
New Artificial Biomimetic Enzyme Analogues Based on Iron(II/III) Schiff Base Complexes: An Effect of (Benz)imidazole Organic Moieties on Phenoxazinone Synthase and DNA Recognition ‡

 , ,
, ,  , ,
, ,  , and
, and
Abstract
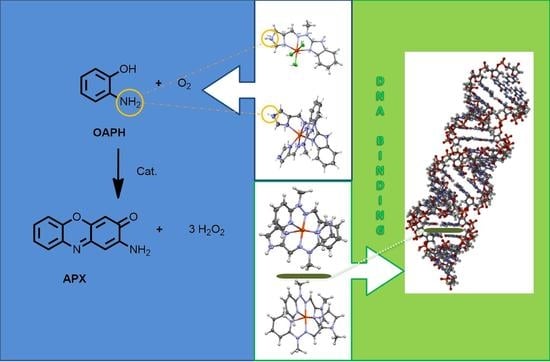
1. Introduction
2. Results and Discussion
2.1. Synthesis and Characterization
2.2. Description of Crystal Structures
2.3. Catalysis and DNA Binding Affinity
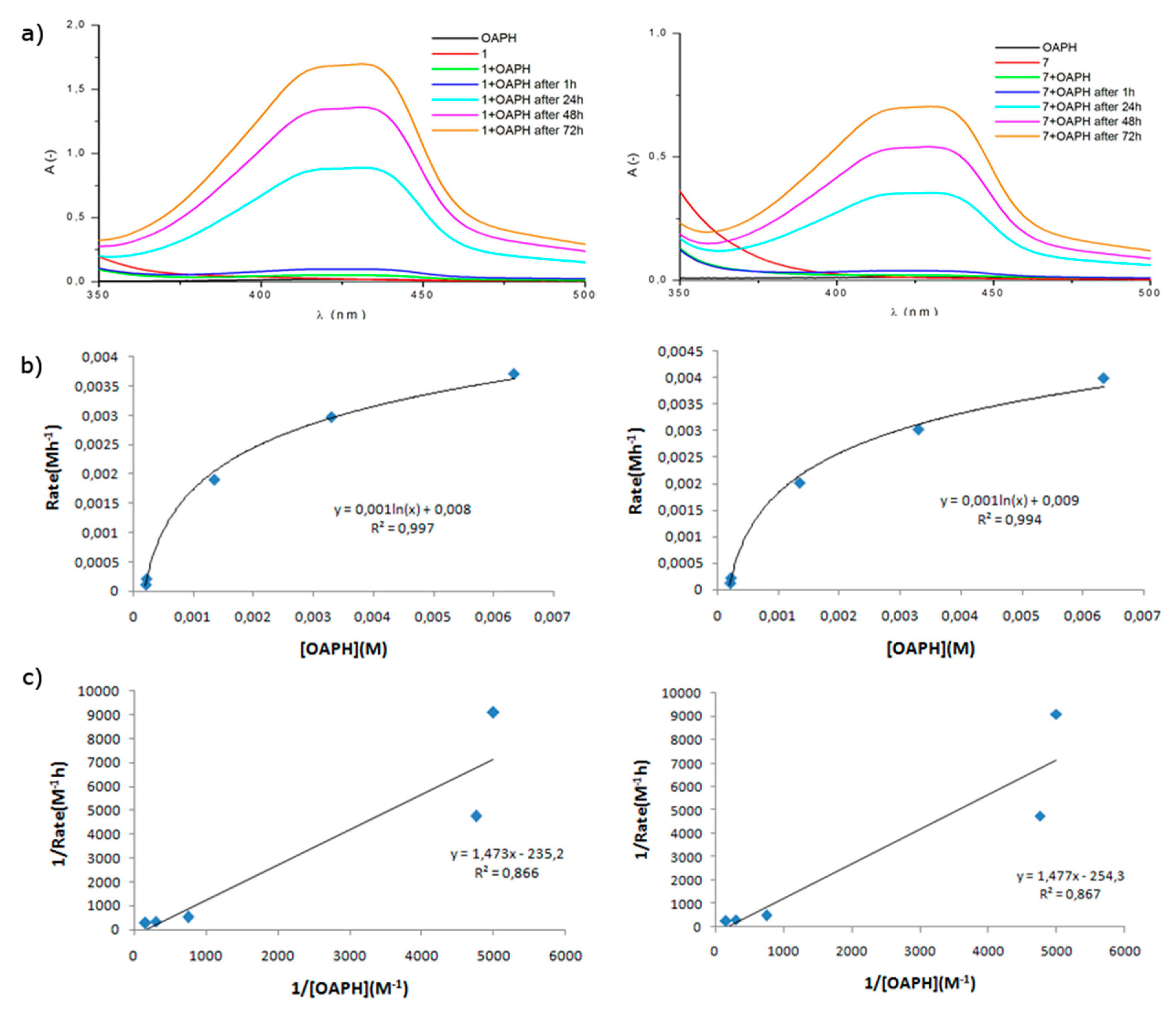
2.3.1. Phenoxazinone Synthase (PHS) Activity
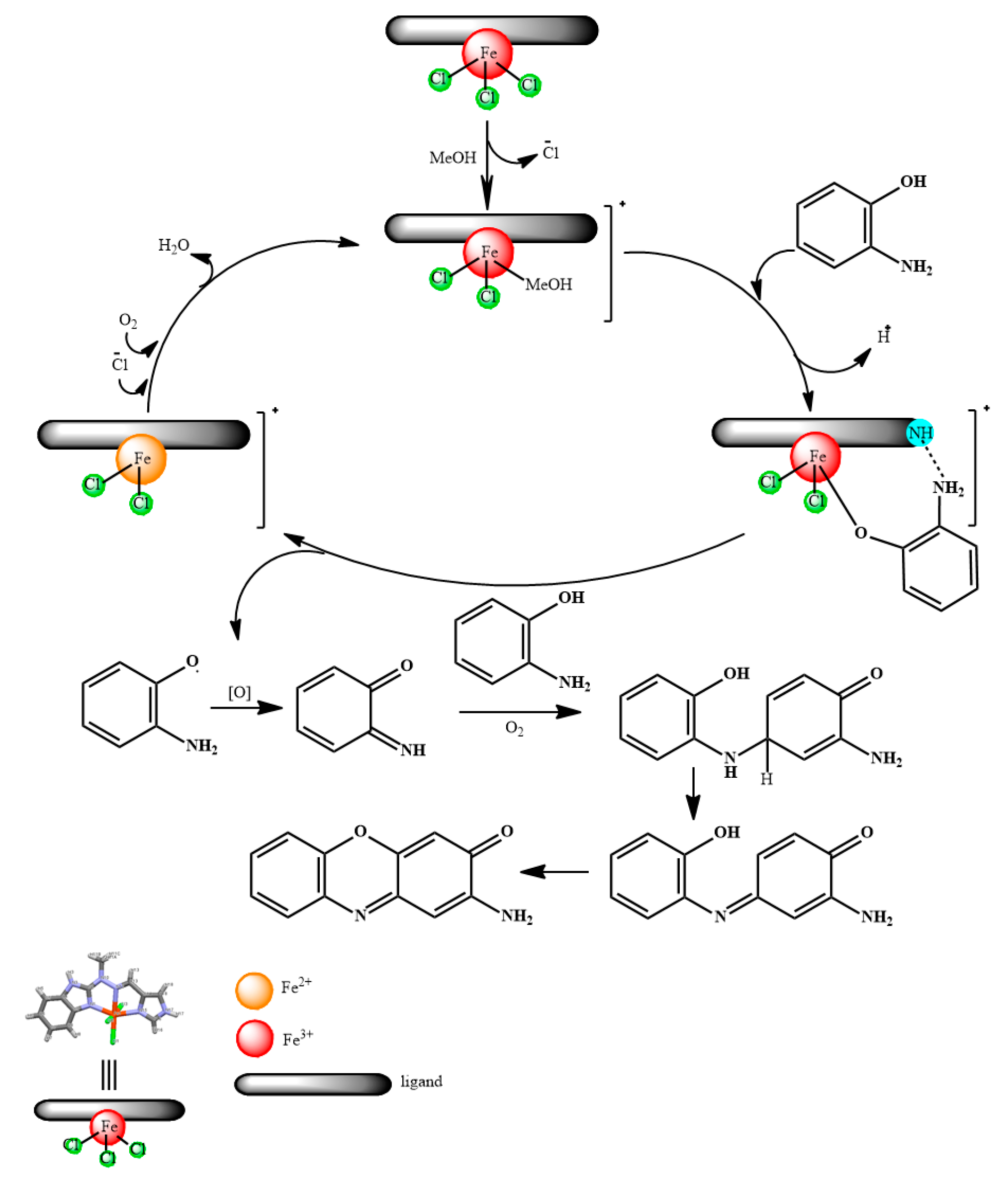
2.3.2. Proposed Mechanism of OAPH Oxidation
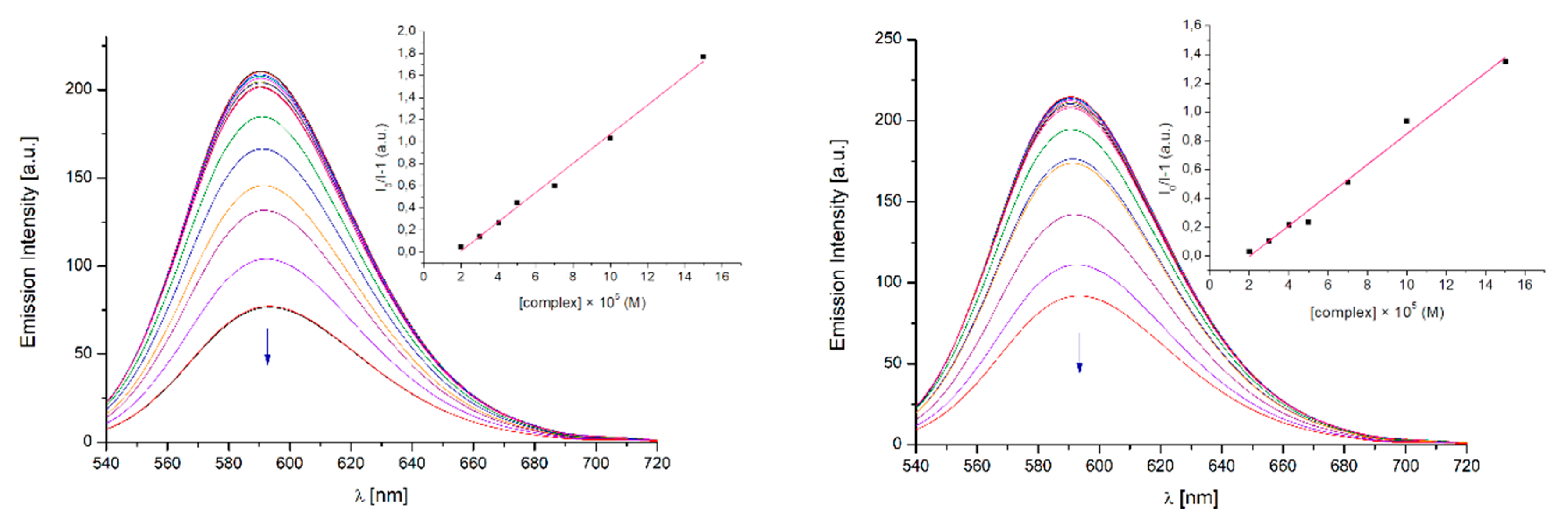
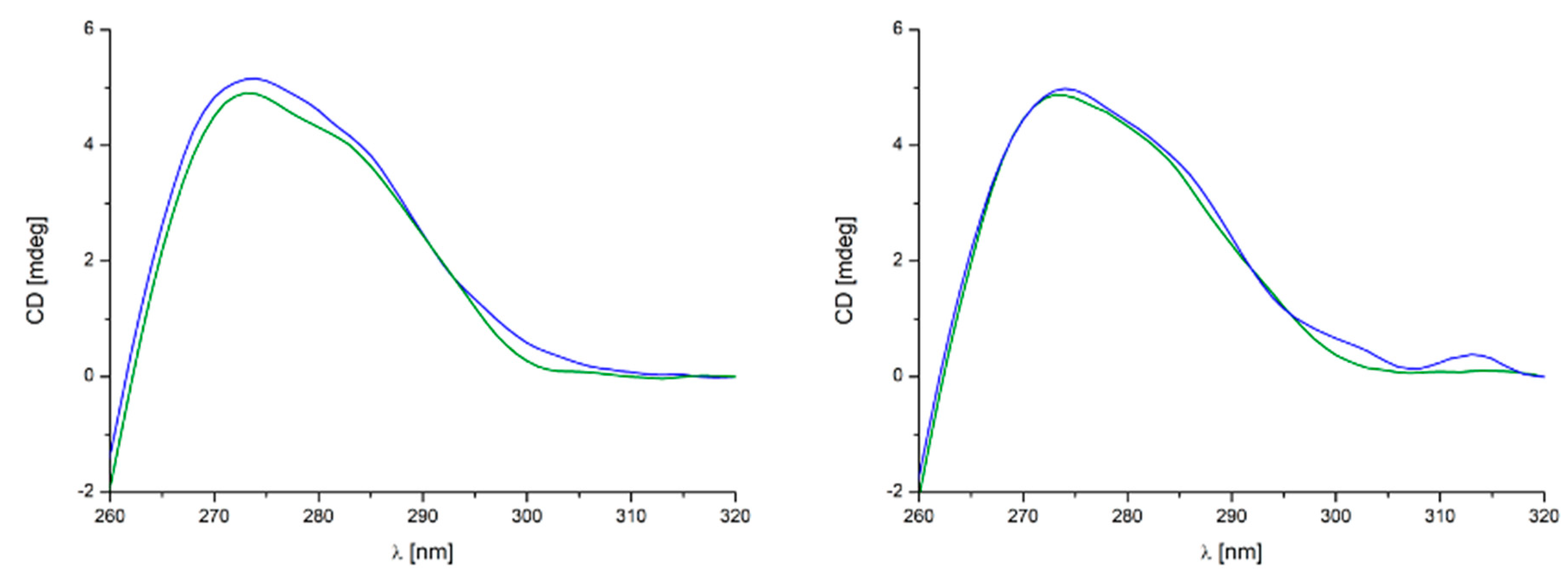
2.3.3. DNA Binding Affinity
Absorption Titration
Competitive Binding Fluorescence Experiment
DNA Binding Investigation via CD
3. Experimental Section
3.1. Materials and Methods
3.2. Synthesis of Ligands
3.2.1. Synthesis of Ligands L1–L3
- -
- for L1: condensation of 4-imidazolecarboxyaldehyde with 2-(1-methyl-hydrazine)-benzimidazole was performed. At two-necked round-bottomed flask 2-(1- methylhydrazine)benzimidazole (1.00 g, 6.16 mmol) was placed under argon atmosphere. The 4-imidazolecarboxyaldehyde (0.591 g, 6.16 mmol) was dissolved in a anhydrous ethanol and was added to the reaction mixture. Yellow suspension was formed and the reaction mixture was stirred for 24 h at 60 °C. Yellow clear solution was cooled to room temperature and the precipitate appeared, which was filtered by vacuum filtration, washed with anhydrous ethanol and dried under vacuum. Yield 80% (1.184 g, 4.9 mmol.)
- -
