
Platycodigenin as Potential Drug Candidate for Alzheimer’s Disease via Modulating Microglial Polarization and Neurite Regeneration


Abstract
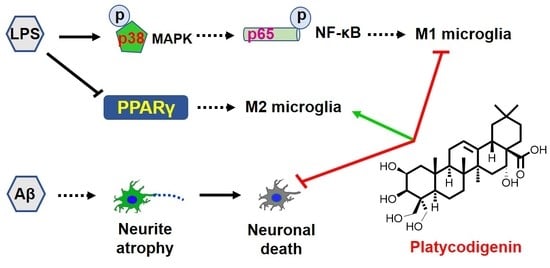
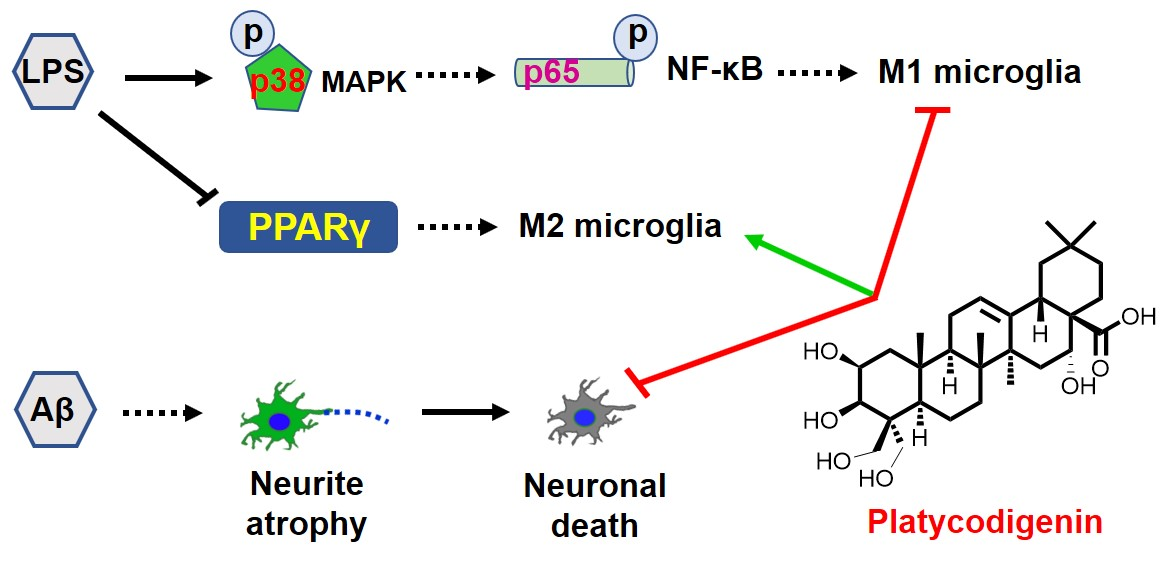{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
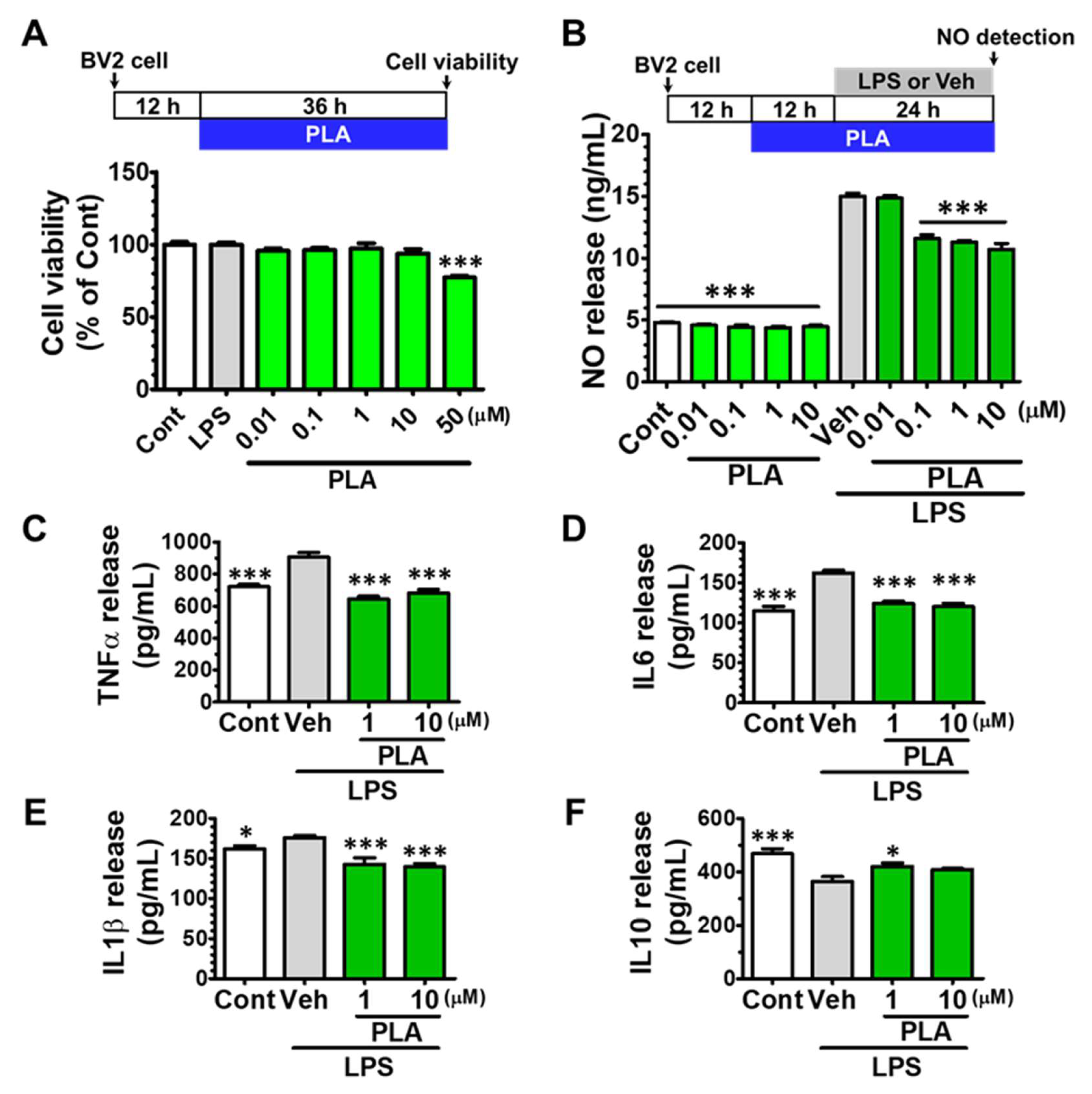
2.1. Platycodigenin Inhibited the Production of NO and Ameliorated the Secretion of Pro-Inflammatory Cytokines
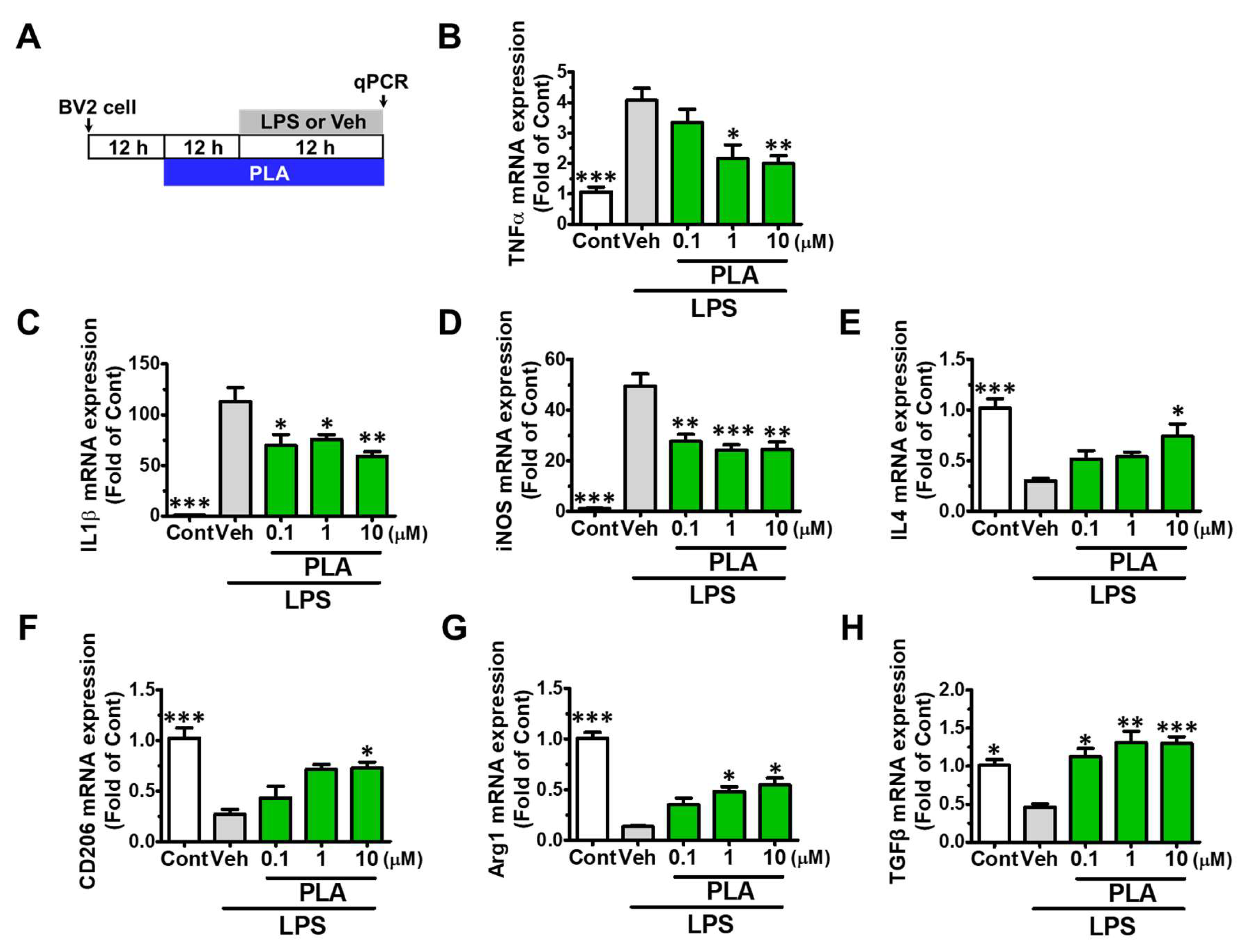
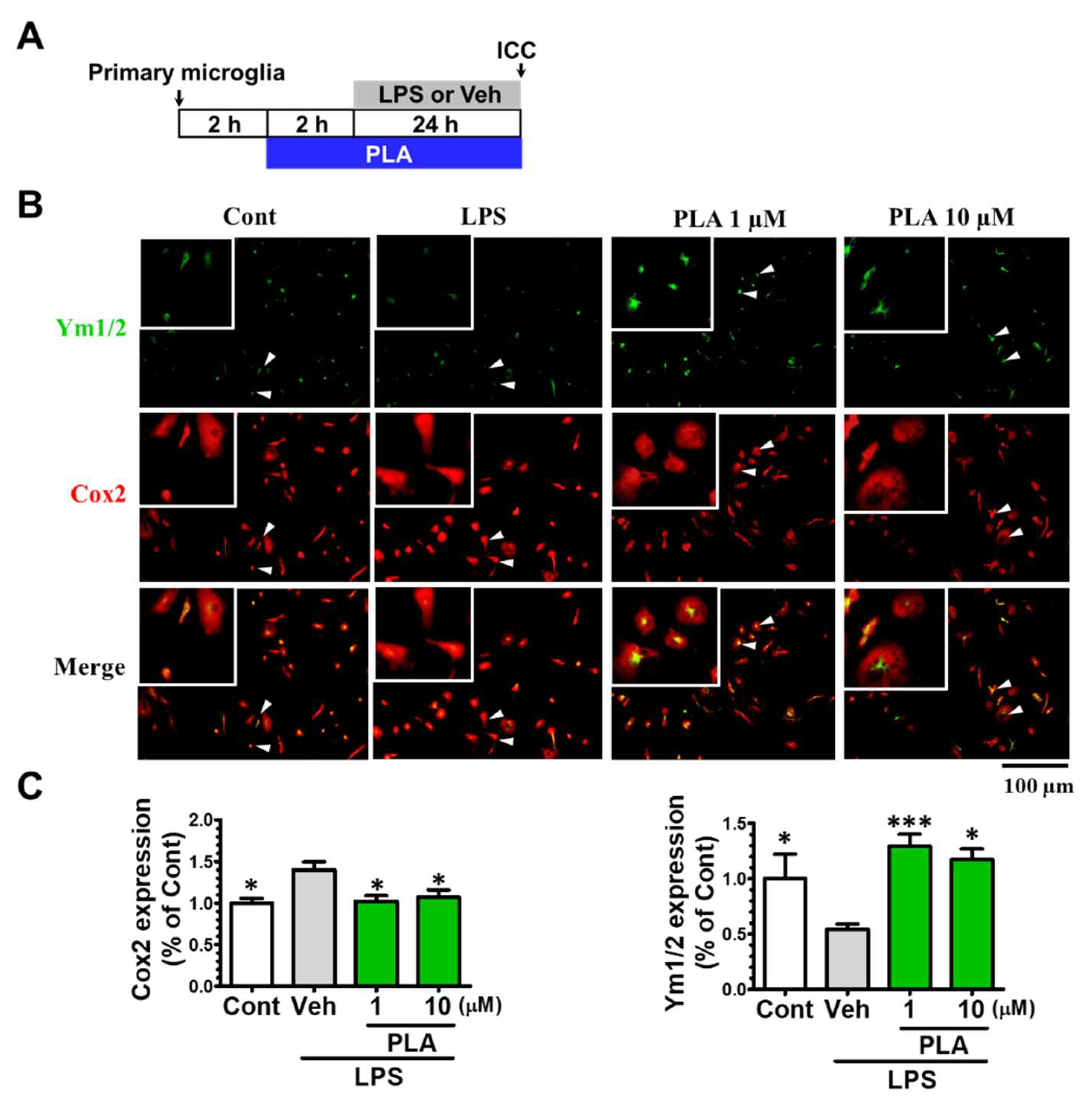
2.2. Platycodigenin Promoted LPS-Induced M1 Phenotype to M2 Microglia Polarization
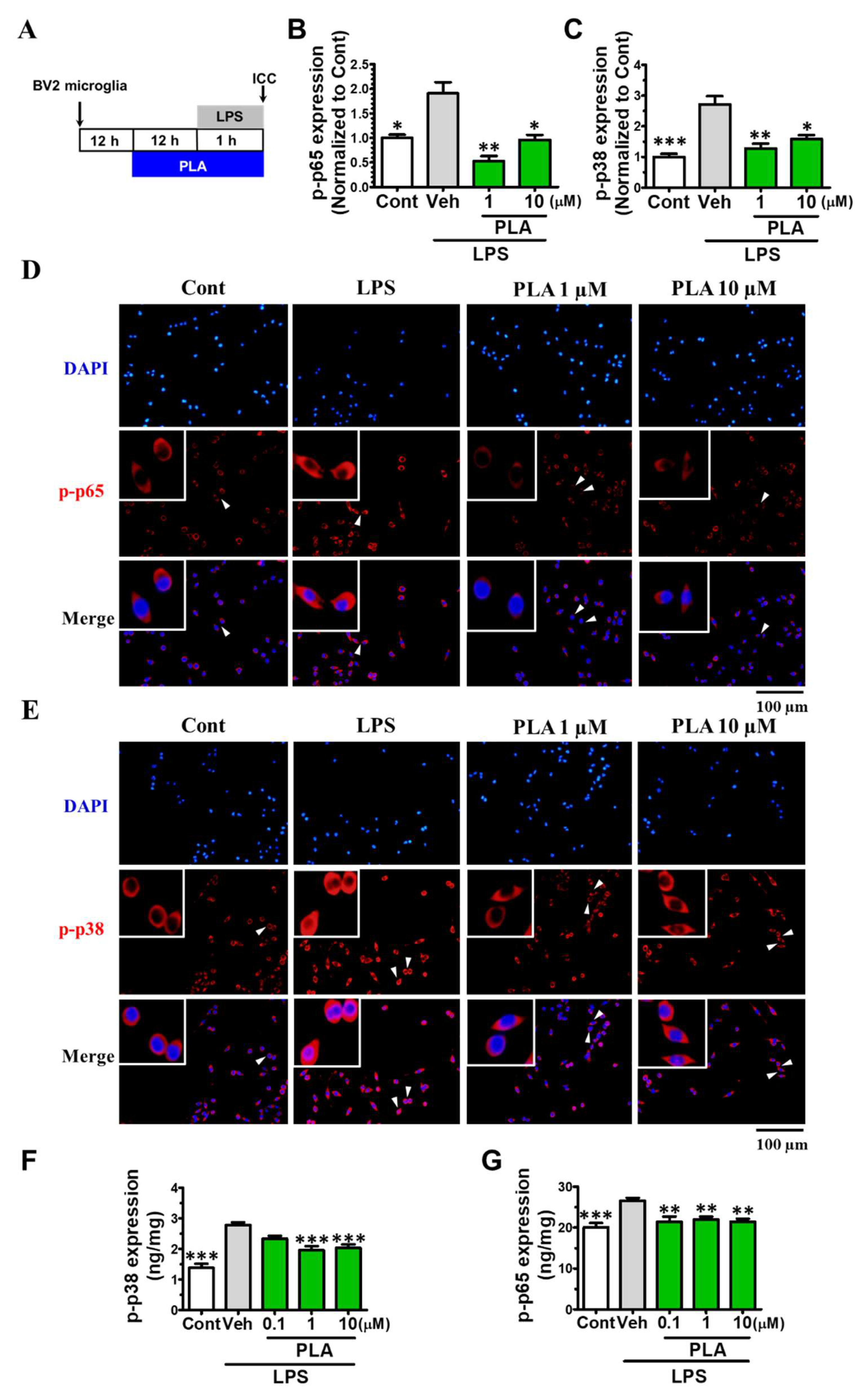
2.3. Platycodigenin Attenuated LPS-Induced Activation of MAPK and NF-κB Signaling Pathways
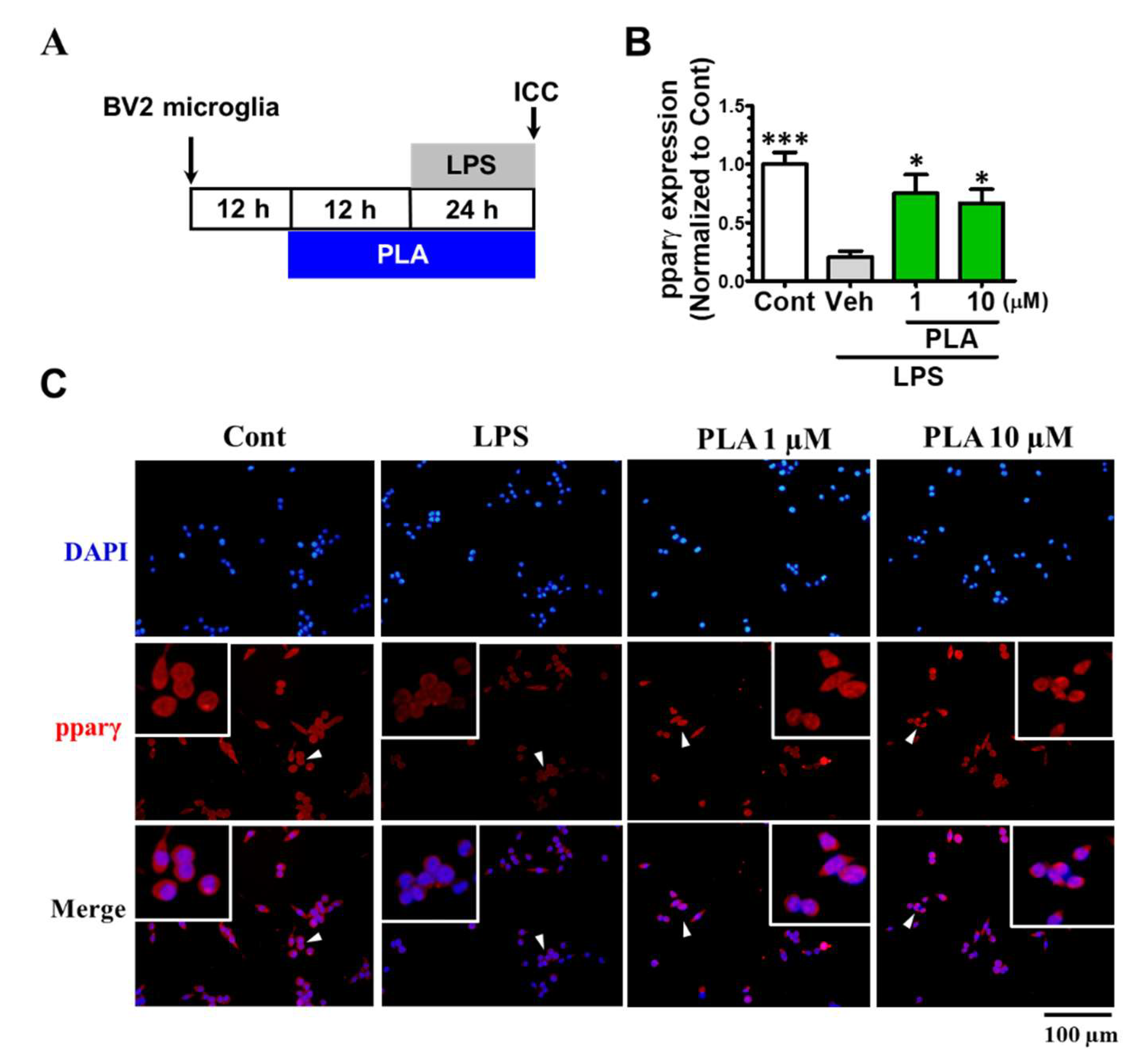
2.4. Platycodigenin as a Potential Agonist for PPARγ
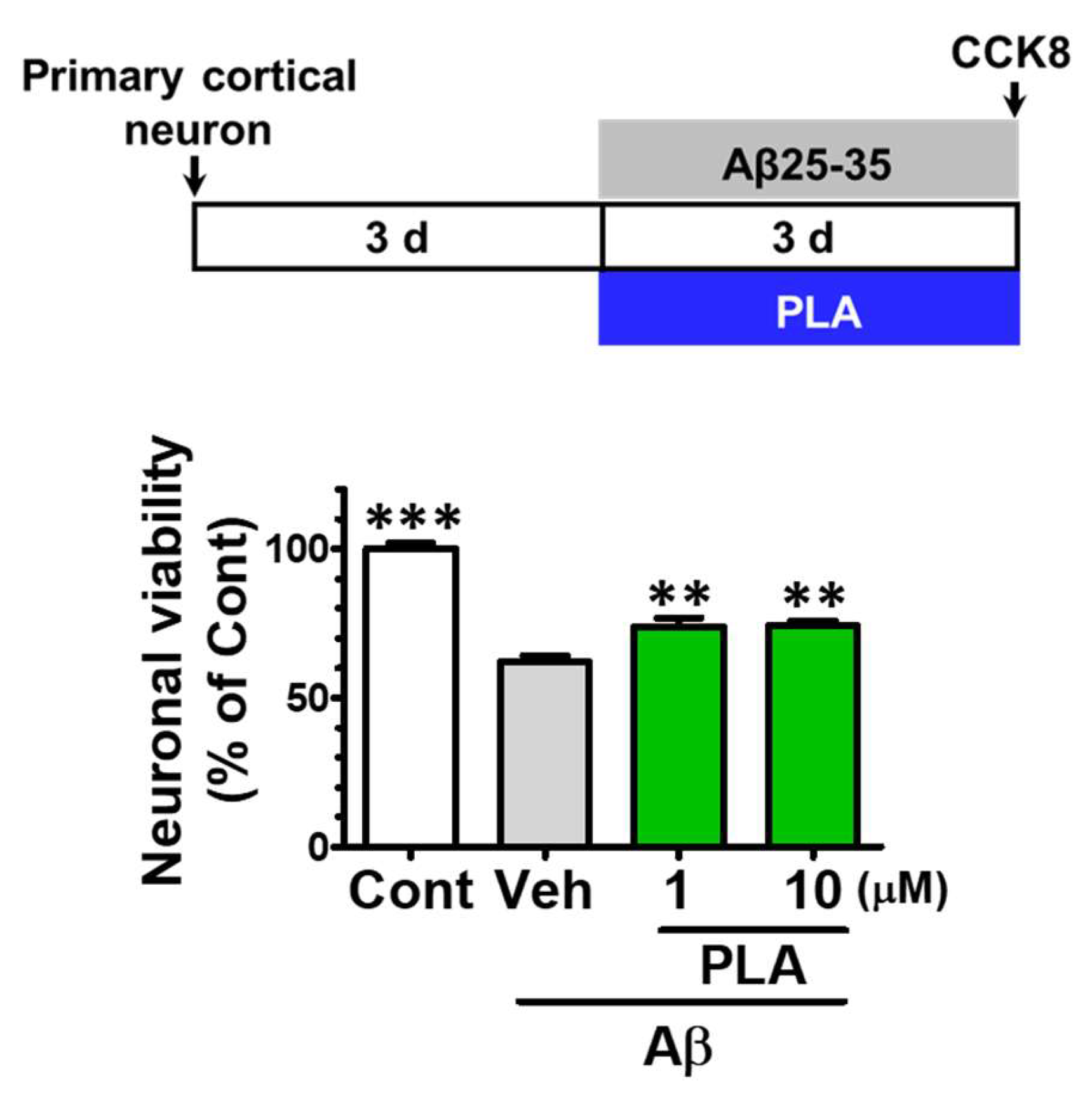
2.5. Platycodigenin Prevents Aβ25-35-Induced Primary Cortical Neuronal Death
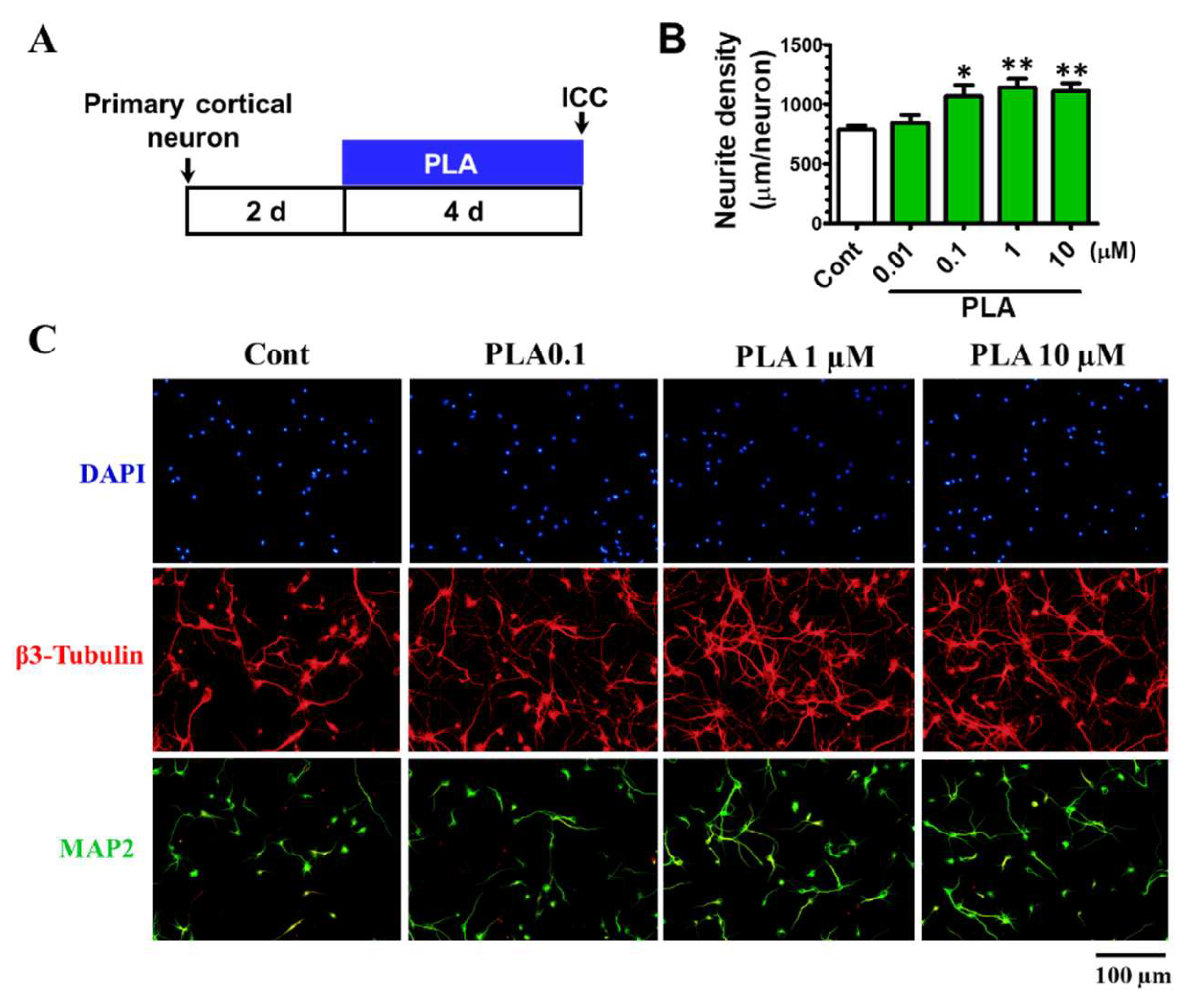
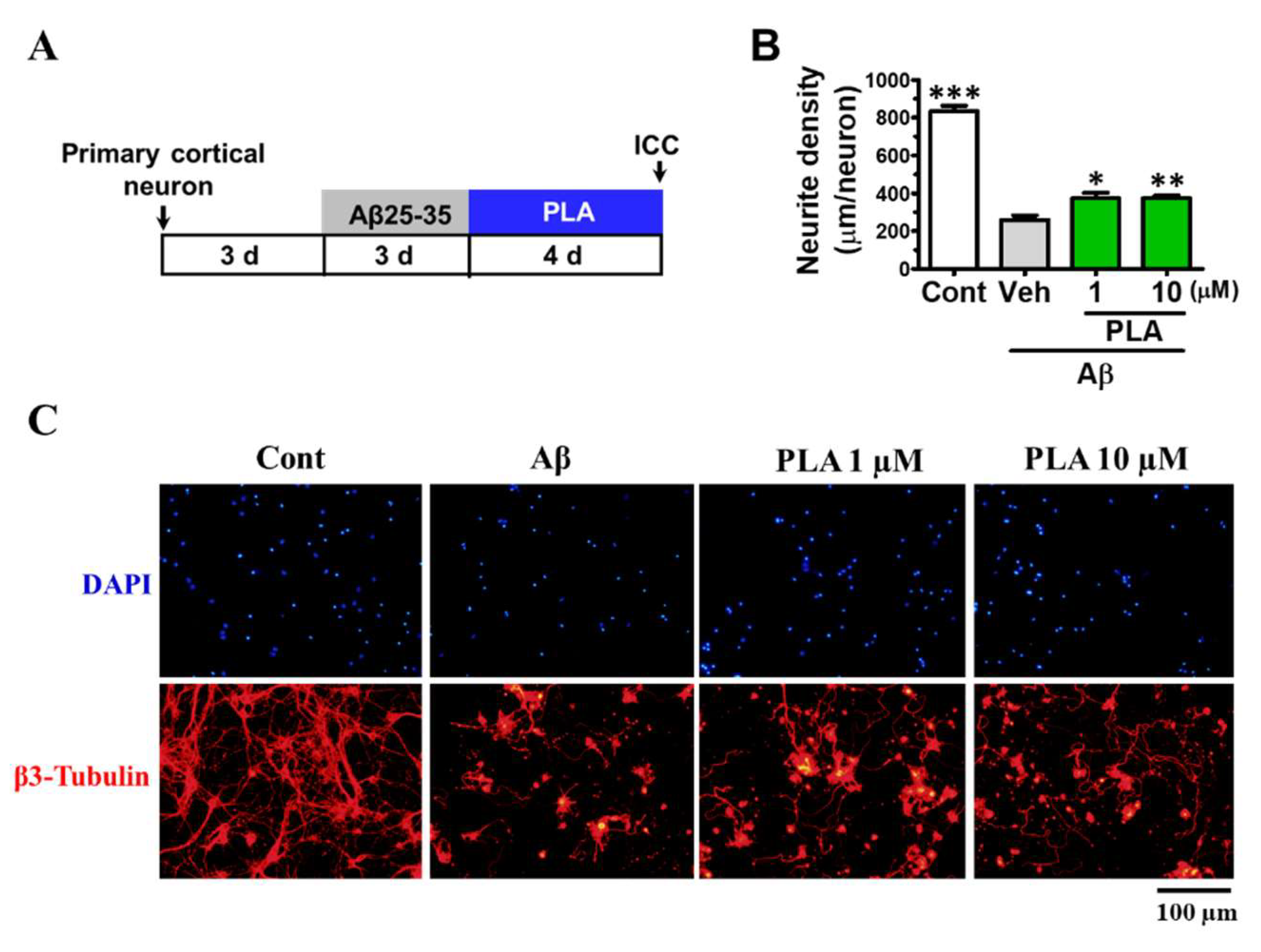
2.6. Platycodigenin Promotes Neurites Regeneration after Aβ25-35-Induced Neurites Atrophy
3. Discussion
4. Materials and Methods
4.1. BV2 Microglia and Primary Cortical Microglia Culture
4.2. Primary Cortical Neuron Culture and Neuronal Viability Assay
4.3. Microglia Viability Assay
4.4. Measurement of Nitric Oxide Levels
4.5. Measurement of TNF-α, IL-6, IL-1β, and IL10
4.6. RNA Isolation and RT-PCR
