4.2. Synthesis
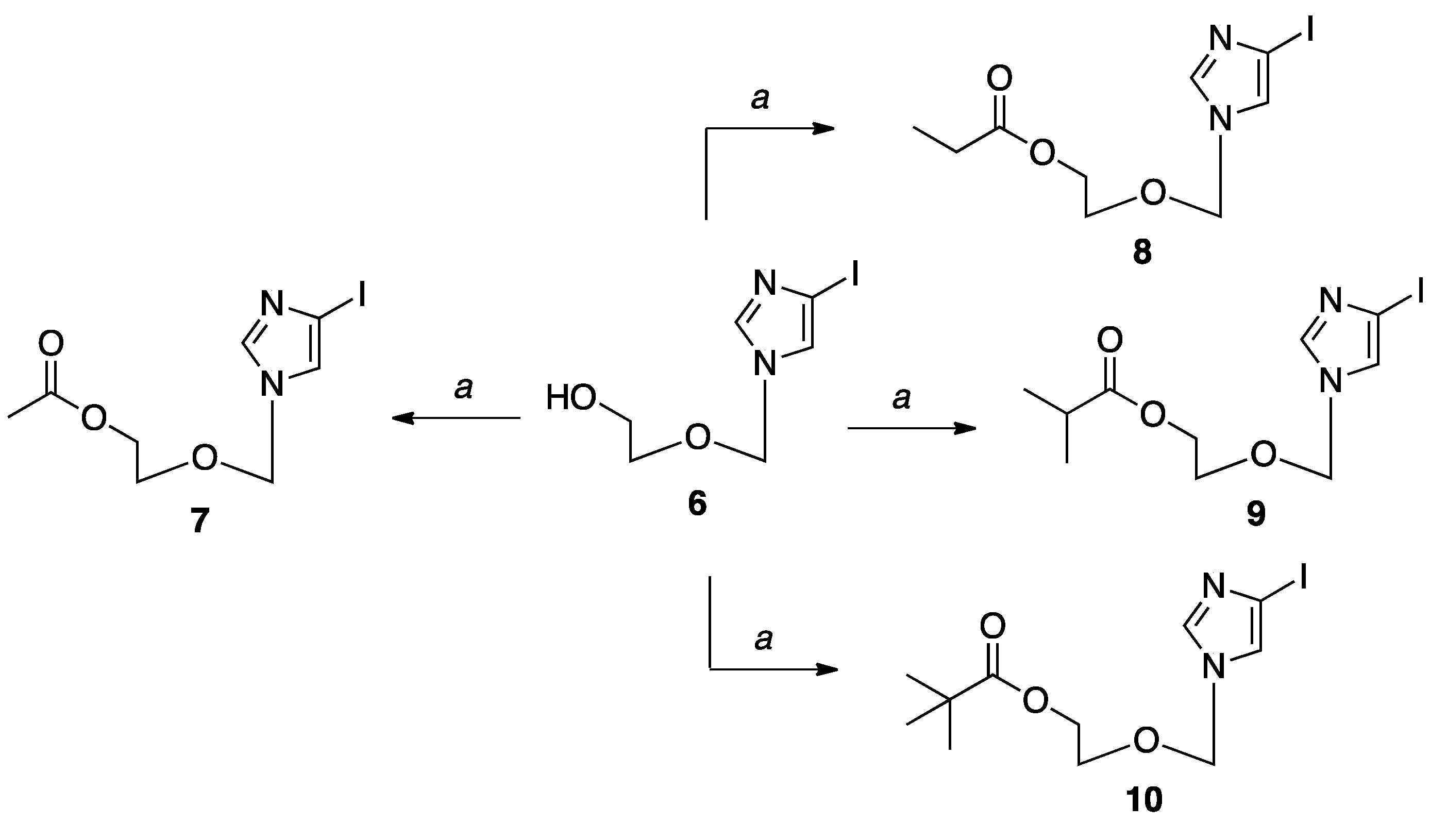
General procedure for the acyl-protected sugars8–10. To a solution of 6 (1.5 g, 5.96 mmol) in anh. CH2Cl2 (100 mL) under a nitrogen balloon was added the corresponding anhydride (2 equivalents), triethylamine (2.50 mL, 17.85 mmol), and DMAP (0.18 g, 1.49 mmol) with stirring at room temperature. The reaction stirred for 3 hours at room temperature; then, the solvent was removed in vacuo. Purification by flash column chromatography on silica gave the products in good yields.
Characterization of 2-((4-iodo-1H-imidazol-1-yl)methoxy)ethyl propionate (8). The anhydride used was propionic anhydride (1.52 mL, 11.90 mmol). The crude mixture was purified by flash column chromatography on silica (0–100% EtOAc in hexanes) to give the product as a clear oil (1.78 g, 99%); Rf = 0.42 (1:1 EtOAc: Hex); 1H-NMR (500 MHz, CDCl3) δ 7.41 (s, 1H), 7.05 (s, 1H), 5.19 (s, 2H), 4.01–4.03 (t, J = 4.7 Hz, 2H), 3.47–3.49 (t, J = 4.6 Hz, 2H), 2.14–2.19 (m, 2H), 0.93–0.96 (t, J = 7.6 Hz, 3H); 13C-NMR (126 MHz, CDCl3) δ 174.43, 139.03, 124.30, 83.12, 76.41, 66.72, 62.80, 27.58, 9.13; MS (ESI+) m/z calcd for C9H13IN2O3 [M + H]+ 325.00 found: 325.01.
Characterization of 2-((4-iodo-1H-imidazol-1-yl)methoxy)ethyl isobutyrate (9). The anhydride used was isobutyric anhydride (1.98 mL, 11.90 mmol). The crude mixture was purified by flash column chromatography on silica (0–100% EtOAc in hexanes) to give the product as a clear oil (2.0 g, 99%); Rf = 0.53 (1:1 EtOAc: Hex;1H-NMR (500 MHz, CDCl3) δ 7.26 (s, 1H), 6.89 (s, 1H), 5.01 (s, 2H), 3.77–3.78 (m, 2H), 3.27–3.28 (m, 2H), 2.14–2.15 (m, 1H), 0.72–0.78 (d, J = 4.4 Hz, 6H); 13C-NMR (101 MHz, CDCl3) δ 176.45, 139.17, 124.53, 82.33, 76.12, 66.70, 62.46, 33.47, 18.79. MS (ESI+) m/z calcd for C10H15IN2O3 [M + Na]+ 361.15 found: 361.03.
Characterization of 2-((4-iodo-1H-imidazol-1-yl)methoxy)ethyl pivalate (10). The anhydride used was trimethylacetic anhydride (2.40 mL, 11.90 mmol). The crude mixture was purified by flash column chromatography on silica (0–100% EtOAc in hexanes) to give the product as a clear oil (1.55 g, 78%); Rf = 0.53 (1:1 EtOAc: Hex; 1H NMR (400 MHz, CDCl3) δ 7.44 (s, 1H), 7.07 (s, 1H), 5.22 (s, 2H), 4.06–4.13 (t, J = 4.1 Hz, 2H), 3.48–3.52 (t, J = 4.1 Hz, 2H), 1.07–1.15(s, 9H); 13C NMR (125 MHz, CDCl3) δ 177.42, 139.53, 130.08, 124.92, 76.02, 66.76, 62.70, 38.35, 28.88. MS (ESI+) m/z calcd for C11H17IN2O3 [M + Na]+ 375.17 found: 375.05.
General procedure for Stille cross coupling. The sugar (1 equivalent) and stannyl reagent (1.2 equivalents) were dried in a three-neck flash in vacuo for two hours, and then were suspended in anh. DMF (100 mL) under direct nitrogen bubbling. CsF (2 equivalents) and CuI (0.4 equivalents) were added, and the reaction was stirred for 10 min. Tetrakis (0.2 equivalents) was then added, and the reaction was heated to 65 °C and stirred with direct nitrogen bubbling for 2.5 h. After completion, the reaction was cooled, and the solvent was removed in vacuo. The crude mixture was then purified three to four times by flash column chromatography on silica to give the pure products.
