Crude Heparin Preparations Unveil the Presence of Structurally Diverse Oversulfated Contaminants


Abstract
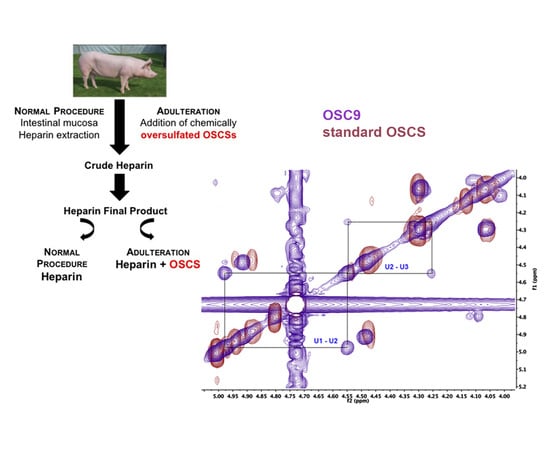
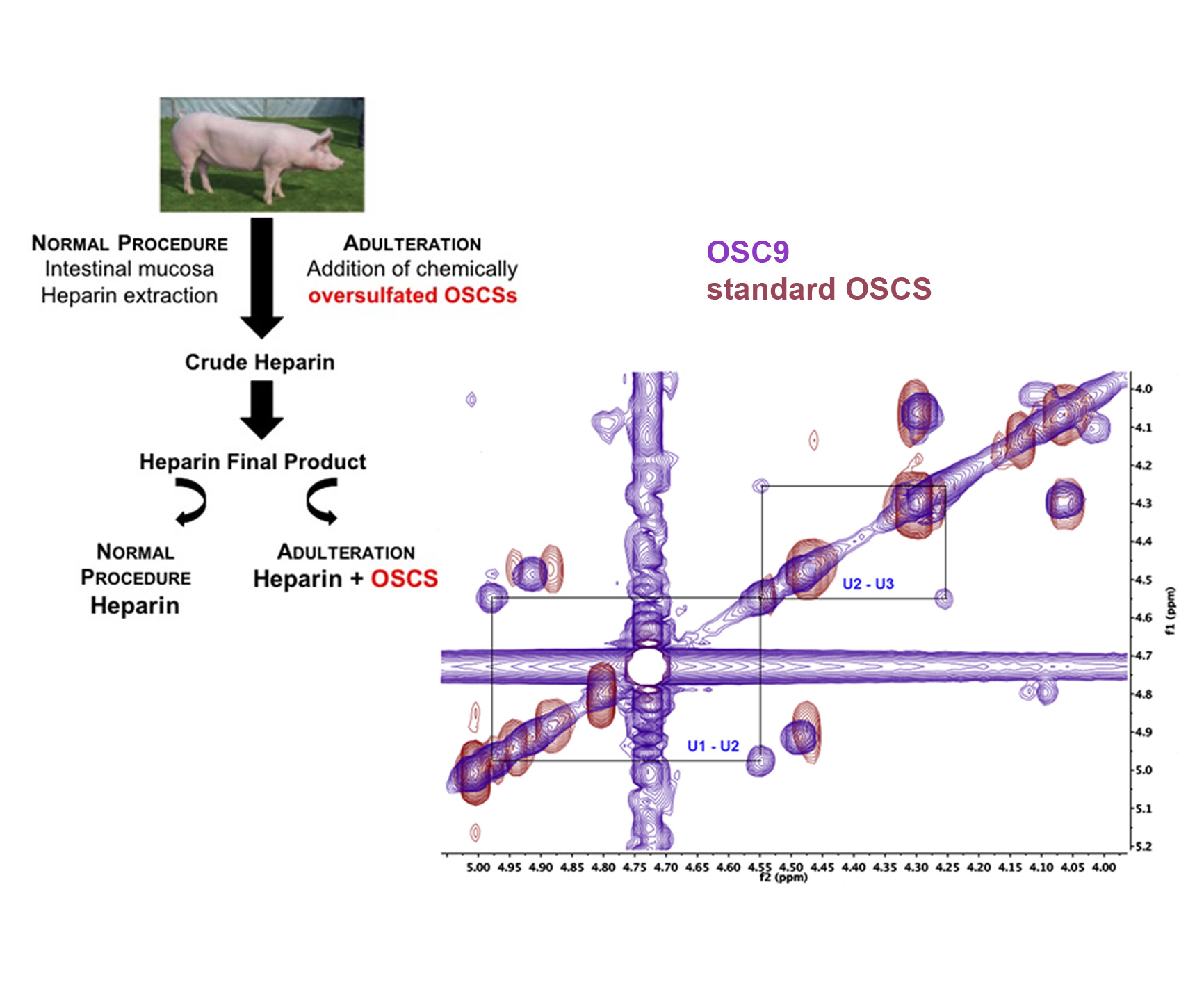{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
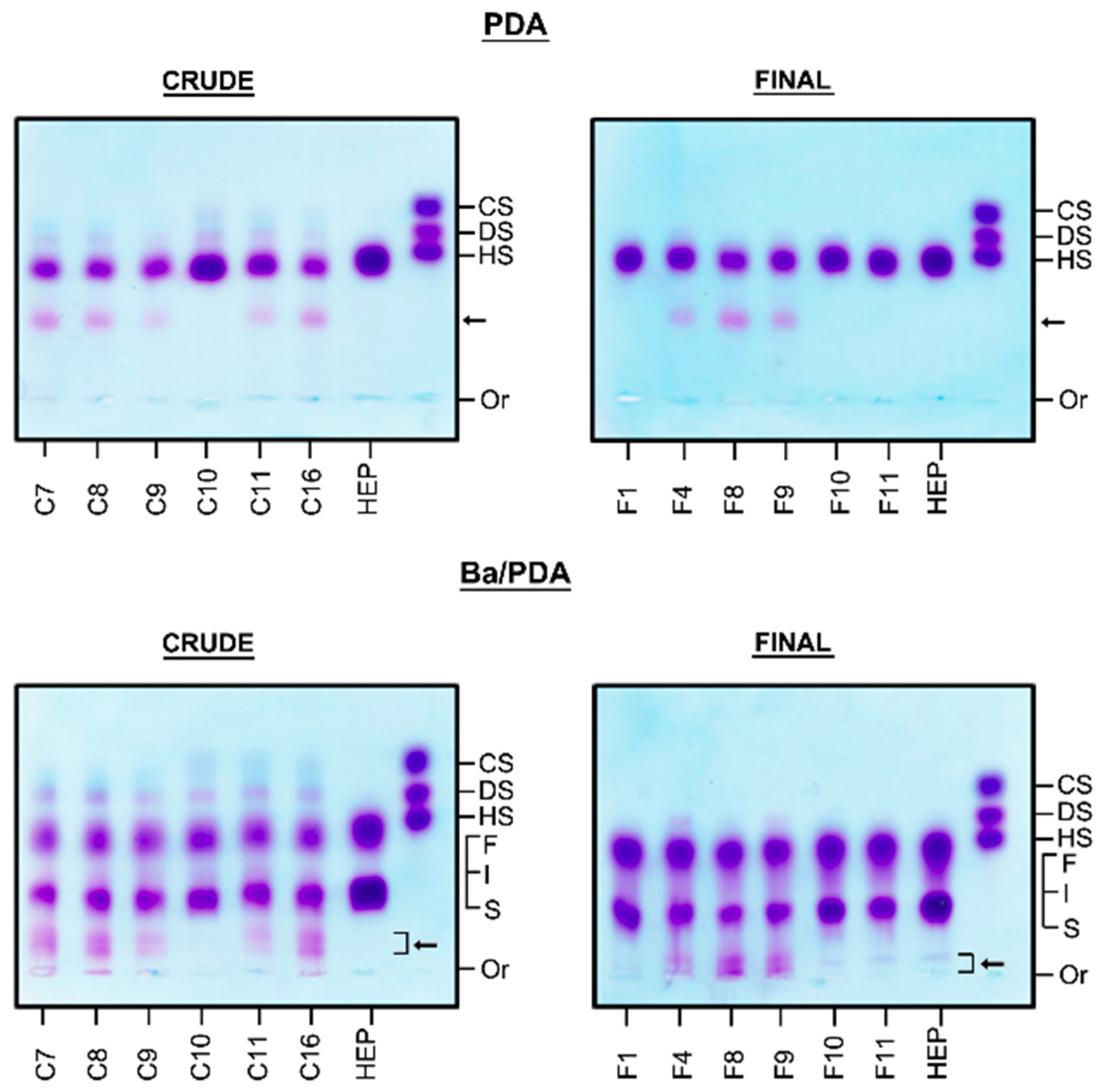