4.7. Immunocytochemistry
4.8. Measurement of Phosphorylated p65 and p38 by ELISA
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Mosser, C.A.; Baptista, S.; Arnoux, I.; Audinat, E. Microglia in CNS development: Shaping the brain for the future. Prog. Neurobiol. 2017, 149, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Orihuela, R.; McPherson, C.A.; Harry, G.J. Microglial M1/M2 polarization and metabolic states. Br. J. Pharm. 2016, 173, 649–665. [Google Scholar] [CrossRef] [PubMed]
- Perry, V.H.; Nicoll, J.A.; Holmes, C. Microglia in neurodegenerative disease. Nat. Rev. Neurol. 2010, 6, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Le, W.D. Differential roles of M1 and M2 microglia in neurodegenerative diseases. Mol. Neurobiol. 2016, 53, 1181–1194. [Google Scholar] [CrossRef] [PubMed]
- Salter, M.W.; Stevens, B. Microglia emerge as central players in brain disease. Nat. Med. 2017, 23, 1018–1027. [Google Scholar] [CrossRef] [PubMed]
- Saijo, K.; Glass, C.K. Microglial cell origin and phenotypes in health and disease. Nat. Rev. Immunol. 2011, 11, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Biswas, S.K.; Galdiero, M.R.; Sica, A.; Locati, M. Macrophage plasticity and polarization in tissue repair and remodeling. J. Pathol. 2013, 229, 176–185. [Google Scholar] [CrossRef]
- Hu, X.; Leak, R.K.; Shi, Y.J.; Suenaga, J.; Gao, Y.; Zheng, P.; Chen, J. Microglial and macrophage polarization—New prospects for brain repair. Nat. Rev. Neurol. 2015, 11, 56–64. [Google Scholar] [CrossRef]