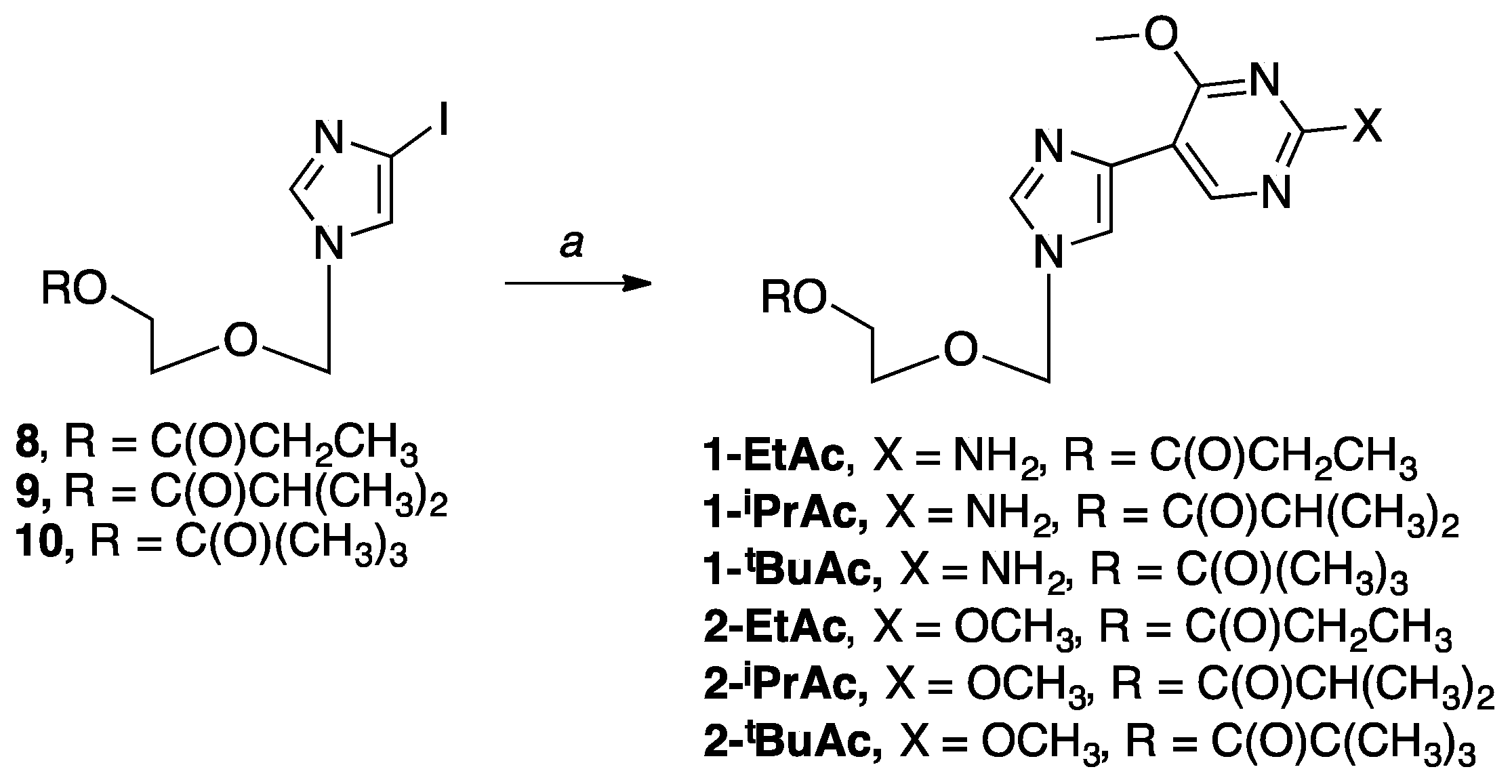
2-((4-(2-Amino-4-methoxypyrimidin-5-yl)-1H-imidazol-1-yl)methoxy)ethyl propionate (1-EtAc). The sugar 8 (0.75 g, 2.31 mmol) and 4-methoxy-5-(tributylstannyl)pyrimidin-2-amine (1.15 g, 2.77 mmol) were used. The product was purified three times by flash column chromatography on silica (0–25% CH3OH in CH2Cl2) to give the product as a white solid (0.041 g, 9%); 1H NMR (500 MHz, CD3OD) δ 8.83 (s, 1H), 7.62 (s, 1H), 7.40 (s, 1H), 5.32 (s, 2H), 4.20–4.21 (t, J = 4.7 Hz, 2H), 4.01 (s, 3H), 3.62–3.64 (t, J = 4.7 Hz, 2H), 2.30–2.35 (m, 2H), 1.09–1.12 (t, J = 7.6 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 174.30, 166.23, 161.08, 154.83, 136.85, 135.41, 117.00, 105.92, 76.46, 66.58, 62.70, 53.66, 27.35, 9.00; Elemental analysis calcd for C14H19N5O4 + 0.25 CH4OH + 0.15 H2O: C, 51.55; H, 6.16; N, 21.09; Found: C, 51.58; H, 6.12, N, 21.06.
2-((4-(2-Amino-4-methoxypyrimidin-5-yl)-1H-imidazol-1-yl)methoxy)ethyl isobutyrate (1-iPrAc). The sugar 9 (0.80 g, 2.37 mmol) and 4-methoxy-5-(tributylstannyl)pyrimidin-2-amine (1.18 g, 2.84 mmol) were used. The product was purified three times by flash column chromatography on silica (0–25% CH3OH in CH2Cl2) to give the product as a pale yellow oil (0.09 g, 12%); 1H NMR (500 MHz, CDCl3) δ 8.80 (s, 1H), 7.60 (s, 1H), 7.39 (s, 1H), 5.31 (s, 2H), 4.17–4.20 (t, J = 5.6 Hz, 2H), 3.98 (s, 3H), 3.59–3.61 (t, J = 5.3 Hz, 2H), 2.48–2.55 (m, 1H), 1.10–1.12 (d, J = 9.2 Hz, 6H); 13C NMR (126 MHz, CDCl3) δ 176.86, 166.42, 161.37, 155.24, 137.35, 135.49, 117.03, 106.04, 76.41, 66.58, 62.66, 53.58, 33.78, 18.96; Elemental analysis calcd for C15H21N5O4 + 0.65 H2O: C, 51.91; H, 6.48; N, 20.18; Found: C, 51.60; H, 6.09; N, 20.18.
2-((4-(2-Amino-4-methoxypyrimidin-5-yl)-1H-imidazol-1-yl)methoxy)ethyl pivalate (1-tBuAc). The sugar 10 (0.75 g, 2.13 mmol) and 4-methoxy-5-(tributylstannyl)pyrimidin-2-amine (1.06 g, 2.56 mmol) were used. The product was purified three times by flash column chromatography on silica (0–20% CH3OH in CH2Cl2) to give the product as a clear oil (0.08 g, 11%); 1H NMR (500 MHz, CDCl3) δ 8.78 (s, 1H), 7.67 (s, 1H), 7.41 (s, 1H), 5.36 (s, 2H), 4.23–4.25 (t, J = 4.0 Hz, 2H), 4.11 (s, 3H), 3.65–3.67 (t, J = 4.0 Hz, 2H), 1.19–1.27 (s, 9H); 13C NMR (126 MHz, CDCl3) δ 176.48, 166.35, 153.71, 137.37, 134.27, 117.33, 104.72, 76.09, 66.45, 62.77, 52.73, 38.31, 26.05; Elemental analysis calcd for C16H23N5O4 + 0.1 CH4OH + 0.95 H2O: C, 52.31; H, 6.90; N, 18.94; Found: C, 52.41; H, 6.74; N, 18.78.
