Leishmania mexicana Trypanothione Reductase Inhibitors: Computational and Biological Studies


Abstract
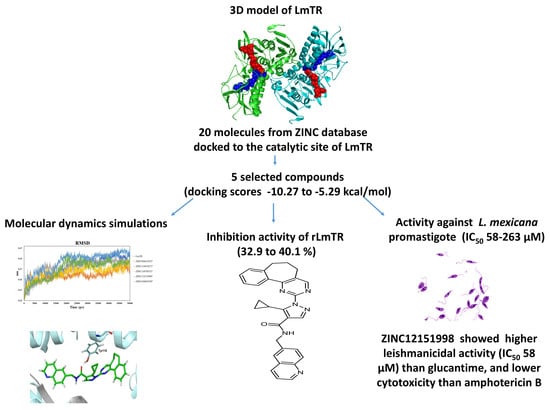
1. Introduction
2. Results and Discussion
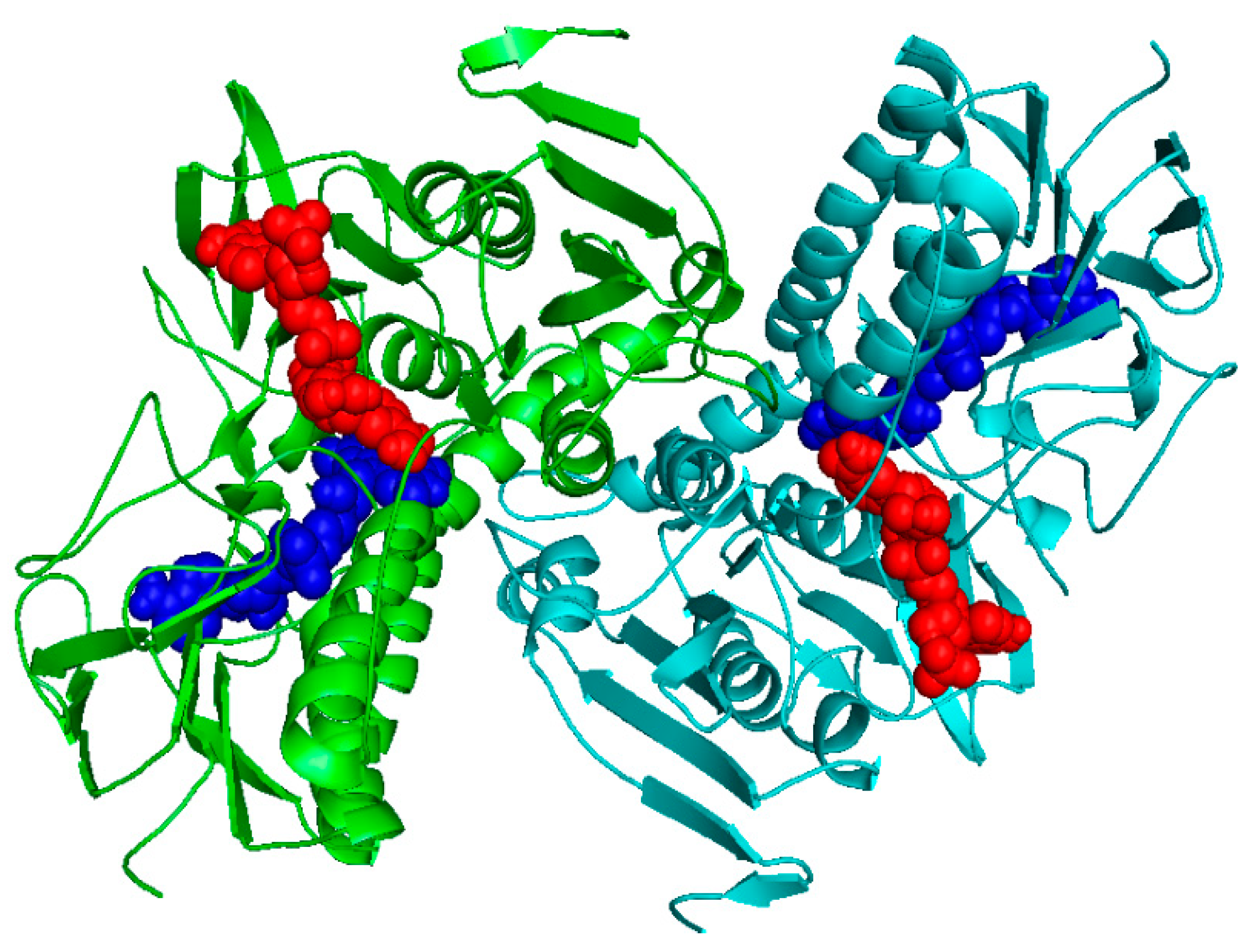
2.1. Homology Modeling
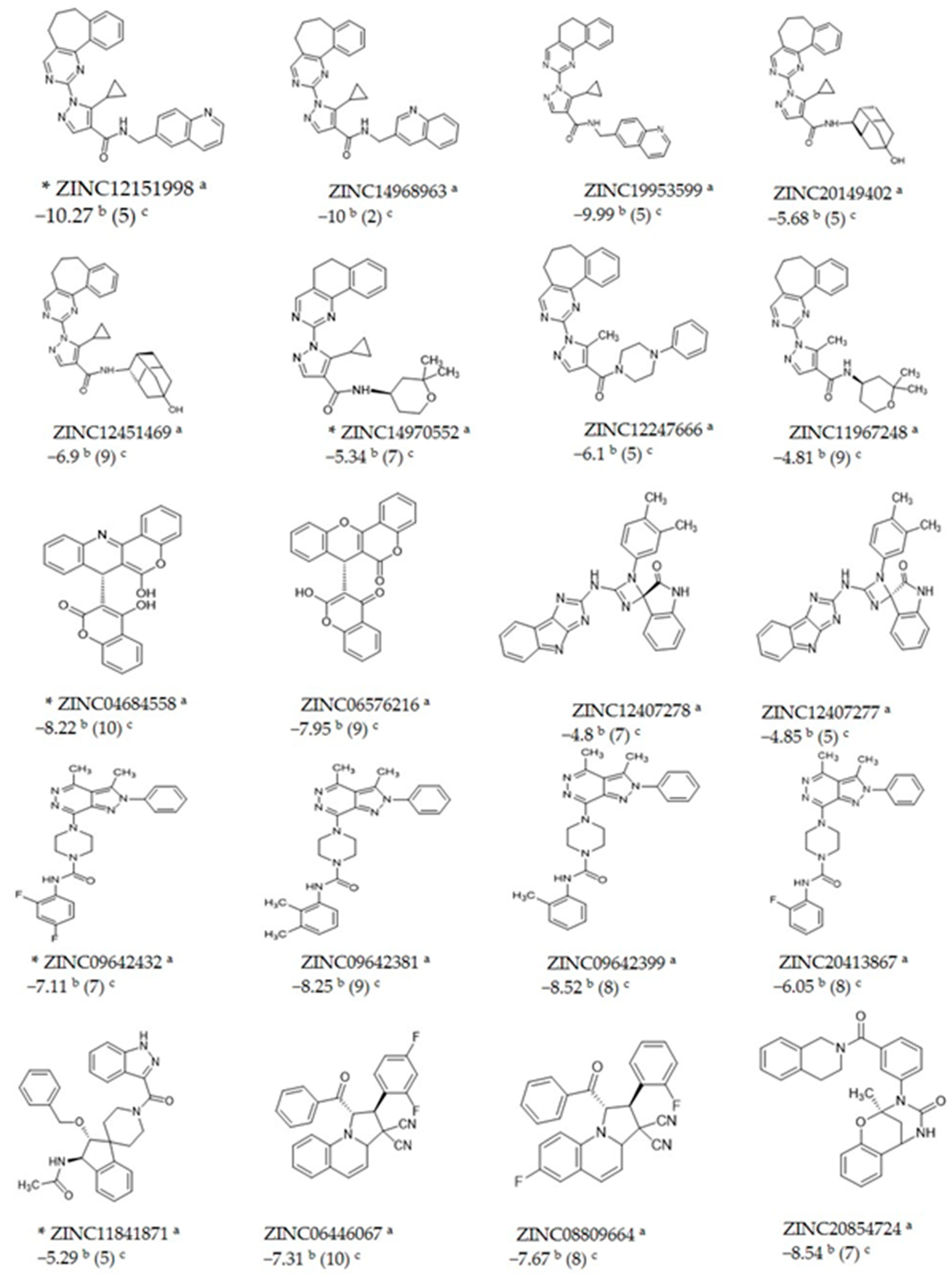
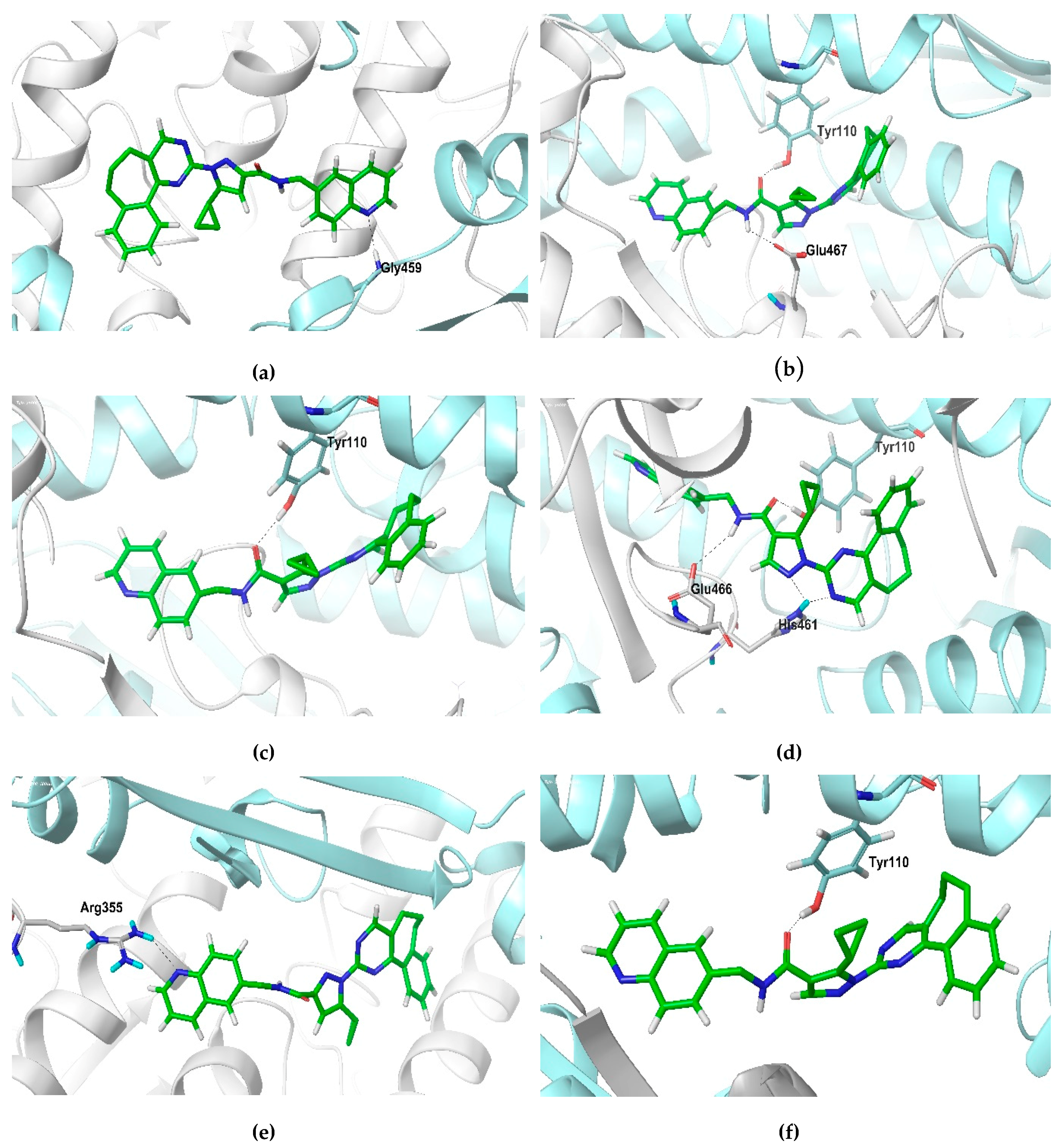
2.2. Docking Studies
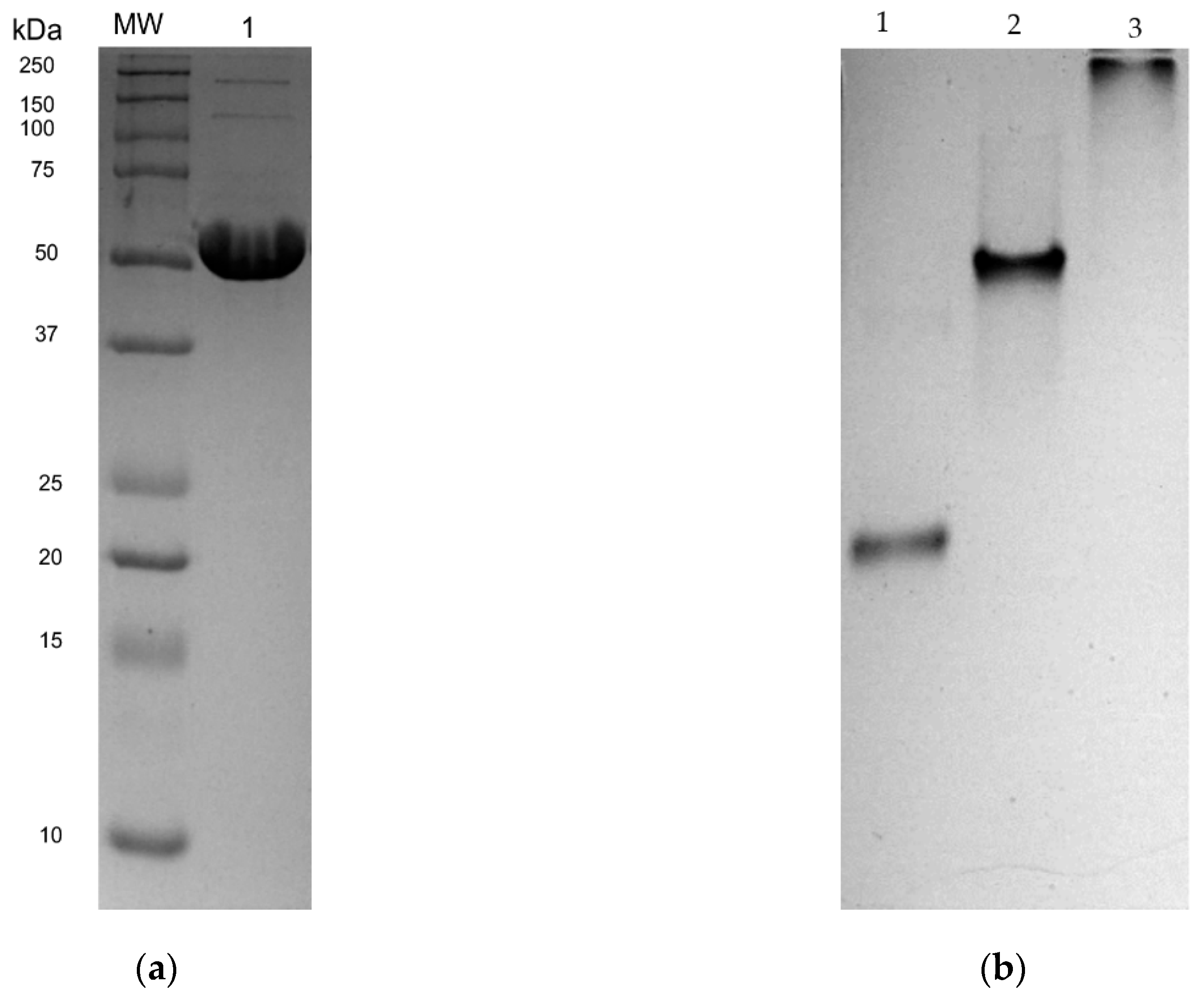
2.3. Expression and Purification of rLmTR
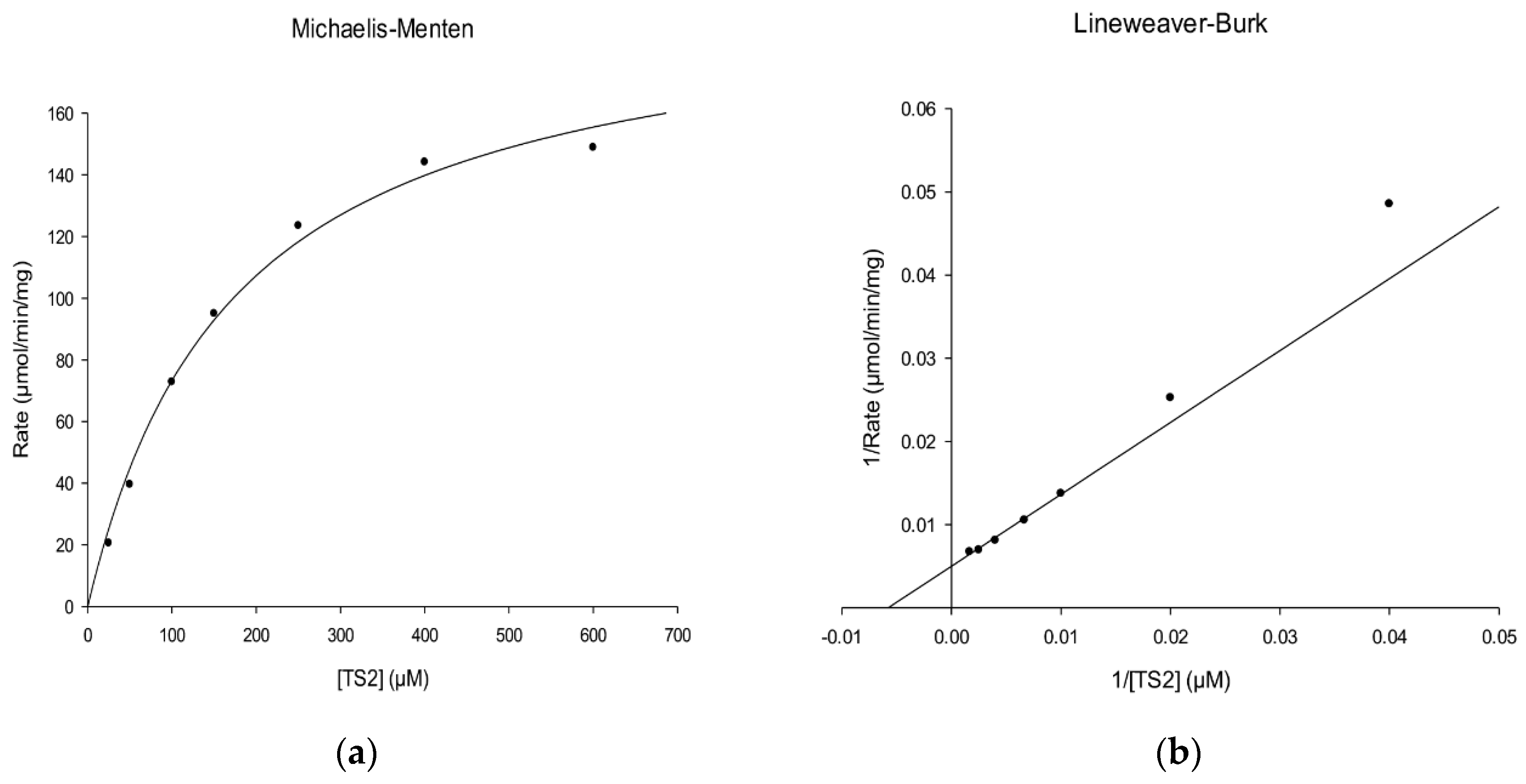
2.4. Kinetic Characterization of rLmTR
2.5. rLmTR Inhibition Assays
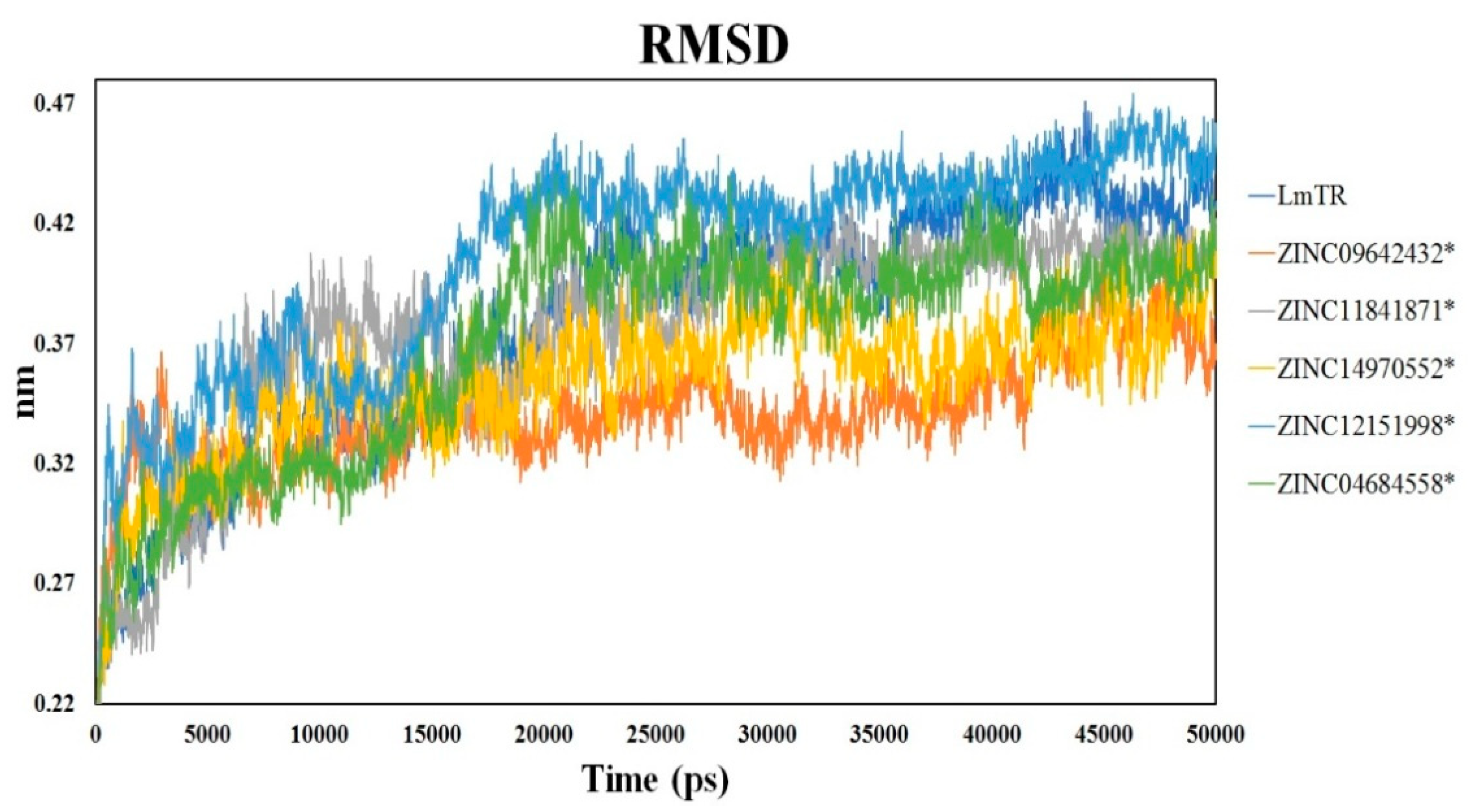
2.6. Molecular Dynamics Simulation
2.7. In Silico Analysis of ADME Properties
2.8. Leishmanicidal and Cytotoxic Activity
3. Materials and Methods
3.1. General Information
3.2. LmTR Homology Model Development and Validation
3.3. Molecular Docking
3.4. Cloning, Expression, and Purification of Trypanothione Reductase from Leishmania mexicana
3.5. Biochemical Characterization of rLmTR
3.6. Inhibition of rLmTR Activity
3.7. Molecular Dynamic Studies
3.8. ADME Characterization of Selected LmTR Inhibitors
3.9. Activity Evaluation against Leishmania mexicana Promastigote
3.10. Macrophage Cytotoxicity Assays
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Turcano, L.; Torrente, E.; Missineo, A.; Andreini, M.; Gramiccia, M.; Di Muccio, T.; Genovese, I.; Fiorillo, A.; Harper, S.; Bresciani, A.; et al. Identification and binding mode of novel Leishmania Trypanothione reductase inhibitor from high throughput screening. PLoS Negl. Trop. Dis. 2018, 12, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Lima, M.I.; Arruda, V.O.; Alves, E.V.; de Azevedo, A.P.; Monteiro, S.G.; Pereira, S.R. Genotoxic effects of the antileishmanial drug Glucantime. Arch. Toxicol. 2010, 84, 227–232. [Google Scholar] [CrossRef]