- for L2: condensation of 2-imidazolecarboxyaldehyde with 2-(1-methyl-hydrazine)-benzimidazole was performed. At two-necked round-bottomed flask 2-(1- methylhydrazine)benzimidazole (1.00 g, 6.16 mmol)was weighed and placed under argon atmosphere. 2-Imidazolecarboxyaldehyde (0.591 g, 6.16 mmol) was dissolved in anhydrous ethanol and was added to the reaction mixture. Yellow suspension was formed, and the reaction mixture was stirred for 24 h at 60 °C. Yellow clear solution was cooled to room temperature and evaporated to dryness. The resulting solid was dissolved in methanol and boiling acetonitrile was added to the flask. Cooling of the reaction mixture resulted in formation of pale pink crystalline product. The precipitate was filtered by vacuum filtration, washed with anhydrous ethanol and dried under vacuum to give 1.24 g (5.16 mmol) of ligand. The supernatant was concentrated to minimal amount of volume and the next part of precipitate was obtained 0.140 g (0.58 mmol). Total yield is 91% (1.38 g, 5.74 mmol).
- -
- for L3: condensation of 1-methyl-2-imidazolecarboxyaldehyde with 2-(1-methyl-hydrazine)-benzimidazole was performed. At two-necked round-bottomed flask 2-(1- methylhydrazine)benzimidazole (1.00 g, 6.16 mmol)was weighed and placed under argon atmosphere. The 1-methyl-2-imidazolecarboxyaldehyde (0.704 g, 6.16 mmol) was dissolved in anhydrous ethanol and was added to the reaction mixture. Yellow suspension was formed and the reaction mixture was stirred for 24 h at 60 °C. Clear yellow solution was cooled to room temperature and kept in fridge. Yellow crystals were filtered by vacuum filtration, washed with anhydrous ethanol and dried under vacuum. Yield is 80% (1.26 g, 4.96 mmol).
3.2.2. Synthesis of Ligands L4–L6
- -