- Miron, V.E.; Boyd, A.; Zhao, J.W.; Yuen, T.J.; Ruckh, J.M.; Shadrach, J.L.; Wijngaarden, P.V.; Wagers, A.J.; Williams, A.; Franklin, R.J.M.; et al. M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat. Neurosci. 2013, 16, 1211–1218. [Google Scholar] [CrossRef]
- Miguel-Álvarez, M.; Santos-Lozano, A.; Sanchis-Gomar, F.; Fiuza-Luces, C.; Pareja-Galeano, H.; Garatachea, N.; Lucia, A. Non-steroidal anti-inflammatory drugs as a treatment for Alzheimer’s disease: A systematic review and meta-analysis of treatment effect. Drugs Aging 2015, 32, 139–147. [Google Scholar] [CrossRef]
- Dickson, T.C.; Vickers, J.C. The morphological phenotype of β-amyloid plaques and associated neuritic changes in Alzheimer’s disease. Neuroscience 2001, 105, 99–107. [Google Scholar] [CrossRef]
- Alobuia, W.M.; Xia, W.; Vohra, B.P. Axon degeneration is key component of neuronal death in amyloid-β toxicity. Neurochem. Int. 2013, 63, 782–789. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Kuboyama, T.; Tohda, C. A systematic strategy for discovering a therapeutic drug for Alzheimer’s disease and its target molecule. Front. Pharm. 2017, 8, 340. [Google Scholar] [CrossRef] [PubMed]
- Tohda, C.; Urano, T.; Umezaki, M.; Nemere, I.; Kuboyama, T. Diosgenin is an exogenous activator of 1,25D3-MARRS/Pdia3/ERp57 and improves Alzheimer’s disease pathologies in 5XFAD mice. Sci. Rep. 2012, 2, 535. [Google Scholar] [CrossRef] [PubMed]
