Miriam, Samuele Fanetti, Naomi Falsini, Giulia Novelli, Juan Casado, Valentín G. Baonza, Mercedes Taravillo, Simon Parsons, Roberto Bini, and Margherita Citroni. 2019. "Linear, Non-Conjugated Cyclic and Conjugated Cyclic Paraphenylene under Pressure" Molecules 24, no. 19: 3496. https://doi.org/10.3390/molecules24193496
APA StylePeña-Álvarez, M., Fanetti, S., Falsini, N., Novelli, G., Casado, J., G. Baonza, V., Taravillo, M., Parsons, S., Bini, R., & Citroni, M. (2019). Linear, Non-Conjugated Cyclic and Conjugated Cyclic Paraphenylene under Pressure. Molecules, 24(19), 3496. https://doi.org/10.3390/molecules24193496