- for L4: condensation of 4-imidazolecarboxyaldehyde with 2-(1-methylhydrazine)pyridine was performed. At two-necked round-bottomed flask the 2-(1-methylhydrazine)pyridine (0.510g, 4.15 mmol) was weighed and placed under argon atmosphere. 4-imidazolecarboxyaldehyde (0.399 g, 4.15 mmol) was dissolved in anhydrous ethanol and was added to the reaction mixture. Yellow solution formed immediately and the reaction mixture was stirred for 24 h at 60 °C. The clear and orange solution was concentrated to minimal amount of volume, then Et2O was added and it was left in the refrigerator. The subsequent pale pink precipitate was filtered by vacuum filtration, washed with anhydrous ethanol and dried under vacuum to give 0.607 g (3.01 mmol) of ligand. 10 mL of Et2O was added to the supernatant and next part of precipitate was obtained 0.053 g (0.27 mmol). Total yield is 79.2% (0.660 g, 3.28 mmol).
- -
- for L5: condensation of 2-imidazolecarboxyaldehyde with 2-(1-methylhydrazine)pyridine was performed. At two-necked round-bottomed flask the 2-(1-methylhydrazine)pyridine (0.525 g, 4.26 mmol) was weighed and placed under argon atmosphere. 2-Imidazolecarboxyaldehyde (0.469 g, 4.26 mmol) was dissolved in anhydrous ethanol and was added to the reaction mixture. Yellow solution formed immediately and the reaction mixture was stirred for 24 h at 60 °C. The clear and yellow solution was concentrated to minimal amount of volume and left in the refrigerator. The subsequent yellow precipitate was filtered by vacuum filtration, washed with anhydrous ethanol and dried in the vacuum to give 0.301 g (1.49 mmol). The supernatant was concentrated to minimal amount of volume and left in the refrigerator. The next part of precipitate was obtained 0.052 g (0.26 mmol). Total yield is 41.1% (0.353 g, 1.75 mmol).