- Moreira, V.R.; de Jesus, L.C.L.; Soares, R.P.; Silva, L.D.M.; Pinto, B.A.S.; Melo, M.N.; Paes, A.M.A.; Pereira, S.R.F. Meglumine Antimoniate (Glucantime) Causes Oxidative Stress-Derived DNA Damage in BALB/c Mice Infected by Leishmania (Leishmania) infantum. Antimicrob. Agents Chemother. 2017, 61, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kapil, S.; Singh, P.K.; Silakari, O. An update on small molecule strategies targeting leishmaniasis. Eur. J. Med. Chem. 2018, 157, 339–367. [Google Scholar] [CrossRef] [PubMed]
- Chawla, B.; Madhubala, R. Drug targets in Leishmania. J. Parasit. Dis. 2010, 34, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Nieto-Meneses, R.; Castillo, R.; Hernández-Campos, A.; Maldonado-Rangel, A.; Matius-Ruiz, J.B.; Trejo-Soto, P.J.; Nogueda-Torres, B.; Dea-Ayuela, M.A.; Bolás-Fernández, F.; Méndez-Cuesta, C.; et al. In vitro activity of new N-benzyl-1H-benzimidazol-2-amine derivatives against cutaneous, mucocutaneous and visceral Leishmania species. Exp. Parasitol. 2018, 184, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Mittal, M.K.; Misra, S.; Owais, M.; Goyal, N. Expression, purification, and characterization of Leishmania donovani trypanothione reductase in Escherichia Coli. Protein Expr. Purif. 2005, 40, 279–286. [Google Scholar] [CrossRef]
- Colotti, G.; Baiocco, P.; Fiorillo, A.; Boffi, A.; Poser, E.; Chiaro, F.D.; Ilari, A. Structural insights into the enzymes of the trypanothione pathway: Targets for antileishmaniasis drugs. Future Med. Chem. 2013, 5, 1861–1875. [Google Scholar] [CrossRef]
- Spinks, D.; Shanks, E.J.; Cleghorn, L.A.; McElroy, S.; Jones, D.; James, D.; Fairlamb, A.H.; Frearson, J.A.; Wyatt, P.G.; Gilbert, I.H. Investigation of trypanothione reductase as a drug target in Trypanosoma brucei. Chem. Med. Chem. 2009, 4, 2060–2069. [Google Scholar] [CrossRef]
- Hamilton, C.J.; Saravanamuthu, A.; Poupat, C.; Fairlamb, A.H.; Eggleston, I.M. Time-dependent inhibitors of trypanothione reductase: Analogues of the spermidine alkaloid lunarine and related natural products. Bioorg. Med. Chem. 2006, 14, 2266–2278. [Google Scholar] [CrossRef]
- Vázquez, K.; Paulino, M.; Salas, C.O.; Zarate-Ramos, J.J.; Vera, B.; Rivera, G. Trypanothione Reductase: A Target for the Development of Anti- Trypanosoma cruzi Drugs. Mini Rev. Med. Chem. 2017, 17, 939–946. [Google Scholar] [CrossRef] [PubMed]