2.1. Identification of Contaminants in Crude Heparins and Final Product by Agarose Gel Electrophoresis
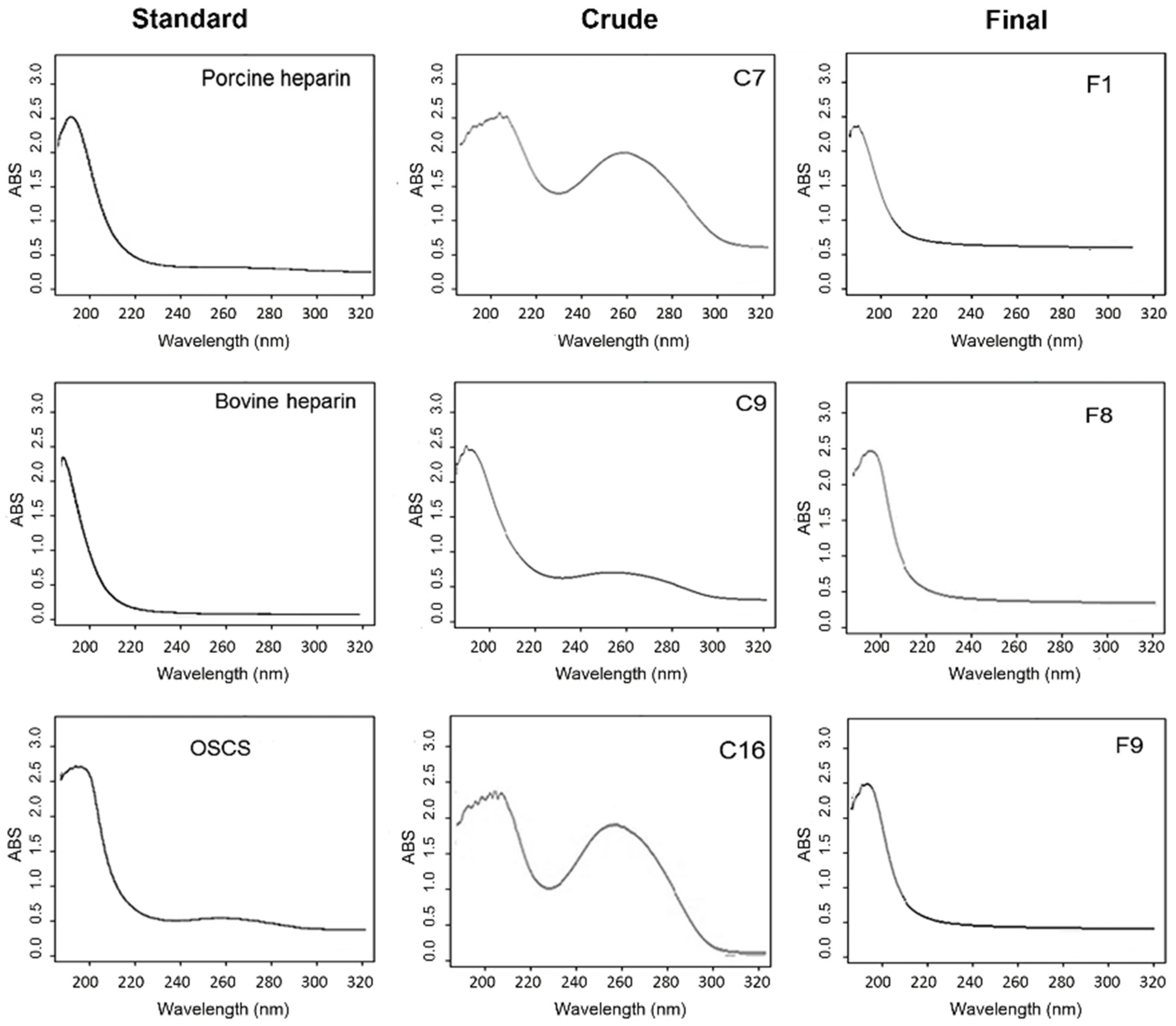