- -
- for L6: synthesized according to our previous work [31]
3.3. Synthesis of Complexes
3.3.1. Synthetic Method for ‘Open’ Complexes
3.3.2. Synthesis Method for ‘Closed’ Complexes
3.4. X-ray Crystallography
3.5. Catalytic Oxidation of 2-Aminophenol
3.6. DNA Binding Assays
3.6.1. Absorption Titration
3.6.2. Competitive Binding Fluorescence Experiment
3.6.3. Circular Dichroism Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Berg, J.M.; Tymoczko, J.L.; Gatto, G.J.; Stryer, L. Biochemistry, 8th ed.; W. H. Freeman and Company: New York, NY, USA, 2015. [Google Scholar]
- Nastri, F.; Chino, M.; Maglio, O.; Bhagi-Damodaran, A.; Lu, Y.; Lombardi, A. Design and engineering of artificial oxygen-activating metalloenzymes. Chem. Soc. Rev. 2016, 45, 5020–5054. [Google Scholar] [CrossRef] [PubMed]
- Whitesides, G.M. Reinventing Chemistry. Angew. Chem. Int. Ed. 2015, 54, 3196–3209. [Google Scholar] [CrossRef] [PubMed]
- Schwizer, F.; Okamoto, Y.; Heinisch, T.; Gu, Y.; Pellizzoni, M.M.; Lebrun, V.; Reuter, R.; Köhler, V.; Lewis, J.C.; Ward, T.R. Artificial Metalloenzymes: Reaction Scope and Optimization Strategies. Chem. Rev. 2018, 118, 142–231. [Google Scholar] [CrossRef] [PubMed]
- Abbaspour, N.; Hurrell, R.; Kelishadi, R. Review on iron and its importance for human health. J. Res. Med. Sci. 2014, 19, 174. [Google Scholar]
- Hohenberger, J.; Ray, K.; Meyer, K. The biology and chemistry of high-valent iron–oxo and iron–nitrido complexes. Nat. Commun 2012, 3, 720. [Google Scholar] [CrossRef] [PubMed]
- Poulos, T.L. Heme Enzyme Structure and Function. Chem. Rev. 2014, 114, 3919–3962. [Google Scholar] [CrossRef] [PubMed]
- Bruijnincx, P.C.A.; van Koten, G.; Klein Gebbink, R.J.M. Mononuclear non-heme iron enzymes with the 2-His-1-carboxylate facial triad: Recent developments in enzymology and modeling studies. Chem. Soc. Rev. 2008, 37, 2716–2744. [Google Scholar] [CrossRef] [PubMed]
- Krainer, F.; Glieder, A. An updated view on horseradish peroxidases: Recombinant production and biotechnological applications. Appl. Microb. Biotechnol. 2015, 99, 1611–1625. [Google Scholar] [CrossRef]
- Denisov, I.G.; Makris, T.M.; Sligar, S.G.; Schlichting, I. Structure and Chemistry of Cytochrome P450. Chem. Rev. 2005, 105, 2253–2278. [Google Scholar] [CrossRef]
- Solomon, E.I.; Brunold, T.C.; Davis, M.I.; Kemsley, J.N.; Lee, S.-K.; Lehnert, N.; Neese, F.; Skulan, A.J.; Yang, Y.-S.; Zhou, J. Geometric and Electronic Structure/Function Correlations in Non-Heme Iron Enzymes. Chem. Rev. 2000, 100, 235–350. [Google Scholar] [CrossRef]
- Tinberg, C.E.; Lippard, S.J. Dioxygen Activation in Soluble Methane Monooxygenase. Acc. Chem. Res. 2011, 44, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Hegg, E.L.; Que, L., Jr. The 2-His-1-Carboxylate Facial Triad—An Emerging Structural Motif in Mononuclear Non-Heme Iron(II) Enzymes. Eur. J. Biochem. 1997, 250, 625–629. [Google Scholar] [CrossRef] [PubMed]
- Solomon, E.I.; Goudarzi, S.; Sutherlin, K.D. O2 Activation by Non-Heme Iron Enzymes. Biochemistry 2016, 55, 6363–6374. [Google Scholar] [CrossRef] [PubMed]
- Tabassum, S.; Amir, S.; Arjmand, F.; Pettinari, C.; Marchetti, F.; Masciocchi, N.; Lupidi, G.; Pettinari, R. Mixed-ligand Cu(II)–vanillin Schiff base complexes; effect of coligands on their DNA binding, DNA cleavage, SOD mimetic and anticancer activity. Eur. J. Med. Chem. 2013, 60, 216–232. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, G.; Ding, W.-d.; O’Brien, L.; Ellestad, G.A. Circular dichroism studies of calicheamicin-DNA interaction: Evidence for calicheamicin-induced DNA conformational change. Tetrahedron 1994, 50, 1341–1349. [Google Scholar] [CrossRef]
- Abdel-Rahman, L.H.; El-Khatib, R.M.; Nassr, L.A.E.; Abu-Dief, A.M. DNA binding ability mode, spectroscopic studies, hydrophobicity, and in vitro antibacterial evaluation of some new Fe(II) complexes bearing ONO donors amino acid Schiff bases. Arab. J. Chem. 2017, 10, S1835–S1846. [Google Scholar] [CrossRef]
- Joseph, J.; Ayisha Bibin Rani, G. Metal based SOD mimetic therapeutic agents: Synthesis, characterization and biochemical studies of metal complexes. Arab. J. Chem. 2017, 10, S1963–S1972. [Google Scholar] [CrossRef]
- Abdel-Rahman, L.H.; Ismail, N.M.; Ismael, M.; Abu-Dief, A.M.; Ahmed, E.A.-H. Synthesis, characterization, DFT calculations and biological studies of Mn(II), Fe(II), Co(II) and Cd(II) complexes based on a tetradentate ONNO donor Schiff base ligand. J. Mol. Struct. 2017, 1134, 851–862. [Google Scholar] [CrossRef]
- Bakshi, R.; Kumar, R.; Mathur, P. Bis-benzimidazole diamide iron(III) complexes as mimics of phenoxazinone synthase. Catal. Commun. 2012, 17, 140–145. [Google Scholar] [CrossRef]
- El-Khalafy, S.H.; Hassanein, M. Oxidation of 2-aminophenol with molecular oxygen and hydrogen peroxide catalyzed by water soluble metalloporphyrins. J. Mol. Catal. A Chem. 2012, 363–364, 148–152. [Google Scholar] [CrossRef]