- Holloway, G.A.; Baell, J.B.; Fairlamb, A.H.; Novello, P.M.; Parisot, J.P.; Richardson, J.; Watson, K.G.; Street, I.P. Discovery of 2-iminobenzimidazoles as a new class of trypanothione reductase inhibitor by high-throughput screening. Bioorg. Med. Chem. Lett. 2007, 17, 1422–1427. [Google Scholar] [CrossRef] [PubMed]
- Colotti, G.; Ilari, A.; Fiorillo, A.; Baiocco, P.; Cinellu, M.A.; Maiore, L.; Scaletti, F.; Gabbiani, C.; Messori, L. Metal-based compounds as prospective antileishmanial agents: Inhibition of trypanothione reductase by selected gold complexes. Chem. Med. Chem. 2013, 8, 1634–1637. [Google Scholar] [CrossRef] [PubMed]
- Ilari, A.; Baiocco, P.; Messori, L.; Fiorillo, A.; Boffi, A.; Gramiccia, M.; Di Muccio, T.; Colotti, G. A gold-containing drug against parasitic polyamine metabolism: The X-ray structure of trypanothione reductase from Leishmania infantum in complex with auranofin reveals a dual mechanism of enzyme inhibition. Amino. Acids 2012, 42, 803–811. [Google Scholar] [CrossRef] [PubMed]
- Baiocco, P.; Colotti, G.; Franceschini, S.; Ilari, A. Molecular basis of antimony treatment in leishmaniasis. J. Med. Chem. 2009, 52, 2603–2612. [Google Scholar] [CrossRef]
- Saccoliti, F.; Angiulli, G.; Pupo, G.; Pescatori, L.; Madia, V.N.; Messore, A.; Colotti, G.; Fiorillo, A.; Scipione, L.; Gramiccia, M.; et al. Inhibition of Leishmania infantum trypanothione reductase by diaryl sulfide derivatives. J. Enzym. Inhib. Med. Chem. 2017, 32, 304–310. [Google Scholar] [CrossRef] [PubMed]
- Baiocco, P.; Poce, G.; Alfonso, S.; Cocozza, M.; Porretta, G.C.; Colotti, G.; Biava, M.; Moraca, F.; Botta, M.; Yardley, V.; et al. Inhibition of Leishmania infantum trypanothione reductase by azole-based compounds: A comparative analysis with its physiological substrate by X-ray crystallography. Chem. Med. Chem. 2013, 8, 1175–1183. [Google Scholar] [CrossRef]