2-((4-(2,4-Dimethoxypyrimidin-5-yl)-1H-imidazol-1-yl)methoxy)ethyl propionate (2-EtAc). The sugar 8 (0.75 g, 2.31 mmol) and 2,4-dimethoxy-5-(tributylstannyl)pyrimidine (1.19 g, 2.78 mmol) were used. The product was purified three times by flash column chromatography on silica (50–100% EtOAc in hexanes) to give the product as a pale yellow oil (0.25 g, 36%); 1H NMR (500 MHz, CDCl3) δ 8.82 (s, 1H), 7.91 (s, 1H), 7.65 (s, 1H), 5.47 (s, 2H), 4.19–4.21 (t, J = 3.5 Hz, 2H), 4.14 (s, 3H), 4.02 (s, 3H), 3.71–3.72 (t, J = 3.6 Hz, 2H), 2.28–2.33 (m, 2H), 1.05–1.08 (t, J = 7.6 Hz, 3H); 13C NMR (126 MHz, CD3OD) δ 174.44, 166.98, 163.58, 154.80, 138.21, 132.99, 118.74, 108.50, 76.34, 66.67, 62.73, 54.08, 53.49, 26.74, 8.01; Elemental analysis calcd for C15H20N4O5 + 0.05 H2O + 0.2 CH2Cl2: C, 51.54; H, 5.83; N, 15.82; Found: C, 51.67; H, 5.64; N, 15.61.
2-((4-(2,4-Dimethoxypyrimidin-5-yl)-1H-imidazol-1-yl)methoxy)ethyl isobutyrate (2-iPrAc). The sugar 9 (0.25 g, 0.74 mmol) and 2,4-dimethoxy-5-(tributylstannyl)pyrimidine (0.38 g, 0.89 mmol) were used. The product was purified twice times by flash column chromatography on silica (25–100% EtOAc in hexanes) to give the product as a light orange oil (0.06 g, 24%); 1H NMR (400 MHz, CDCl3) δ 8.96 (s, 1H), 7.61 (s, 1H), 7.43 (s, 1H), 5.32 (s, 2H), 4.14–4.19 (t, J = 3.4 Hz, 2H), 4.03 (s, 3H), 3.95 (s, 3H), 3.55–3.57 (t, J = 3.4 Hz, 2H), 2.45–2.47 (m, 1H), 1.05–1.09 (d, J = 6.9 Hz, 6H); 13C NMR (101 MHz, CDCl3) δ 176.95, 166.89, 163.73, 155.65, 137.12, 134.63, 117.99, 109.21, 76.91, 66.71, 62.61, 54.86, 54.12, 33.84, 18.95; Elemental analysis calcd for C16H22N4O5: C, 54.85; H, 6.33; N, 15.99; Found: C, 54.56; H, 6.50; N, 15.81.
2-((4-(2,4-Dimethoxypyrimidin-5-yl)-1H-imidazol-1-yl)methoxy)ethyl pivalate (2-tBuAc). The sugar 10 (0.30 g, 0.85 mmol) and 2,4-dimethoxy-5-(tributylstannyl)pyrimidine (0.44 g, 1.02 mmol) were used. The product was purified twice by flash column chromatography on silica (50–100% EtOAc in hexanes) to give the product as a light orange oil (0.16 g, 51%); 1H NMR (400 MHz, CDCl3) δ 9.05 (s, 1H), 7.95 (s, 1H), 7.53 (s, 1H), 5.42 (s, 2H), 4.22–4.23 (t, J = 4.7 Hz, 2H), 4.12 (s, 3H), 4.04 (s, 3H), 3.65–3.68 (t, J = 4.6 Hz, 2H), 1.18–1.20 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 178.40, 167.03, 163.64, 154.57, 137.83, 133.21, 118.62, 108.77, 76.15, 66.55, 62.74, 54.04, 53.44, 38.30, 26.06; Elemental analysis calcd for C17H24N4O5 + 0.35 CH3OH + 0.05 H2O: C, 55.35; H, 6.83; N, 14.88; Found: C, 55.36; H, 6.75; N, 14.83.
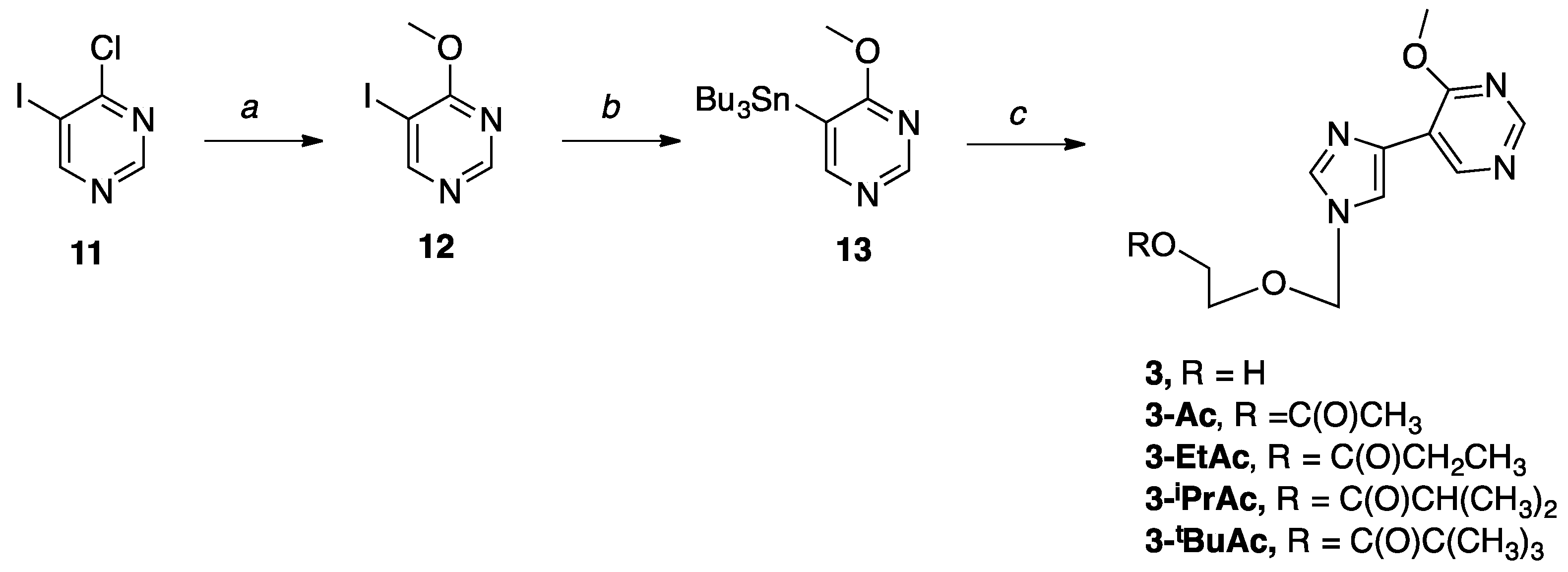
5-Iodo-4-methoxypyrimidine (12). To a solution of 4-chloro-5-iodopyrimidine (0.5 g, 2.08 mmol) in anh. CH3OH (20 mL) under nitrogen was added NaOMe (30% by weight in CH3OH, 0.53 mL, 2.88 mmol) at room temperature. Then, the reaction was stirred at room temperature under nitrogen for 6 hours. Upon completion, the solvent was removed in vacuo to give a yellow solid. The crude mixture was purified by flash column chromatography on silica (0–50% EtOAc in hexanes) to give the product as a white solid (0.45 g, 92%); Rf = 0.86 (1:4 EtOAc: Hex); 1H NMR (500 MHz, CDCl3) δ 8.79 (s, 1H), 8.72 (s, 1H), 4.10 (s, 3H); 13C NMR (126 MHz, CDCl3) δ 167.1, 163.9, 157.4, 80.1, 54.8; MS (ESI+) m/z calcd for C5H5IN2O [M + H]+ 236.95, found: 236.56.