- Gorlovoy, P.; Larionov, S.; Pham, T.T.; Neumann, H. Accumulation of tau induced in neurites by microglial proinflammatory mediators. Faseb J. 2009, 23, 2502–2513. [Google Scholar] [CrossRef] [PubMed]
- Luzina, I.G.; Keegan, A.D.; Heller, N.M.; Rook, G.A.W.; Shea-Donohue, T.; Atamas, S.P. Regulation of inflammation by interleukin-4: A review of “alternatives”. J. Leukoc. Biol. 2012, 92, 753–764. [Google Scholar] [CrossRef] [PubMed]
- Vergès, B. Clinical interest of PPARs ligands. Diabetes Metab. 2004, 30, 7–12. [Google Scholar] [CrossRef]
- Nick, J.A.; Young, S.K.; Brown, K.K.; Avdi, N.J.; Arndt, P.G.; Suratt, B.T.; Janes, M.S.; Henson, P.M.; Worthen, G.S. Role of p38 mitogen-activated protein kinase in a murine model of pulmonary inflammation. J. Immunol. 2000, 164, 2151–2159. [Google Scholar] [CrossRef]
- Ji, H.; Wang, H.; Zhang, F.; Li, X.; Xiang, L.; Shen, A.G. PPARγ agonist pioglitazone inhibits microglia inflammation by blocking p38 mitogen-activated protein kinase signaling pathways. Inflamm. Res. 2010, 59, 921–929. [Google Scholar] [CrossRef]
- Chiang, M.C.; Cheng, Y.C.; Chen, H.M.; Liang, Y.J.; Yen, C.H. Rosiglitazone promotes neurite outgrowth and mitochondrial function in N2A cells via PPARgamma pathway. Mitochondrion 2014, 14, 7–17. [Google Scholar] [CrossRef]