- da Silva, A.D.; Dos Santos, J.A.; Machado, P.A.; Alves, L.A.; Laque, L.C.; de Souza, V.C.; Coimbra, E.S.; Capriles, P.V. Insights about resveratrol analogues against trypanothione reductase of Leishmania braziliensis: Molecular modeling, computational docking and in vitro antileishmanial studies. J. Biomol. Struct. Dyn. 2018, 37, 2960–2969. [Google Scholar] [CrossRef]
- Ochoa, R.; Watowich, S.J.; Flórez, A.; Mesa, C.V.; Robledo, S.M.; Muskus, C. Drug search for leishmaniasis: A virtual screening approach by grid computing. J. Comput. Aided Mol. Des. 2016, 30, 541–552. [Google Scholar] [CrossRef]
- Benkert, P.; Künzli, M.; Schwede, T. QMEAN server for protein model quality estimation. Nucleic Acids Res. 2009, 37, 510–514. [Google Scholar] [CrossRef]
- Vriend, G. WHAT IF: A molecular modeling and drug design program. J. Mol. Graph. 1990, 8, 52–56. [Google Scholar] [CrossRef]
- DeLano, W.L. The PyMOL Molecular Graphics System; DeLano Scientific LLC: South San Francisco, CA, USA, 2002. [Google Scholar]
- Bond, C.S.; Zhang, Y.; Berriman, M.; Cunningham, M.L.; Fairlamb, A.H.; Hunter, W.N. Crystal structure of Trypanosoma cruzi trypanothione reductase in complex with trypanothione, and the structure-based discovery of new natural product inhibitors. Structure 1999, 7, 81–89. [Google Scholar] [CrossRef]
- Villalobos-Rocha, J.C.; Sánchez-Torres, L.; Nogueda-Torres, B.; Segura-Cabrera, A.; García-Pérez, C.A.; Bocanegra-García, V.; Palos, I.; Monge, A.; Rivera, G. Anti-Trypanosoma cruzi and anti-leishmanial activity by quinoxaline-7-carboxylate 1,4-di-N-oxide derivatives. Parasitol. Res. 2014, 113, 2027–2035. [Google Scholar] [CrossRef] [PubMed]
- Pandey, R.K.; Sharma, D.; Bhatt, T.K.; Sundar, S.; Prajapati, V.K. Developing imidazole analogues as potential inhibitor for Leishmania donovani trypanothione reductase: Virtual screening, molecular docking, dynamics and ADMET approach. J. Biomol. Struct. Dyn. 2015, 33, 2541–2553. [Google Scholar] [CrossRef]
- Mutlu, O. Molecular modeling, structural analysis and identification of ligand binding sites of trypanothione reductase from Leishmania mexicana. J. Vector Borne Dis. 2013, 50, 38–44. [Google Scholar]