- Hassanein, M.; Abdo, M.; Gerges, S.; El-Khalafy, S. Study of the oxidation of 2-aminophenol by molecular oxygen catalyzed by cobalt(II) phthalocyaninetetrasodiumsulfonate in water. J. Mol. Catal. A Chem. 2008, 287, 53–56. [Google Scholar] [CrossRef]
- Panja, A.; Shyamal, M.; Saha, A.; Kanti Mandal, T. Methylene bridge regulated geometrical preferences of ligands in cobalt(III) coordination chemistry and phenoxazinone synthase mimicking activity. Dalton Trans. 2014, 43, 5443–5452. [Google Scholar] [CrossRef] [PubMed]
- Simándi, L.I.; Simándi, T.M.; May, Z.; Besenyei, G. Catalytic activation of dioxygen by oximatocobalt(II) and oximatoiron(II) complexes for catecholase-mimetic oxidations of o-substituted phenols. Coord. Chem. Rev. 2003, 245, 85–93. [Google Scholar] [CrossRef]
- Adhikary, J.; Chakraborty, A.; Dasgupta, S.; Chattopadhyay, S.K.; Kruszynski, R.; Trzesowska-Kruszynska, A.; Stepanović, S.; Gruden-Pavlović, M.; Swart, M.; Das, D. Unique mononuclear MnII complexes of an end-off compartmental Schiff base ligand: Experimental and theoretical studies on their bio-relevant catalytic promiscuity. Dalton Trans. 2016, 45, 12409–12422. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, B.; Maji, M.; Biswas, B. Catalytic aspects of a copper(II) complex: Biological oxidase to oxygenase activity. J. Chem. Sci. 2017, 129, 1–11. [Google Scholar] [CrossRef]
- Horváth, T.; Kaizer, J.; Speier, G. Functional phenoxazinone synthase models: Kinetic studies on the copper-catalyzed oxygenation of 2-aminophenol. J. Mol. Catal. A Chem. 2004, 215, 9–15. [Google Scholar] [CrossRef]
- Szávuly, M.; Csonka, R.; Speier, G.; Barabás, R.; Giorgi, M.; Kaizer, J. Oxidation of 2-aminophenol by iron(III) isoindoline complexes. J. Mol. Catal. A Chem. 2014, 392, 120–126. [Google Scholar] [CrossRef]
- Fik, M.A.; Löffler, M.; Weselski, M.; Kubicki, M.; Korabik, M.J.; Patroniak, V. New Fe(II) complexes with Schiff base ligand: Synthesis, spectral characterization, magnetic studies and thermal stability. Polyhedron 2015, 102, 609–614. [Google Scholar] [CrossRef]
- Gorczyński, A.; Pakulski, D.; Szymańska, M.; Kubicki, M.; Bułat, K.; Łuczak, T.; Patroniak, V. Electrochemical deposition of the new manganese(II) Schiff-base complex on a gold template and its application for dopamine sensing in the presence of interfering biogenic compounds. Talanta 2016, 149, 347–355. [Google Scholar] [CrossRef]
- Gorczyński, A.; Zaranek, M.; Witomska, S.; Bocian, A.; Stefankiewicz, A.R.; Kubicki, M.; Patroniak, V.; Pawluć, P. The cobalt(II) complex of a new tridentate Schiff-base ligand as a catalyst for hydrosilylation of olefins. Catal. Commun. 2016, 78, 71–74. [Google Scholar] [CrossRef]
- Fik, M.A.; Czepa, W.; Kubicki, M.; Patroniak, V. Formation of non-covalent porous framework with Fe ions and N2O Schiff base ligand: structural and thermal studies. Supramol. Chem. 2017, 29, 643–650. [Google Scholar] [CrossRef]
- Czepa, W.; Fik, M.A.; Witomska, S.; Kubicki, M.; Consiglio, G.; Pawluć, P.; Patroniak, V. Simple Schiff-Base Cu(II) Complexes as Efficient Catalysts for Benzyl Alcohol Oxidation. ChemistrySelect 2018, 3, 9504–9509. [Google Scholar] [CrossRef]
- Marcinkowski, D.; Fik, M.A.; Łuczak, T.; Kubicki, M.; Patroniak, V. New Mn(II) complexes with benzoxazole-based ligands: Synthesis, structure and their electrochemical behavior. Polyhedron 2018, 141, 125–132. [Google Scholar] [CrossRef]
- Gorczyński, A.; Marcinkowski, D.; Kubicki, M.; Löffler, M.; Korabik, M.; Karbowiak, M.; Wiśniewski, P.; Rudowicz, C.; Patroniak, V. New field-induced single ion magnets based on prolate Er(iii) and Yb(iii) ions: tuning the energy barrier Ueff by the choice of counterions within an N3-tridentate Schiff-base scaffold. Inorg. Chem. Front. 2018, 5, 605–618. [Google Scholar] [CrossRef]
- Gorczyński, A.; Kubicki, M.; Szymkowiak, K.; Łuczak, T.; Patroniak, V. Utilization of a new gold/Schiff-base iron(iii) complex composite as a highly sensitive voltammetric sensor for determination of epinephrine in the presence of ascorbic acid. RSC Adv. 2016, 6, 101888–101899. [Google Scholar] [CrossRef]
- Wałęsa-Chorab, M.; Gorczyński, A.; Kubicki, M.; Hnatejko, Z.; Patroniak, V. Self-assembly of a tridentate Schiff-base ligand with Zn(II) in the presence of lanthanides: Novel crystal structures and spectroscopic properties. Polyhedron 2012, 31, 51–57. [Google Scholar] [CrossRef]
- Fik, M.A.; Gorczyński, A.; Kubicki, M.; Hnatejko, Z.; Fedoruk-Wyszomirska, A.; Wyszko, E.; Giel-Pietraszuk, M.; Patroniak, V. 6,6″-Dimethyl-2,2′:6′,2″-terpyridine revisited: New fluorescent silver(I) helicates with in vitro antiproliferative activity via selective nucleoli targeting. Eur. J. Med. Chem. 2014, 86, 456–468. [Google Scholar] [CrossRef]
- Adamski, A.; Kruszka, D.; Dutkiewicz, Z.; Kubicki, M.; Gorczyński, A.; Patroniak, V. Novel family of fused tricyclic [1,4]diazepines: Design, synthesis, crystal structures and molecular docking studies. Tetrahedron 2017, 73, 3377–3386. [Google Scholar] [CrossRef]