- Saha, R.N.; Jana, M.; Pahan, K. MAPK p38 regulates transcriptional activity of NF-kappaB in primary human astrocytes via acetylation of p65. J. Immunol. 2007, 179, 7101–7109. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Xu, Y.; Wang, Y.; Wang, Y.J.; He, L.; Jiang, Z.Z.; Huang, Z.J.; Liao, H.; Li, J.; Saavedra, J.M.; et al. Telmisartan prevention of LPS-induced microglia activation involves M2 microglia polarization via CaMKKβ-dependent AMPK activation. Brain Behav. Immun. 2015, 50, 298–313. [Google Scholar] [CrossRef] [PubMed]
- Bouhlel, M.A.; Derudas, B.; Rigamonti, E.; Dievart, R.; Brozek, J.; Haulon, S.; Zawadzki, C.; Jude, B.; Torpier, G.; Marx, N.; et al. PPARgamma activation primes human monocytes into alternative M2 macrophages with anti-inflammatory properties. Cell Metab. 2007, 6, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Kuboyama, T.; Tohda, C. Naringenin promotes microglial M2 polarization and Aβ degradation enzyme expression. Phytother. Res. 2019, 33, 1114–1121. [Google Scholar] [CrossRef] [PubMed]
- Son, H.; Park, Y.H.; Lee, S.I.; Yang, H.D.; Moon, H.I. Neuroprotective activity of triterpenoid saponins from Platycodi radix against glutamate-induced toxicity in primary cultured rat cortical cells. Molecules 2007, 12, 1147–1152. [Google Scholar] [CrossRef]