- Saravanamuthu, A.; Vickers, T.J.; Bond, C.S.; Peterson, M.R.; Hunter, W.N.; Fairlamb, A.H. Two interacting binding sites for quinacrine derivatives in the active site of trypanothione reductase: A template for drug design. J. Biol. Chem. 2004, 279, 29493–29500. [Google Scholar] [CrossRef]
- Patterson, S.; Alphey, M.S.; Jones, D.C.; Shanks, E.J.; Street, I.P.; Frearson, J.A.; Wyatt, P.G.; Gilbert, I.H.; Fairlamb, A.H. Dihydroquinazolines as a novel class of Trypanosoma brucei trypanothione reductase inhibitors: Discovery, synthesis, and characterization of their binding mode by protein crystallography. J. Med. Chem. 2011, 54, 6514–6530. [Google Scholar] [CrossRef]
- Garforth, J.; Yin, H.; McKie, J.H.; Douglas, K.T.; Fairlamb, A.H. Rational design of selective ligands for trypanothione reductase from Trypanosoma cruzi. Structural effects on the inhibition by dibenzazepines based on imipramine. J. Enzym. Inhib. 1997, 12, 161–173. [Google Scholar] [CrossRef]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014, 42, 320–324. [Google Scholar] [CrossRef]
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinform. 2008, 9, 40. [Google Scholar] [CrossRef]
- Biasini, M.; Bienert, S.; Waterhouse, A.; Arnold, K.; Studer, G.; Schmidt, T.; Kiefer, F.; Cassarino, T.G.; Bertoni, M.; Bordoli, L.; et al. SWISS-MODEL: Modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014, 42, 252–258. [Google Scholar] [CrossRef] [PubMed]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J. The Phyre web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [PubMed]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [PubMed]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J. Using ZINC to acquire a virtual screening library. Curr. Protoc. Bioinformatics 2008, 14, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Maestro, version 9.6; Schrödinger, LLC: New York, NY, USA, 2014.
- Sanner, M.F. Python: A programming language for software integration and development. J. Mol. Graph. Model. 1999, 17, 57–61. [Google Scholar] [PubMed]