- Adamski, A.; Fik, M.A.; Kubicki, M.; Hnatejko, Z.; Gurda, D.; Fedoruk-Wyszomirska, A.; Wyszko, E.; Kruszka, D.; Dutkiewicz, Z.; Patroniak, V. Full characterization and cytotoxic activity of new silver(i) and copper(i) helicates with quaterpyridine. New J. Chem. 2016, 40, 7943–7957. [Google Scholar] [CrossRef]
- Chatterjee, S.; Sukul, D.; Banerjee, P.; Adhikary, J. Phenoxazinone synthase activity of two iron(III) complexes comprising the same Schiff base ligand: Biomimetic functional model and mechanistic investigation. Inorg. Chim. Acta 2018, 474, 105–112. [Google Scholar] [CrossRef]
- Liu, X.-F.; Xiang, L.; Zhou, Q.; Carralot, J.-P.; Prunotto, M.; Niederfellner, G.; Pastan, I. Actinomycin D enhances killing of cancer cells by immunotoxin RG7787 through activation of the extrinsic pathway of apoptosis. Proc. Natl. Acad. Sci. USA 2016, 113, 201611481. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.-F.; Wang, Y.-S.; Li, C.; Wei, G.-J.; Chen, R.; Dong, D.-M.; Yao, M. Actinomycin D inhibits cell proliferations and promotes apoptosis in osteosarcoma cells. Int. J. Clin. Exp. Med. 2015, 8, 1904–1911. [Google Scholar] [PubMed]
- Lohani, N.; Singh, H.; Moganty, R. Structural aspects of the interaction of anticancer drug Actinomycin-D to the GC rich region of hmgb1 gene. Int. J. Biol. Macromol. 2016, 87, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Panja, A.; Jana, N.C.; Brandão, P. Influence of the first and second coordination spheres on the diverse phenoxazinone synthase activity of cobalt complexes derived from a tetradentate Schiff base ligand. New J. Chem. 2017, 41, 9784–9795. [Google Scholar] [CrossRef]
- Panja, A. Syntheses and structural characterizations of cobalt(II) complexes with N4-donor Schiff base ligands: Influence of methyl substitution on structural parameters and on phenoxazinone synthase activity. Polyhedron 2014, 80, 81–89. [Google Scholar] [CrossRef]
- Mahato, M.; Mondal, D.; Nayek, H.P. Syntheses, Structures and Phenoxazinone Synthase Activities of Two Cobalt(III) Complexes. ChemistrySelect 2016, 1, 6777–6782. [Google Scholar] [CrossRef]
- Strekowski, L.; Wilson, B. Noncovalent interactions with DNA: An overview. Mut. Res. 2007, 623, 3–13. [Google Scholar] [CrossRef]
- Wolfe, A.; Shimer, G.H.; Meehan, T. Polycyclic aromatic hydrocarbons physically intercalate into duplex regions of denatured DNA. Biochemistry 1987, 26, 6392–6396. [Google Scholar] [CrossRef]
- Li, L.; Guo, Q.; Dong, J.; Xu, T.; Li, J. DNA binding, DNA cleavage and BSA interaction of a mixed-ligand copper(II) complex with taurine Schiff base and 1,10-phenanthroline. J. Photochem. Photobiol. B 2013, 125, 56–62. [Google Scholar] [CrossRef]
- Woody, R.W. Circular dichroism. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 1995; Volume 246, pp. 34–71. [Google Scholar]
- Liu, X.-W.; Li, J.; Li, H.; Zheng, K.-C.; Chao, H.; Ji, L.-N. Synthesis, characterization, DNA-binding and photocleavage of complexes [Ru(phen)2(6-OH-dppz)]2+ and [Ru(phen)2(6-NO2-dppz)]2+. J. Inorg. Biochem. 2005, 99, 2372–2380. [Google Scholar] [CrossRef]
- Abdel-Rahman, L.H.; Abu-Dief, A.M.; El-Khatib, R.M.; Abdel-Fatah, S.M. Some new nano-sized Fe(II), Cd(II) and Zn(II) Schiff base complexes as precursor for metal oxides: Sonochemical synthesis, characterization, DNA interaction, in vitro antimicrobial and anticancer activities. Bioorg. Chem. 2016, 69, 140–152. [Google Scholar] [CrossRef] [PubMed]
- Sirajuddin, M.; Ali, S.; Badshah, A. Drug–DNA interactions and their study by UV–Visible, fluorescence spectroscopies and cyclic voltametry. J. Photochem. Photobiol. B Biol. 2013, 124, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Mishra, M.; Tiwari, K.; Shukla, S.; Mishra, R.; Singh, V.P. Synthesis, structural investigation, DNA and protein binding study of some 3d-metal complexes with N′-(phenyl-pyridin-2-yl-methylene)-thiophene-2-carboxylic acid hydrazide. Spectrochim. Acta A 2014, 132, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Raman, N.; Pothiraj, K.; Baskaran, T. DNA interaction, antimicrobial, electrochemical and spectroscopic studies of metal(II) complexes with tridentate heterocyclic Schiff base derived from 2′-methylacetoacetanilide. J. Mol. Struct. 2011, 1000, 135–144. [Google Scholar] [CrossRef]
- Marmur, J. A procedure for the isolation of deoxyribonucleic acid from micro-organisms. J. Mol. Biol. 1961, 3, 208–218. [Google Scholar] [CrossRef]
- Reichmann, M.E.; Rice, S.A.; Thomas, C.A.; Doty, P. A Further Examination of the Molecular Weight and Size of Desoxypentose Nucleic Acid. J. Am. Chem. Soc. 1954, 76, 3047–3053. [Google Scholar] [CrossRef]
- Rigaku, O.D. CrysAlis PRO (Version 1.171.38.34c). 2015. Available online: https://www.rigaku.com/en/products/smc/crysalis (accessed on 12 April 2019).