- Choi, J.H.; Yoo, K.Y.; Park, O.K. Platycodin D and 2″-O-acetyl-polygalacin D2 isolated from Platycodon grandiflorum protect ischemia/reperfusion injury in the gerbil hippocampus. Brain Res. 2009, 1279, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, Y.L.; Yang, D.W.; Zhang, C.H.; Zhang, N.; Li, M.H.; Liu, Y.Z. Platycodon grandiflorus—an ethnopharmacological, phytochemical and pharmacological review. J. Ethnopharmacol. 2015, 164, 147–161. [Google Scholar] [CrossRef]
- Jang, K.J.; Kim, H.K.; Han, M.H.; Oh, Y.N.; Yoon, H.M.; Chung, Y.H.; Kim, G.Y.; Hwang, H.J.; Kim, B.W.; Choi, Y.H. Anti-inflammatory effects of saponins derived from the roots of Platycodon grandiflorus in lipopolysaccharide‑stimulated BV2 microglial cells. Int. J. Mol. Med. 2013, 31, 1357–1366. [Google Scholar] [CrossRef]
- Fu, Y.; Xin, Z.; Liu, B.; Wang, J.; Wang, J.; Zhang, X.; Wang, Y.; Li, F. Platycodin D inhibits inflammatory response in lps-stimulated primary rat microglia cells through activating LXRα-ABCA1 signaling pathway. Front. Immunol. 2018, 8, 1929. [Google Scholar] [CrossRef]
- Zhang, W.Z.; Hou, J.A.; Yan, X.T.; Leng, J.; Li, R.Y.; Zhang, J.; Xing, J.J.; Chen, C.; Wang, Z.; Li, W. Platycodon grandiflorum saponins ameliorate cisplatin-induced acute nephrotoxicity through the nf-κb-mediated inflammation and pi3k/akt/apoptosis signaling pathways. Nutrients 2018, 10, 1328. [Google Scholar] [CrossRef]