2.2. Scanning UV Spectroscopy Detects Contaminants in Heparin Preparations
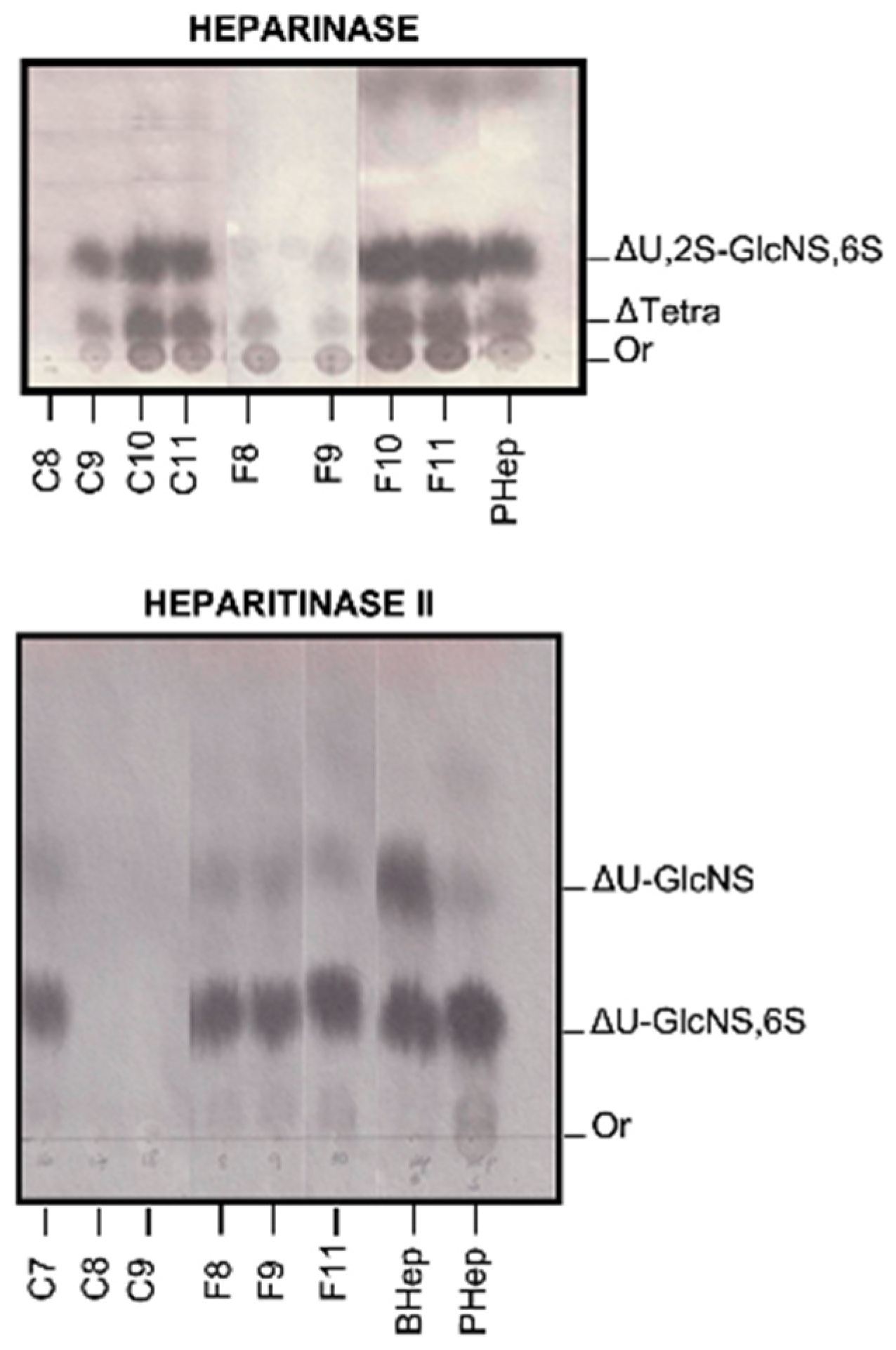