- Gasteiger, J.; Marsili, M. A new model for calculating atomic charges in molecules. Tetrahedron Lett. 1978, 19, 3181–3184. [Google Scholar] [CrossRef]
- Muñoz-Cruz, S.; Gomez-García, A.; Matadamas-Martínez, F.; Alvarado-Torres, J.A.; Meza-Cervantez, P.; Arriaga-Pizano, L.; Yépez-Mulia, L. Giardia lamblia: Identification of molecules that contribute to direct mast cell activation. Parasitol. Res. 2018, 117, 2555–2567. [Google Scholar] [CrossRef]
- Cunningham, M.L.; Zvelebil, M.J.; Fairlamb, A.H. Mechanism of inhibition of trypanothione reductase and glutathione reductase by trivalent organic arsenials. Eur. J. Biochem. 1994, 221, 285–295. [Google Scholar] [CrossRef]
- Jockers-Scherübl, M.C.; Schirmer, R.H.; Krauth-Siegel, R.L. Trypanothione reductase from Trypanosoma cruzi. Catalytic properties of the enzyme and inhibition studies with trypanocidal compounds. Eur. J. Biochem. 1989, 180, 267–272. [Google Scholar] [CrossRef]
- Lineweaver, H.; Burk, D. The determination of enzyme dissociation constants. J. Am. Chem. Soc. 1934, 56, 658–666. [Google Scholar] [CrossRef]
- Schuèttelkopf, A.W.; Van Aalten, D.M. PRODRG: A tool for high-throughput crystallography of protein–ligand complexes. Acta. Cryst. D. Biol. Cryst. 2004, 60, 1355–1363. [Google Scholar] [CrossRef] [PubMed]
- Scott, W.; van Gunsteren, W. The GROMOS software package for biomolecular simulations. METECC 1995, 95, 397–434. [Google Scholar]
- Berendsen, H.J.; Postma, J.V.; van Gunsteren, W.F.; DiNola, A.; Haak, J. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Pastor, R.W.; Brooks, B.R.; Szabo, A. An analysis of the accuracy of Langevin and molecular dynamics algorithms. Mol. Phys. 1988, 65, 1409–1419. [Google Scholar] [CrossRef]
- Lagorce, D.; Bouslama, L.; Becot, J.; Miteva, M.A.; Villoutreix, B.O. FAF-Drugs4: Free ADME-tox filtering computations for chemical biology and early stages drug discovery. Bioinformatics. 2017, 33, 3658–3660. [Google Scholar] [CrossRef]
- Sander, T.; Freyss, J.; Von Korff, M.; Rufener, C. DataWarrior: An open-source program for chemistry aware data visualization and analysis. J. Chem. Inf. Model. 2015, 55, 460–473. [Google Scholar] [CrossRef]