- Jang, E.; Lee, S.; Kim, J.H.; Kim, J.H.; Seo, J.W.; Lee, W.H.; Mori, K.; Nakao, K.; Suk, K. Secreted protein lipocalin-2 promotes microglial M1 polarization. Faseb J. 2013, 27, 1176–1190. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, S.R.; Federoff, H.J. Targeting microglial activation states as a therapeutic avenue in parkinson’s disease. Front. Aging Neurosci. 2017, 9, 176. [Google Scholar] [CrossRef] [PubMed]
- Bi, C.L.; Wang, H.; Wang, Y.J.; Sun, J.; Dong, J.S.; Meng, X.; Li, J.J. Selenium inhibits Staphylococcus aureus-induced inflammation by suppressing the activation of the NF-kappaB and MAPK signalling pathways in RAW264.7 macrophages. Eur. J. Pharm. 2016, 780, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Ahmadian, M.; Suh, J.M.; Hah, N.; Liddle, C.; Atkins, A.R.; Downes, M.; Evans, R.M. PPARγ signaling and metabolism: The good, the bad and the future. Nat. Med. 2013, 19, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Perry, V.H.; Holmes, C. Microglial priming in neurodegenerative disease. Nat. Rev. Neurol. 2014, 10, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, V.; Bates, T.E.; Stella, A.M. NO synthase and NO-dependent signal pathways in brain aging and neurodegenerative disorders: The role of oxidant/antioxidant balance. Neurochem. Res. 2000, 25, 1315–1341. [Google Scholar] [CrossRef] [PubMed]
- Warburton, E.C.; Brown, M.W. Neural circuitry for rat recognition memory. Behav. Brain Res. 2015, 285, 131–139. [Google Scholar] [CrossRef]
- Jorge, J.P.; Lennart, M. Amyloid-β induced neuronal dysfunction in Alzheimer’s disease: From synapses toward neural networks. Nat. Neurosci. 2010, 13, 812–818. [Google Scholar]