4-Methoxy-5-(tributylstannyl)pyrimidine (13). To a solution of 12 (0.89 g, 3.79 mmol) in anh. DMF (50 mL) under direct nitrogen bubble was added bis(tributyltin) (2.29 mL, 4.55 mmol) and Pd2dba3•CHCl3 (0.39 g, 0.38 mmol). Then, the reaction was stirred at 65 °C for 2.5 h. After completion, the reaction was cooled to room temperature, and the solvent was removed in vacuo to give a black oil. The crude mixture was resuspended in EtOAc (100 mL) and filtered over a celite pad. The filtrated was concentrated in vacuo to give a brown oil. Then, the crude product was purified by flash column chromatography on silica (0–70% EtOAc in hexanes) to give the pure product as a pale yellow oil (1.40 g, 93%); Rf = 0.64 (1:1 EtOAc: Hex); 1H NMR (500 MHz, CDCl3) δ 8.46 (s, 1H), 8.11 (s, 1H), 3.65 (s, 3H), 1.20–1.31 (m, 6H), 1.03–1.06 (m, 6H), 0.81–0.89 (m, 6H), 0.58–0.62 (m, 9H); 13C NMR (126 MHz, CDCl3) δ 188.5, 163.3, 158.5, 128.7, 53.1, 28.9, 27.3, 13.6, 9.6; MS (ESI+) m/z calcd for C17H32N2OSn [M + H]+ 401.15, found: 401.18.
2-((4-(4-Methoxypyrimidin-5-yl)-1H-imidazol-1-yl)methoxy)ethan-1-ol (3). The sugar 6 (0.35 g, 1.31 mmol) and stannyl pyrimidine 13 (0.63 g, 1.57 mmol) were used. The product was purified three times by flash column chromatography on silica (0–25% CH3OH in CH2Cl2) to give the product as a shiny white solid (0.135 g, 41%); 1H NMR (500 MHz, CD3OD) δ 9.05 (s, 1H), 8.62 (s, 1H), 7.97 (s, 1H), 7.83 (s, 1H), 5.51 (s, 2H), 4.16 (s, 3H), 3.67–3.69 (t, J = 5.0 Hz, 2H), 3.57–3.59 (t, J = 4.4 Hz, 2H); 13C NMR (126 MHz, CD3OD) δ 164.99, 154.93, 151.84, 138.32, 132.41, 120.82, 115.24, 76.54, 70.09, 60.51, 53.41; Elemental analysis calcd for C11H14N4O3: C, 52.79; H, 5.64; N, 22.39. Found: C, 52.62, H, 5.62, N, 22.17.
2-((4-(4-Methoxypyrimidin-5-yl)-1H-imidazol-1-yl)methoxy)ethyl acetate (3-Ac). The sugar 7 (0.35 g, 1.13 mmol) and stannyl pyrimidine 13 (0.54 g, 1.35 mmol) were used. The product was purified three times by flash column chromatography on silica (0–20% CH3OH in CH2Cl2) to give the product as a pale yellow-white solid (0.093 g, 30%); 1H NMR (500 MHz, CDCl3) δ 9.05 (s, 1H), 8.65 (s, 1H), 7.65 (s, 1H), 7.60 (s, 1H), 5.33 (s, 2H), 4.13–4.15 (t, J = 4.8 Hz, 2H), 4.06 (s, 3H), 3.58–3.60 (t, J = 4.6 Hz, 2H), 1.98 (s, 3H); 13C NMR (126 MHz, CDCl3) δ 170.89, 164.37, 155.43, 153.56, 137.72, 134.37, 121.32, 115.17, 76.55, 66.90, 62.88, 54.44, 21.43; Elemental analysis calcd for C13H16N4O4: C, 53.42; H, 5.52; N, 19.17. Found: C, 53.14, H, 5.56, N, 18.88.
2-((4-(4-Methoxypyrimidin-5-yl)-1H-imidazol-1-yl)methoxy)ethyl propionate (3-EtAc). The sugar 8 (0.35 g, 1.08 mmol) and stannyl pyrimidine 13 (0.52 g, 1.30 mmol) were used. The product was purified three times by flash column chromatography on silica (0–10% CH3OH in CH2Cl2) to give the product as a pale yellow oil (0.15 g, 45%); 1H NMR (400 MHz, CDCl3) δ 9.19 (s, 1H), 8.60 (s, 1H), 7.63 (s, 1H), 7.57 (s, 1H), 5.31 (s, 2H), 4.13–4.14 (t, J = 5.0 Hz, 2H), 4.05 (s, 3H), 3.57–3.59 (t, J = 4.6 Hz, 2H), 2.22–2.26 (m, 2H), 1.01–1.05 (t, J = 7.8 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 174.27, 164.74, 155.58, 153.57, 137.50, 134.17, 119.98, 115.11, 76.62, 66.79, 62.66, 54.05, 27.38, 9.03; Elemental analysis calcd for C14H18N4O4 + 0.1% CH3OH + 0.5% H2O: C, 53.17; H, 6.14; N, 17.59. Found: C, 52.99, H, 5.95, N, 17.40.
2-((4-(4-Methoxypyrimidin-5-yl)-1H-imidazol-1-yl)methoxy)ethyl isobutyrate (3-iPrAc). The sugar 9 (0.35 g, 1.04 mmol) and stannyl pyrimidine 13 (0.50 g, 1.24 mmol) were used. The product was purified three times by flash column chromatography on silica (first purification 40–100% EtOAc in hexanes, subsequent purifications 0–5% CH3OH in CH2Cl2) to give the product as a pale yellow oil (0.12 g, 37%); 1H NMR (400 MHz, CDCl3) δ 9.24 (s, 1H), 8.63 (s, 1H), 7.63 (s, 1H), 7.57 (s, 1H), 5.31 (s, 2H), 4.12–4.14 (t, J = 9.6 Hz, 2H), 4.04 (s, 3H), 3.57–3.58 (t, J = 5.0 Hz, 2H), 2.44–2.47 (m, 1H), 1.04–1.06 (d, J = 6.9 Hz, 6H); 13C NMR (126 MHz, CDCl3) δ 177.42, 165.12, 155.43, 153.66, 137.54, 134.37, 120.02, 115.26, 76.59, 66.34, 62.43, 54.04, 34.19, 19.99; Elemental analysis calcd for C15H20N4O4 + 0.3% H2O: C, 55.31; H, 6.37; N, 17.20. Found: C, 55.29, H, 6.33, N, 17.09.
2-((4-(4-Methoxypyrimidin-5-yl)-1H-imidazol-1-yl)methoxy)ethyl pivalate (3-tBuAc). The sugar 10 (0.38 g, 1.06 mmol) and stannyl pyrimidine 13 (0.51 g, 1.28 mmol) were used. The product was purified three times by flash column chromatography on silica (first purification 25–100% EtOAc in hexanes, subsequent purifications 0–5–10% CH3OH in CH2Cl2) to give the product as a clear oil (0.042 g, 12%); 1H NMR (500 MHz, CD3OD) δ 9.07 (s, 1H), 8.65 (s, 1H), 7.98 (s, 1H), 7.86 (s, 1H), 5.50 (s, 2H), 4.19–4.21 (m, 2H), 4.17 (s, 3H), 3.71–3.73 (m, 2H), 1.15 (s, 9H); 13C NMR (126 MHz, CD3OD) δ 178.41, 165.02, 155.01, 151.91, 138.36, 132.60, 120.77, 115.23, 76.21, 66.61, 62.73, 53.43, 38.30, 26.04; Elemental analysis calcd for C16H22N4O4 + 0.35 CH3OH + 0.1 H2O: C, 56.53; H, 6.85; N, 16.13; Found: C, 56.52; H, 6.83; N, 16.09.