- Sheldrick, G. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. A 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G. Crystal structure refinement with SHELXL. Acta Crystallogr. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Shivakumar, L.; Shivaprasad, K.; Revanasiddappa, H.D. Synthesis, spectroscopic characterization, antimicrobial, DNA binding and oxidative-induced DNA cleavage activities: New oxovanadium(IV) complexes of 2-(2-hydroxybenzylideneamino)isoindoline-1,3-dione. Spectrochim. Acta A 2012, 97, 659–666. [Google Scholar] [CrossRef]
- Sabolová, D.; Kožurková, M.; Plichta, T.; Ondrušová, Z.; Hudecová, D.; Šimkovič, M.; Paulíková, H.; Valent, A. Interaction of a copper(II)–Schiff base complexes with calf thymus DNA and their antimicrobial activity. Int. J. Biol. Macromol. 2011, 48, 319–325. [Google Scholar] [CrossRef]
- Baguley, B.C.; Le Bret, M. Quenching of DNA-ethidium fluorescence by amsacrine and other antitumor agents: A possible electron-transfer effect. Biochemistry 1984, 23, 937–943. [Google Scholar] [CrossRef] [PubMed]
- Fik, M.A.; Gorczyński, A.; Kubicki, M.; Hnatejko, Z.; Wadas, A.; Kulesza, P.J.; Lewińska, A.; Giel-Pietraszuk, M.; Wyszko, E.; Patroniak, V. New vanadium complexes with 6,6″-dimethyl-2,2′:6′,2″-terpyridine in terms of structure and biological properties. Polyhedron 2015, 97, 83–93. [Google Scholar] [CrossRef]
- Gorczyński, A.; Harrowfield, J.M.; Patroniak, V.; Stefankiewicz, A.R. Quaterpyridines as Scaffolds for Functional Metallosupramolecular Materials. Chem. Rev. 2016, 116, 14620–14674. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds: L1–L6, 1–12 are available from the authors. |












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Vmax (10−3Ms−1) | KM (10−3M) | kcat (h−1) |
|---|---|---|---|
| [Fe(L1)Cl3] (1) | 3.71 | 1.45 | 185.25 |
| [Fe(L2)Cl3] (2) | 2.55 | 2.16 | 127.30 |
| [Fe(L3)Cl3] (3) | 3.45 | 1.95 | 172.30 |
| [Fe(L4)Cl3] (4) | 2.68 | 2.02 | 134.05 |
| [Fe(L5)Cl3] (5) | 2.07 | 2.02 | 103.34 |
| [Fe(L6)Cl3] (6) | 3.01 | 2.19 | 150.49 |
| [Fe(L1)2](OTf)2 (7) | 3.98 | 1.56 | 199.40 |
| [Fe(L2)2](OTf)2 (8) | 1.96 | 2.08 | 97.68 |
| [Fe(L3)2](OTf)2 (9) | 3.68 | 1.86 | 183.80 |
| [Fe(L4)2](OTf)2 (10) | 0.86 | 1.79 | 42.76 |
| [Fe(L5)2](OTf)2 (11) | 1.30 | 1.89 | 65.14 |
| [Fe(L6)2](OTf)2 (12) | 2.13 | 2.08 | 106.53 |
| Catalyst | Solvent | kcat (h−1) | Ref. |
|---|---|---|---|
| [Fe(L1)Cl3] (1) | Methanol | 185.25 | This work |
| [Fe(L1)2](OTf)2 (7) | Methanol | 199.40 | This work |
| [FeCl2(La)] | DMF | 137.0 | [28] |
| [Fe(Lb)Cl3] | Methanol | 56.0 | [20] |
| [{Fe(Lc)(4,4′-byp)ClO4}]n | Methanol | 32.36 | [42] |
| [FeLcCl]2 | Methanol | 196.18 | [42] |
| [Co(Ld)(N3)3] | Methanol | 33.26 | [23] |
| [Co(Le)(N3)2] | Methanol | 54.0 | [45] |
| [Co(Lf)Cl(H2O)]Cl·H2O | Methanol | 13.68 | [46] |
| [LgCo(Lh)2]ClO4 | Methanol | 11.48 | [47] |
| Complex | Ligand (mg) | Fe(II/III) Salt (mg) | Precipitation Color/Solid | Yield a | Crystals From | |
|---|---|---|---|---|---|---|
| mg | % | |||||
| 1 | 26.7 | 30.0 | very dark green | 50.4 | 88.9 | iPr2O |
| 2 | 402.9 | 453.6 | dark green | 693.7 | 81 | tBuOMe |
| 3 | 60.0 | 63.8 | dark green | 66.2 | 67.3 | - |
| 4 | 22.3 | 30.0 | dark green | 46.8 | 89.4 | iPr2O |
| 5 | 23.9 | 30.0 | black | 47.3 | 87.7 | - |
| 6 | 21.9 | 30.0 | dark green | 39.8 | 76.8 | iPr2O |
| 7 | 70.0 | 51.6 | cinnamon | 87.9 | 72.3 | Et2O |
| 8 | 70.0 | 51.6 | hot chocolate | 104.0 | 72.3 | iPr2O |
| 9 | 70.0 | 48.7 | raspberry | 89.0 | 75.0 | tBuOMe |
| 10 | 70.0 | 61.6 | red | 121.0 | 91.9 | Et2O |
| 11 | 70.0 | 61.6 | warm brown | 117.0 | 88.8 | - |
| 12 | 55.0 | 45.3 | dark chocolate | 90.0 | 89.2 | PhMe |
| Compound | 1 | 2 | 4 | 6 | 7 | 8 | 9 | 10 | 12 |
|---|---|---|---|---|---|---|---|---|---|
| Formula | C12H12Cl3FeN6 | C13H16Cl2FeN6O+·Cl− | C10H11Cl3FeN5 | C11H13Cl3FeN5 | C24H24FeN122+·2(CF3SO3)−·C4H10O | C24H24FeN122+·2(CF3SO3)−·solvent | C26H28FeN122+·2(CF3SO3) −·C5H12O | C20H22FeN102+·2(CF3SO3)−·C4H10O | C22H26FeN102+·2(CF3SO3)−·CH4O |
| FeL1Cl3 | [FeL2Cl2(CH3OH)]+·Cl− | FeL4Cl3 | FeL6Cl3 | (FeL12)2+·2(CF3SO3)−·C4H10O | (FeL22)2+·2(CF3SO3)−·solvent | (FeL32)2+·2(CF3SO3)−·C5H12O | (FeL42)2+·2(CF3SO3)−·C4H10O | (FeL62)2+·2(CF3SO3)−·CH3OH | |