- Drwal, M.N.; Banerjee, P.; Dunkel, M.; Wettig, M.R.; Preissner, R. ProTox: A web server for the in silico prediction of rodent oral toxicity. Nucleic Acids Res. 2014, 42, 53–58. [Google Scholar] [CrossRef]
- Bilbao-Ramos, P.; Sifontes-Rodríguez, S.; Dea-Ayuela, M.A.; Bolás-Fernández, F. A fluorometric method for evaluation of pharmacological activity against intracellular Leishmania amastigotes. J. Microbiol. Methods 2012, 89, 8–11. [Google Scholar] [CrossRef]
Sample Availability: Not available. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Server | RMSD * | Q-MEAN4 ** | Ramachandran Plot (%) | |
|---|---|---|---|---|
| F | P | |||
| SWISS-MODEL | 0.063 | −0.17 | 96.4% | 99.7% |
| I-Tasser | 0.257 | −3.34 | 89.0% | 96.9% |
| Phyre2 | 0.269 | −1.20 | 91.51% | 99.8% |
| Compounds | % Inhibition |
|---|---|
| ZINC14970552 (300 µM) | 35.7 |
| ZINC09642432 (300 µM) | 40.1 |
| ZINC04684558 (300 µM) | 39.5 |
| ZINC11841871 (150 µM) | 39.5 |
| ZINC12151998 (100 µM) | 32.9 |
| Compound | Time (ns) | ||||||
|---|---|---|---|---|---|---|---|
| Active Site | 0 | 10 | 20 | 30 | 40 | 50 | |
| ZINC14970552 | 1 | A: Met333 | - | A: Tyr198 | - | - | A: Tyr198 |
| 2 | B: Glu18 | - | - | - | - | - | |
| ZINC09642432 | 1 | - | - | - | - | - | - |
| 2 | - | B: Leu167 B: Gly168 | B: Leu167 | - | - | B: Leu167 | |
| ZINC04684558 | 1 | A: Tyr110 B: His461 | B: Asn402 | - | - | - | - |
| 2 | B: Val58 | - | B: Ser109 | B: Ser109 | B: Ser109 | B: Ser109 A: Glu466 | |
| ZINC11841871 | 1 | B: His461 | B: Ser470 B: His461 | A: Val58 | A: Val58 | A: Ser14 | B: Arg472 |
| 2 | B: Tyr110 | B: Tyr110 | B: Tyr110 | - | - | - | |
| ZINC12151998 | 1 | - | B: Gly459 | - | - | - | A: Arg355 |
| 2 | - | - | B: Tyr110 A: Glu467 | B: Tyr110 | B: Tyr110 A: Glu466 A: His461 | B: Tyr110 | |
| Molecule | MW a | HBD a | HBA a | LogP a | LD50 b (mg/kg) | Toxicity Class b | Toxicity Targets b | Mutagenicity c | Tumorigenicity c | Drug Likeness d |
|---|---|---|---|---|---|---|---|---|---|---|