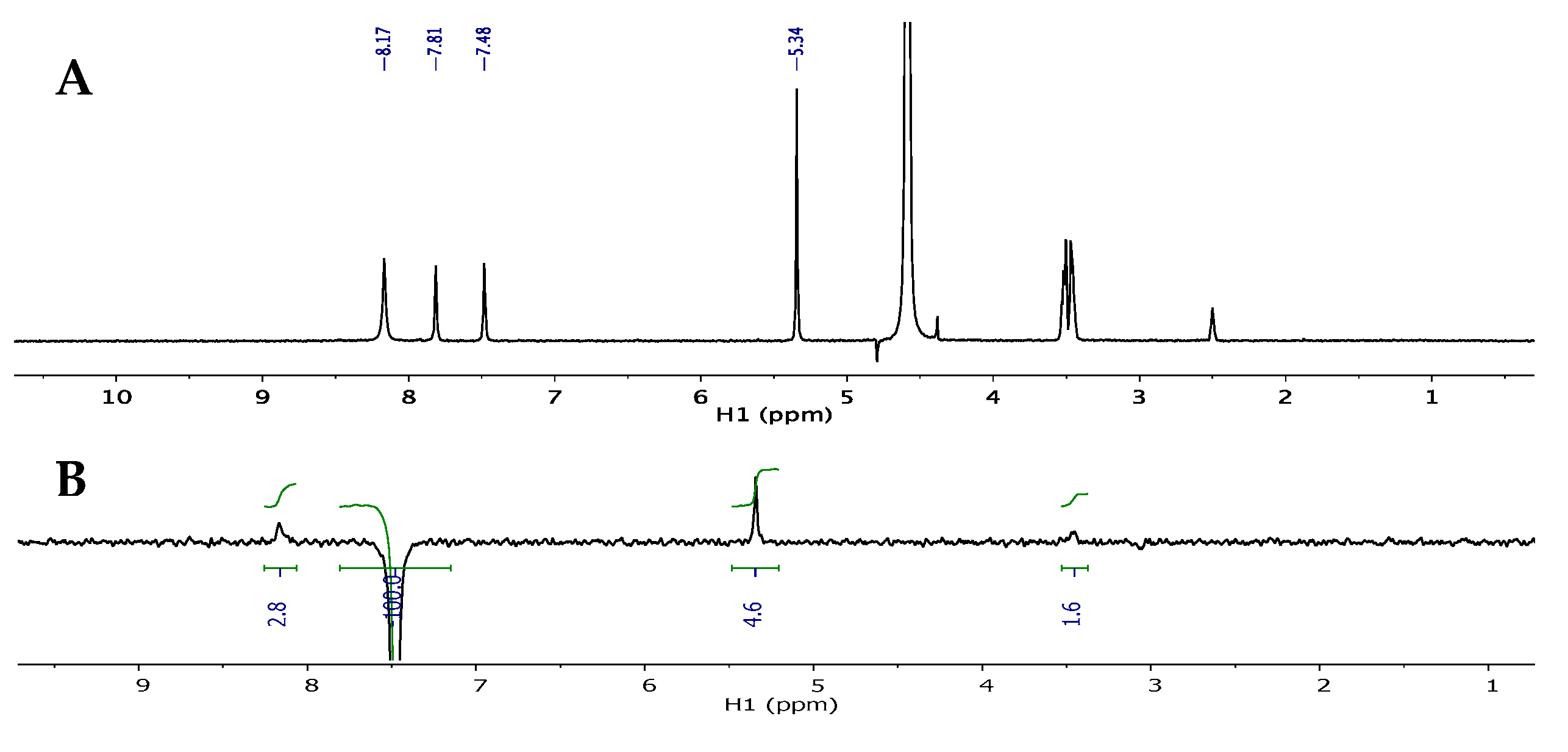
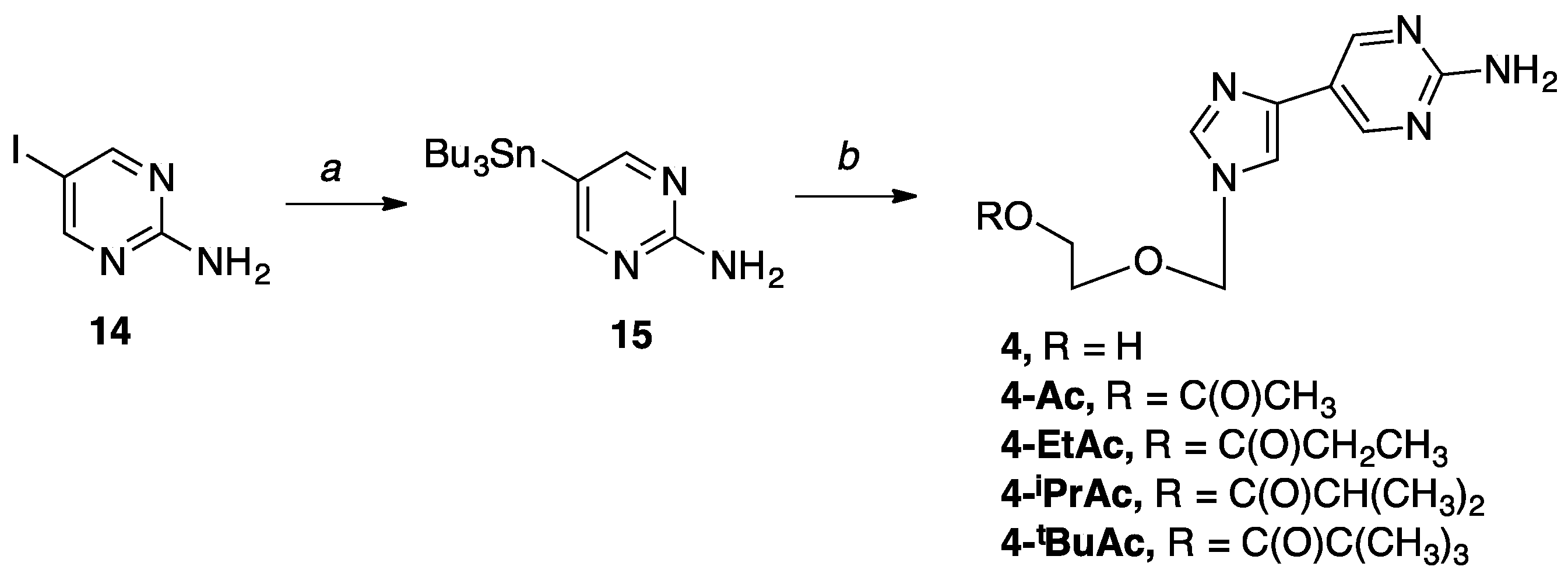
5-(Tributylstannyl)pyrimidin-2-amine (15). To a solution of 2-amino-5-iodopyrimidine (0.50 g, 2.26 mmol) in anh. DMF (100 mL) under direct nitrogen bubbling was added bis(tributyltin) (1.37 mL, 2.71 mmol) and Pd2dba3•CHCl3 (0.23 g, 0.23 mmol). Then, the reaction was heated to 65 °C with stirring for 3 h under direct nitrogen bubbling. After completion, the solvent was removed in vacuo to give a black thick oil. The crude mixture was resuspended in EtOAc and filtered over a celite pad; then, the filtrate was concentrated in vacuo to give a dark liquid. Purification was performed using flash column chromatography on silica (0–100% EtOAc in hexanes) to give the pure product as a yellow oil (0.75 g, 86%); Rf = 0.76 (1:1 EtOAc: Hex); 1H NMR (500 MHz, CDCl3) δ 8.17 (s, 2H), 6.08 (br, 2H), 1.45–1.51 (m, 6H), 1.39–1.44 (m, 6H), 1.21–1.29 (m, 6H), 0.83–1.06 (m, 9H); 13C NMR (126 MHz, CDCl3) δ 164.32, 163.16, 119.22, 28.84, 27.44, 13.57, 8.09; MS (ESI+) m/z calcd for C16H31N3Sn [M + H]+ 386.15, found: 386.19.
2-((4-(2-Aminopyrimidin-5-yl)-1H-imidazol-1-yl)methoxy)ethan-1-ol (4). The sugar 6 (0.44 g, 1.63 mmol) and stannyl pyrimidine 15 (0.75 g, 1.95 mmol) were used. The product was purified four times by flash column chromatography on silica (0–30% CH3OH in CH2Cl2) to give the product as a pale pink solid (0.19 g, 50%); 1H NMR (500 MHz, CD3OD) δ 8.64 (s, 2H), 7.91 (s, 1H), 7.62 (s, 1H), 5.48 (s, 2H), 3.67–3.69 (t, J = 4.9 Hz, 2H), 3.56–3.58 (t, J = 4.6 Hz, 2H); 13C NMR (126 MHz, CD3OD) δ 163.2, 154.8, 138.5, 137.0, 117.6, 114.4, 77.4, 70.9, 60.2; Elemental analysis calcd for C10H13N5O2: C, 51.06; H, 5.57; N, 29.77. Found: C, 51.01, H, 5.47, N, 29.83.