| Formula weight | 402.48 | 434.52 | 363.44 | 377.46 | 908.66 | 834.54 | 950.74 | 830.58 | 816.56 |
| Crystal system | triclinic | monoclinic | triclinic | monoclinic | monoclinic | monoclinic | triclinic | monoclinic | orthorhombic |
| Space group | P-1 | P21/n | P-1 | P21/c | C2/c | P21/c | P-1 | P21/c | Pca21 |
| a (Å) | 7.459(2) | 7.6840(4) | 7.5198(4) | 7.2144(2) | 23.1912(8) | 12.9879(7) | 12.4297(8) | 15.5382(2) | 22.3563(6) |
| b (Å) | 9.055(2) | 13.9429(6) | 8.1040(6) | 12.4684(3) | 17.0021(5) | 13.0532(8) | 13.0407(7) | 12.55315(14) | 8.3830(2) |
| c (Å) | 12.460(3) | 16.8684(6) | 12.4702(10) | 16.7244(4) | 22.3107(8) | 21.9024(19) | 13.4814 (7) | 18.1225 (2) | 17.5420(6) |
| α (°) | 75.69(2) | 90 | 75.205(6) | 90 | 90 | 90 | 88.870(4) | 90 | 90 |
| β (°) | 87.94(2) | 98.281(4) | 84.655(5) | 99.730(2) | 116.163(4) | 91.711(6) | 74.131(5) | 91.2230(13) | 90 |
| γ (°) | 66.12(3) | 90 | 66.519(6) | 90 | 90 | 90 | 75.262(5) | 90 | 90 |
| V(Å3) | 743.7(4) | 1788.39(14) | 673.87(9) | 1482.75(7) | 7895.8(5) | 3711.5(4) | 2029.9(2) | 3534.05(7) | 3287.60(16) |
| Z | 2 | 4 | 2 | 4 | 8 | 4 | 2 | 4 | 4 |
| Dx(g cm−3) | 1.797 | 1.614 | 1.791 | 1.691 | 1.529 | 1.493 | 1.556 | 1.561 | 1.650 |
| F(000) | 406 | 884 | 366 | 764 | 3728 | 1696 | 980 | 1704 | 1672 |
| μ (mm−1) | 13.136 | 1.305 | 14.392 | 1.553 | 0.577 | 0.604 | 0.565 | 0.634 | 0.680 |
| Reflections: | |||||||||
| collected | 1450 | 7518 | 4550 | 6161 | 18337 | 13346 | 30279 | 15200 | 17885 |
| unique (Rint) | 917 (0.042) | 3576 (0.024) | 2666 (0.054) | 3106 (0.022) | 7987 (0.031) | 6526 (0.059) | 7129 (0.083) | 7095 (0.015) | 5695 (0.029) |
| with I>2σ(I) | 716 | 2958 | 2319 | 2688 | 6243 | 3623 | 5726 | 6301 | 5323 |
| R(F) [I>2σ(I)] | 0.078 | 0.033 | 0.075 | 0.029 | 0.058 | 0.081 | 0.059 | 0.030 | 0.063 |
| wR(F2) [I>2σ(I)] | 0.233 | 0.081 | 0.196 | 0.059 | 0.174 | 0.153 | 0.156 | 0.072 | 0.165 |
| R(F) [all data] | 0.104 | 0.044 | 0.084 | 0.037 | 0.077 | 0.146 | 0.070 | 0.035 | 0.067 |
| wR(F2) [all data] | 0.233 | 0.087 | 0.204 | 0.063 | 0.188 | 0.174 | 0.169 | 0.075 | 0.169 |
| Goodness of fit | 1.05 | 1.05 | 1.14 | 1.04 | 1.24 | 1.02 | 1.07 | 1.02 | 1.08 |
| max/min Δρ (e·Å−3) | 0.77/−0.98 | 0.50/−0.29 | 1.75/−0.79 | 0.32/−0.31 | 1.68/−0.67 | 0.87/−0.45 | 1.14/−0.62 | 0.34/−0.39 | 0.83/−0.92 |
| CCDC number | 1851825 | 1851824 | 1851829 | 1851826 | 1871758 | 1871759 | 1871760 | 1871763 | 1871761 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bocian, A.; Szymańska, M.; Brykczyńska, D.; Kubicki, M.; Wałęsa-Chorab, M.; Roviello, G.N.; Fik-Jaskółka, M.A.; Gorczyński, A.; Patroniak, V. New Artificial Biomimetic Enzyme Analogues Based on Iron(II/III) Schiff Base Complexes: An Effect of (Benz)imidazole Organic Moieties on Phenoxazinone Synthase and DNA Recognition ‡. Molecules 2019, 24, 3173. https://doi.org/10.3390/molecules24173173
Bocian A, Szymańska M, Brykczyńska D, Kubicki M, Wałęsa-Chorab M, Roviello GN, Fik-Jaskółka MA, Gorczyński A, Patroniak V. New Artificial Biomimetic Enzyme Analogues Based on Iron(II/III) Schiff Base Complexes: An Effect of (Benz)imidazole Organic Moieties on Phenoxazinone Synthase and DNA Recognition ‡. Molecules. 2019; 24(17):3173. https://doi.org/10.3390/molecules24173173
Chicago/Turabian StyleBocian, Aleksandra, Martyna Szymańska, Daria Brykczyńska, Maciej Kubicki, Monika Wałęsa-Chorab, Giovanni N. Roviello, Marta A. Fik-Jaskółka, Adam Gorczyński, and Violetta Patroniak. 2019. "New Artificial Biomimetic Enzyme Analogues Based on Iron(II/III) Schiff Base Complexes: An Effect of (Benz)imidazole Organic Moieties on Phenoxazinone Synthase and DNA Recognition ‡" Molecules 24, no. 17: 3173. https://doi.org/10.3390/molecules24173173
APA StyleBocian, A., Szymańska, M., Brykczyńska, D., Kubicki, M., Wałęsa-Chorab, M., Roviello, G. N., Fik-Jaskółka, M. A., Gorczyński, A., & Patroniak, V. (2019). New Artificial Biomimetic Enzyme Analogues Based on Iron(II/III) Schiff Base Complexes: An Effect of (Benz)imidazole Organic Moieties on Phenoxazinone Synthase and DNA Recognition ‡. Molecules, 24(17), 3173. https://doi.org/10.3390/molecules24173173