| ZINC11841871 | 494.5 | 7 | 2 | 3.6 | 1000 | 4 | none | none | none | 0.68 |
| ZINC12151998 | 486.5 | 7 | 1 | 4.8 | 500 | 4 | none | high | none | 0.76 |
| ZINC14970552 | 443.5 | 7 | 1 | 3.7 | 1000 | 4 | none | none | none | 1.09 |
| ZINC04684558 | 409.3 | 6 | 2 | 4.9 | 1600 | 4 | none | none | none | 0.08 |
| ZINC09642432 | 463.4 | 8 | 1 | 2.9 | 1870 | 4 | none | none | none | 0.69 |
| Compounds | IC50 (µM) | CC50 (µM) | SI * |
|---|---|---|---|
| ZINC04684558 | 263 | 338 | 1.2 |
| ZINC12151998 | 58 | 53 | 0.9 |
| ZINC14970552 | 147 | 219 | 1.4 |
| ZINC09642432 | 222 | 335 | 1.5 |
| ZINC11841871 | 159 | 103 | 0.6 |
| Glucantime | >273.2 | >273.2 | 1.0 |
| Amphotericin B | 0.11 | 7.4 | 67 |
| Miltefosine | 2.98 | 141 | 47 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matadamas-Martínez, F.; Hernández-Campos, A.; Téllez-Valencia, A.; Vázquez-Raygoza, A.; Comparán-Alarcón, S.; Yépez-Mulia, L.; Castillo, R. Leishmania mexicana Trypanothione Reductase Inhibitors: Computational and Biological Studies. Molecules 2019, 24, 3216. https://doi.org/10.3390/molecules24183216
Matadamas-Martínez F, Hernández-Campos A, Téllez-Valencia A, Vázquez-Raygoza A, Comparán-Alarcón S, Yépez-Mulia L, Castillo R. Leishmania mexicana Trypanothione Reductase Inhibitors: Computational and Biological Studies. Molecules. 2019; 24(18):3216. https://doi.org/10.3390/molecules24183216
Chicago/Turabian StyleMatadamas-Martínez, Félix, Alicia Hernández-Campos, Alfredo Téllez-Valencia, Alejandra Vázquez-Raygoza, Sandra Comparán-Alarcón, Lilián Yépez-Mulia, and Rafael Castillo. 2019. "Leishmania mexicana Trypanothione Reductase Inhibitors: Computational and Biological Studies" Molecules 24, no. 18: 3216. https://doi.org/10.3390/molecules24183216
APA StyleMatadamas-Martínez, F., Hernández-Campos, A., Téllez-Valencia, A., Vázquez-Raygoza, A., Comparán-Alarcón, S., Yépez-Mulia, L., & Castillo, R. (2019). Leishmania mexicana Trypanothione Reductase Inhibitors: Computational and Biological Studies. Molecules, 24(18), 3216. https://doi.org/10.3390/molecules24183216