2-((4-(2-Aminopyrimidin-5-yl)-1H-imidazol-1-yl)methoxy)ethyl acetate (4-Ac). The sugar 7 (0.48 g, 1.54 mmol) and stannyl pyrimidine 15 (0.71 g, 1.86 mmol) were used. The product was purified three times by flash column chromatography on silica (0–30% CH3OH in CH2Cl2) to give the product as a white solid (0.10 g, 24%); 1H NMR (400 MHz, CD3OD) δ 8.71 (s, 2H), 7.70 (s, 1H), 7.29 (s, 1H), 5.38 (s, 2H), 4.22–4.24 (t, J = 4.6 Hz, 2H), 3.66–3.68 (t, J = 4.6 Hz, 2H), 2.02 (s, 3H); 13C NMR (126 MHz, CD3OD) δ 171.5, 162.6, 154.6, 138.9, 136.8,117.2, 114.3, 75.6, 67.0, 63.1, 19.2; Elemental analysis calcd for C12H15N5O3 + 0.1 CH3OH + 0.05 CH2Cl2: C, 51.25; H, 5.49; N, 24.60. Found: C, 51.21, H, 5.47, N, 24.54.
2-((4-(2-Aminopyrimidin-5-yl)-1H-imidazol-1-yl)methoxy)ethyl propionate (4-EtAc). The sugar 8 (0.30 g, 0.93 mmol) and stannyl pyrimidine 15 (0.43 g, 1.11 mmol) were used. The product was purified four times by flash column chromatography on silica (0–20% CH3OH in CH2Cl2) to give the product as a white solid (0.09 g, 26%); 1H NMR (400 MHz, CD3OD) δ 8.60 (s, 2H), 7.87 (s, 1H), 7.57 (s, 1H), 5.42 (s, 2H), 4.15–4.17 (t, J = 4.6 Hz, 2H), 3.67–3.69 (t, J = 4.6 Hz, 2H), 2.24–2.28 (m, 2H), 1.02–1.06 (t, J = 7.8 Hz, 3H); 13C NMR (101 MHz, CD3OD) δ 174.58, 162.29, 154.81, 138.47, 136.81, 117.60, 114.45, 76.38, 66.75, 62.79, 26.78, 8.03; Elemental analysis calcd for C13H17N5O3 + 0.05 CH3OH + 0.95 H2O: C, 50.56 H, 6.21; N, 22.59. Found: C, 50.45, H, 6.07, N, 22.49.
2-((4-(2-Aminopyrimidin-5-yl)-1H-imidazol-1-yl)methoxy)ethyl isobutyrate (4-iPrAc). The sugar 9 (0.45 g, 1.33 mmol) and stannyl pyrimidine 15 (0.61 g, 1.60 mmol) were used. The product was purified four times by flash column chromatography on silica (0–20% CH3OH in CH2Cl2) to give the product as a pink solid (0.07 g, 17%); 1H NMR (400 MHz, CD3OD) δ 8.59 (s, 2H), 7.87 (s, 1H), 7.56 (s, 1H), 5.42 (s, 2H), 4.15–4.17 (t, J = 4.6 Hz, 2H), 3.67–3.69 (t, J = 4.6 Hz, 2H), 2.46–2.50 (m, 1H), 1.06–1.08 (d, J = 6.8 Hz, 6H); 13C NMR (101 MHz, CD3OD) δ 177.42, 162.32, 155.05, 138.65, 136.98, 117.69, 114.61, 76.41, 66.72, 62.80, 33.55, 18.45; Elemental analysis calcd for C14H19N5O3: C, 55.07 H, 6.27; N, 22.94. Found: C, 55.02, H, 6.13, N, 22.73.
2-((4-(2-Aminopyrimidin-5-yl)-1H-imidazol-1-yl)methoxy)ethyl pivalate (4-tBuAc). The sugar 10 (0.30 g, 0.85 mmol) and stannyl pyrimidine 15 (0.39 g, 1.02 mmol) were used. The product was purified twice by flash column chromatography on silica (0–20% CH3OH in CH2Cl2) to give the product as a white solid (0.06 g, 20%); 1H NMR (400 MHz, CDCl3) δ 8.63 (s, 2H), 7.63 (s, 1H), 7.22 (s, 1H), 5.48 (br, 2H), 5.30 (s, 2H), 4.15–4.16 (t, J = 3.7 Hz, 2H), 3.58–3.60 (t, J = 3.7 Hz, 2H), 1.09–1.11 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 178.40, 162.17, 155.12, 138.17, 137.85, 118.49, 113.33, 76.58, 66.82, 62.64, 38.78, 27.17; Elemental analysis calcd for C15H21N5O3: C, 56.41; H, 6.63; N, 21.93. Found: C, 56.11, H, 6.47, N, 21.65.
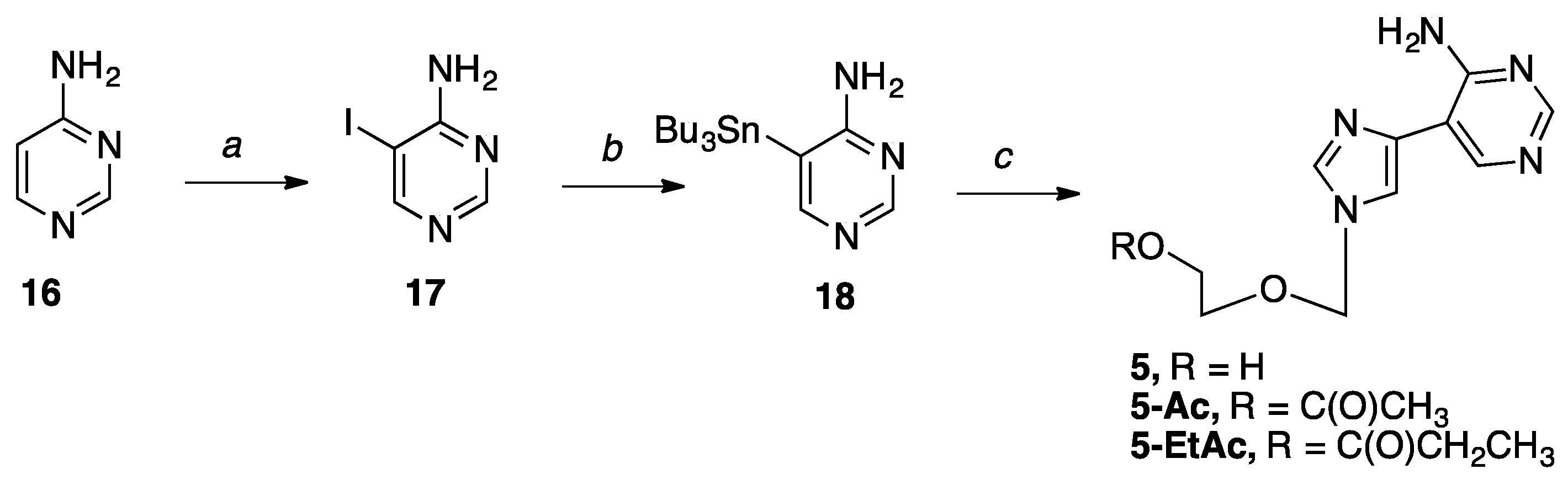