- Kuboyama, T.; Wahane, S.; Huang, Y.; Zhou, X.; Wong, J.K.; Koemeter-Cox, A.; Martini, M.; Friedel, R.H.; Zou, H.Y. HDAC3 inhibition ameliorates spinal cord injury by immunomodulation. Sci. Rep. 2017, 7, 8641. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from authors. |








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Z.; Liu, B.; Yang, L.-e.; Zhang, C. Platycodigenin as Potential Drug Candidate for Alzheimer’s Disease via Modulating Microglial Polarization and Neurite Regeneration. Molecules 2019, 24, 3207. https://doi.org/10.3390/molecules24183207
Yang Z, Liu B, Yang L-e, Zhang C. Platycodigenin as Potential Drug Candidate for Alzheimer’s Disease via Modulating Microglial Polarization and Neurite Regeneration. Molecules. 2019; 24(18):3207. https://doi.org/10.3390/molecules24183207
Chicago/Turabian StyleYang, Zhiyou, Baiping Liu, Long-en Yang, and Cai Zhang. 2019. "Platycodigenin as Potential Drug Candidate for Alzheimer’s Disease via Modulating Microglial Polarization and Neurite Regeneration" Molecules 24, no. 18: 3207. https://doi.org/10.3390/molecules24183207
APA StyleYang, Z., Liu, B., Yang, L.-e., & Zhang, C. (2019). Platycodigenin as Potential Drug Candidate for Alzheimer’s Disease via Modulating Microglial Polarization and Neurite Regeneration. Molecules, 24(18), 3207. https://doi.org/10.3390/molecules24183207