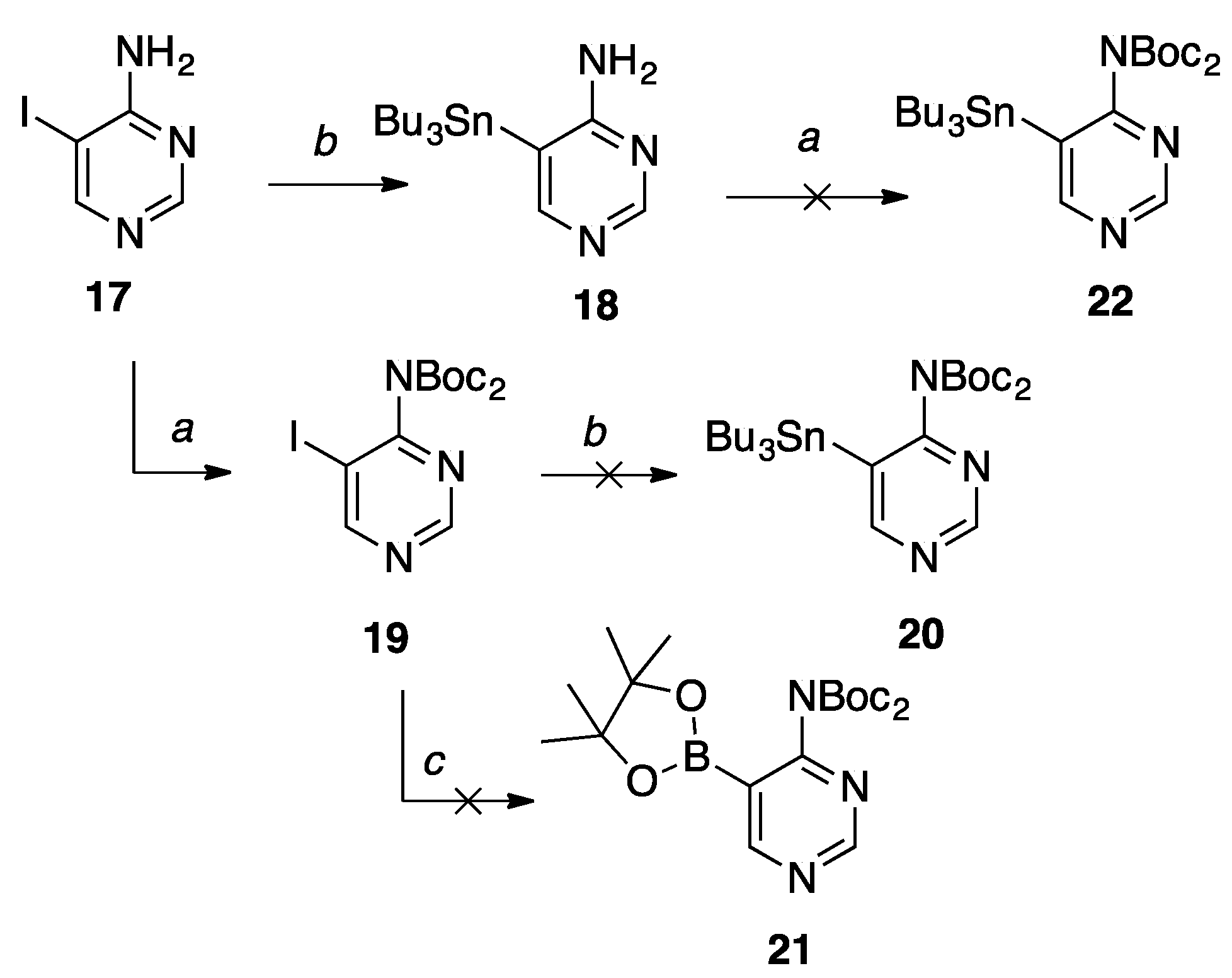
5-Iodopyrimidin-4-amine (17).14 To a solution of 4-aminopyrimidine (0.80 g, 8.5 mmol) in glacial AcOH (20 mL) under nitrogen was added N-iodosuccinimide (2.30 g, 10.20 mmol) with stirring. The reaction refluxed at 80 °C for two hours. Upon completion, the reaction was cooled to room temperature, and the solvent was removed in vacuo. The resulting yellow solid was resuspended in EtOAc, quenched with NaHCO3 (50 mL), washed with Na2S2O3 (50 mL), and the organic layer was dried with MgSO4 and gravity filtered to give a yellow solid. Then, the crude product was purified by flash column chromatography on silica (0–10% CH3OH in CH2Cl2) to give the pure product as a pale yellow solid (1.5 g, 80%); Rf = 0.48 (1:1 EtOAc: Hex); Spectral data is in agreement with literature values.14
5-(tributylstannyl)pyrimidin-4-amine (18).14 To a solution of 17 (0.75 g, 3.39 mmol) in anh. DMF (50 mL) under direct bubbling nitrogen was added bis(tributyl)tin (2.05 mL, 4.07 mmol) and Pd2dba3•CHCl3 (0.35 g, 0.34 mmol) with stirring. Then, the reaction was heated to 65 °C for 2.5 h. Upon completion, the reaction was cooled to room temperature and the solvent was removed in vacuo. Then, the resulting oil was resuspended in EtOAc, filtered over a celite pad, and the filtrate was concentrated in vacuo to give a yellow oil. Then, the crude product was purified by flash column chromatography on silica (10 -60% EtOAc in hexanes) to give the pure product as a pale yellow solid (0.80 g, 61%); Rf = 0.65 (1:1 EtOAc: Hex); Spectral data is in agreement with literature values.14
2-((4-(4-Aminopyrimidin-5-yl)-1H-imidazol-1-yl)methoxy)ethan-1-ol (5). The sugar 6 (0.25 g, 0.93 mmol) and stannyl pyrimidine 18 (0.43 g, 1.12 mmol) were used. The product was purified five times by flash column chromatography on silica (0–15% CH3OH in CH2Cl2) to give the product as a pale pink solid (0.03 g, 11%); 1H NMR (500 MHz, CD3OD) δ 8.44 (s, 1H), 8.29 (s, 1H), 7.93 (s, 1H), 7.78 (s, 1H), 5.50 (s, 2H), 3.67–3.69 (m, 2H), 3.56–3.58 (m, 2H); 13C NMR (126 MHz, CD3OD) δ 160.25, 155.37, 150.03, 137.43, 136.56, 116.33, 110.63, 76.53, 70.11, 60.50. Elemental analysis calcd for C10H13N5O2 + 0.05 CH2Cl2: C, 50.40; H, 5.41; N, 29.24; Found: C, 50.55; H, 5.47; N, 29.10.
2-((4-(4-Aminopyrimidin-5-yl)-1H-imidazol-1-yl)methoxy)ethyl acetate (5-Ac). The sugar 7 (0.27 g, 0.87 mmol) and stannyl pyrimidine 18 (0.40 g, 1.04 mmol) were used. The product was purified five times by flash column chromatography on silica (0–15% CH3OH in CH2Cl2) to give the product as a pink solid (0.03 g, 11%); 1H NMR (500 MHz, DMSO-d6) δ 8.52 (s, 1H), 8.26 (s, 1H), 8.00 (s, 1H), 7.91 (s, 1H), 5.45 (s, 2H), 4.10–4.12 (t, J = 4.7 Hz, 2H), 3.65–3.67 (t, J = 4.7 Hz, 2H), 2.01 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 171.13, 159.92, 156.76, 151.23, 138.36, 133.76, 114.90, 109.90, 76.58, 66.57, 62.71, 20.42; Elemental analysis calcd for C12H15N5O3 + 0.55 CH2Cl2: C, 46.52; H, 5.01; N, 21.62; Found: C, 46.42; H, 4.87; N, 21.60.
2-((4-(4-Aminopyrimidin-5-yl)-1H-imidazol-1-yl)methoxy)ethyl propionate (5-EtAc). The sugar 8 (0.27 g, 0.84 mmol) and stannyl pyrimidine 18 (0.39 g, 1.01 mmol) were used. The product was purified four times by flash column chromatography on silica (0–10% CH3OH in CH2Cl2) to give the product as a clear oil (0.015 g, 6.1%); 1H NMR (500 MHz, CD3OD) δ 8.45 (s, 1H), 8.29 (s,1H), 7.93 (s, 1H), 7.79 (s, 1H), 5.49 (s, 2H), 4.19–4.21 (m, 2H), 3.72–3.74 (m, 2H), 2.29–2.34 (q, J = 7.6 Hz, 2H), 1.06–1.09 (t, J = 7.6 Hz, 3H); 13C NMR (126 MHz, CD3OD) δ 174.53, 160.25, 155.41, 150.04, 137.43, 136.69, 116.28, 110.56, 76.31, 66.73, 62.71, 26.70, 7.93; Elemental analysis cald for C13H17N5O3 + 0.1 CH3OH + 0.4 H2O: C, 52.12; H, 6.08; N, 23.21; Found: C, 52.26; H, 5.82; N, 23.03.
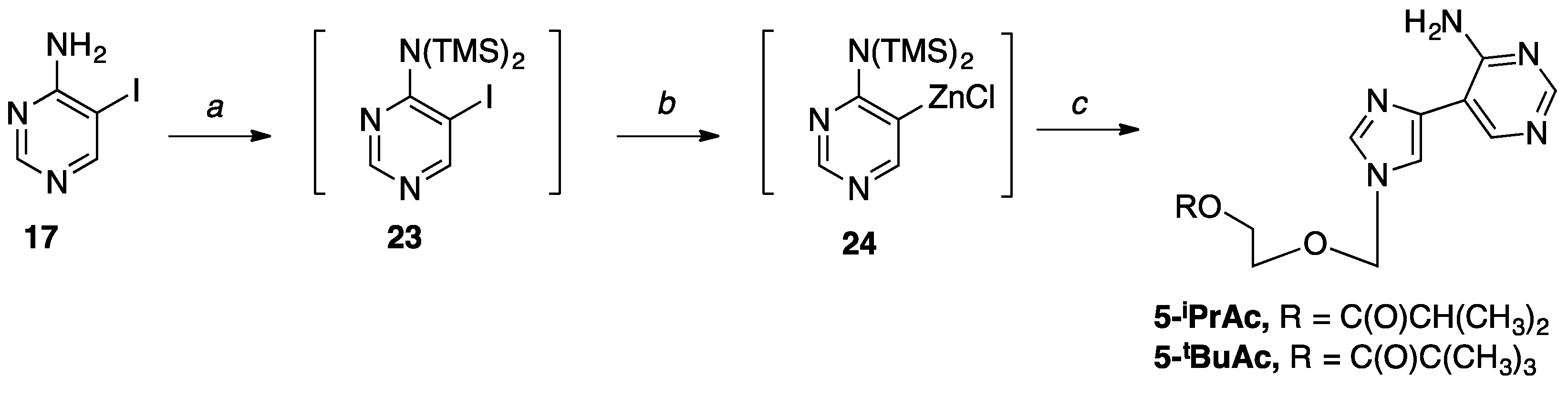
2-((4-(4-Aminopyrimidin-5-yl)-1H-imidazol-1-yl)methoxy)ethyl isobutyrate (5-iPrAc). A solution of 17 (0.2 g, 0.91 mmol) in anh. THF (20 mL) was cooled to −78 °C in an acetone/CO2(s) bath under nitrogen. EtMgBr (0.33 mL, 1.0 mmol) was added dropwise and the reaction was stirred for two minutes, followed by the addition of TMSCl (0.14 mL, 1.09 mmol), after which the reaction was stirred for 5 min. Another addition of EtMgBr (0.33 mL, 1.0 mmol) was added dropwise, and the reaction was stirred for 2 min; then, another addition of TMSCl (0.14 mL, 1.09 mmol) was added, and the reaction was stirred for 5 min. A final addition of EtMgBr (0.33 mL, 1.0 mmol) was added, and the reaction stirred for 10 min; then, ZnCl2 (1.81 mL, 1.81 mmol) was added dropwise. The solution was stirred at −78 °C for 10 min, and then warmed to room temperature and stirred for 2 h. The organozinc mixture was then added dropwise to a solution of 9 (0.34 g, 1.0 mmol), Pd(PPh3)4 (0.11 g, 0.09 mmol), and CuI (0.01 g, 0.05 mmol) in anh. THF (10 mL). Then, the reaction stirred under nitrogen for 18 hours at room temperature. Upon completion, the reaction was quenched with saturated ethylenediaminetetraacetic acid (EDTA) (10 mL), after which it was concentrated in vacuo, extracted three times with CH2Cl2, washed with brine, and the organics were dried with MgSO4. After gravity filtration, the solvent was removed in vacuo to give a dark oil. Then, the crude mixture was purified twice via flash column chromatography on silica (0–10% CH3OH (10% NH4OH) in DCM) to give the product as a pink oil (0.16 g, 48%); 1H NMR (500 MHz, CD3OD) δ 8.45 (s, 1H), 8.29 (s, 1H), 7.94 (s, 1H), 7.80 (s, 1H), 5.50 (s, 2H), 4.20–4.21 (m, 2H), 3.72–3.74 (m, 2H), 2.52–2.53 (m, 1H), 1.11–1.13 (d, J = 7.0 Hz, 6H); 13C NMR (126 MHz, (CD3)3CO) δ 176.26, 160.13, 156.41, 151.08, 137.78, 137.24, 115.69, 109.97, 76.43, 66.84, 62.68, 33.54, 18.42; Elemental analysis calcd for C14H19N5O3 + 0.75 H2O: C, 52.74; H, 6.48; N, 21.96; Found: C, 52.55; H, 6.21; N, 21.82.
2-((4-(4-Aminopyrimidin-5-yl)-1H-imidazol-1-yl)methoxy)ethyl pivalate (5-tBuAc). A solution of 17 (0.2 g, 0.90 mmol) in anh. THF (15 mL) was cooled to −78 °C in an acetone/CO2(s) bath under nitrogen. EtMgBr (0.33 mL, 1.0 mmol) was added dropwise, and the reaction was stirred for two minutes, followed by the addition of TMSCl (0.14 mL, 1.09 mmol) and the reaction was stirred for another 5 min. Another addition of EtMgBr (0.33 mL, 1.0 mmol) was added dropwise, after which the reaction stirred for 2 min; then, another addition of TMSCl (0.14 mL, 1.09 mmol) was added, and the reaction was stirred for 5 min. A final addition of EtMgBr (0.33 mL, 1.0 mmol) was added, and the reaction stirred for 10 min; then, ZnCl2 (1.81 mL, 1.81 mmol) was added dropwise. The solution was stirred at −78 °C for 10 min, and then warmed to room temperature and stirred for 2 h. Then, the organozinc mixture was added dropwise to a solution of 10 (0.35 g, 1.0 mmol), Pd(PPh3)4 (0.01 g, 0.09 mmol), and CuI (0.01 g, 0.04 mmol) in anh. THF (10 mL). Then, the reaction was stirred under nitrogen for 18 hours at room temperature. Upon completion, the reaction was quenched with saturated EDTA (10 mL), after which it was concentrated in vacuo, extracted three times with CH2Cl2, washed with brine, and the organics were dried with MgSO4. After gravity filtration, the solvent was removed in vacuo to give a dark oil. Then, the crude mixture was purified twice via flash column chromatography on silica (0–10% CH3OH (10% NH4OH) in DCM) to give the product as a light pink oil (0.03, 9.4%); 1H NMR (500 MHz, CD3OD) δ 8.54 (s, 1H), 8.30 (s, 1H), 7.98 (s, 1H), 7.87 (s, 1H), 5.58 (s, 2H), 4.19–4.21 (m, 2H), 3.72–3.78 (m, 2H), 1.16 (s, 9H); 13C NMR (126 MHz, CDCl3) δ 179.09, 159.91, 156.79, 151.26, 138.42, 136.21, 114.81, 109.90, 76.66, 66.93, 62.52, 38.74, 27.71; Elemental analysis calcd for C14H19N5O3 + 0.05 CH3OH + 0.8 H2O: C, 54.01; H, 6.53; N, 20.50.


 ,
, 














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}