2.3. Degradation with Lyases–Heparinase and Heparitinase II
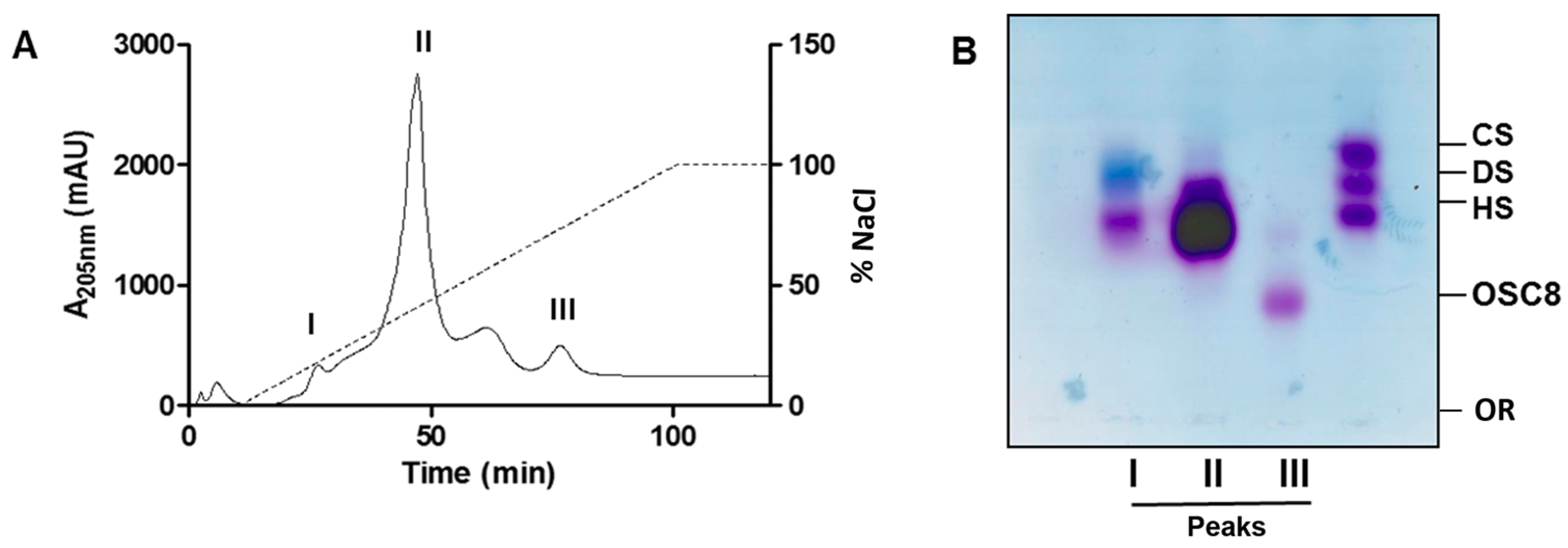
2.4. Isolation of the Contaminants
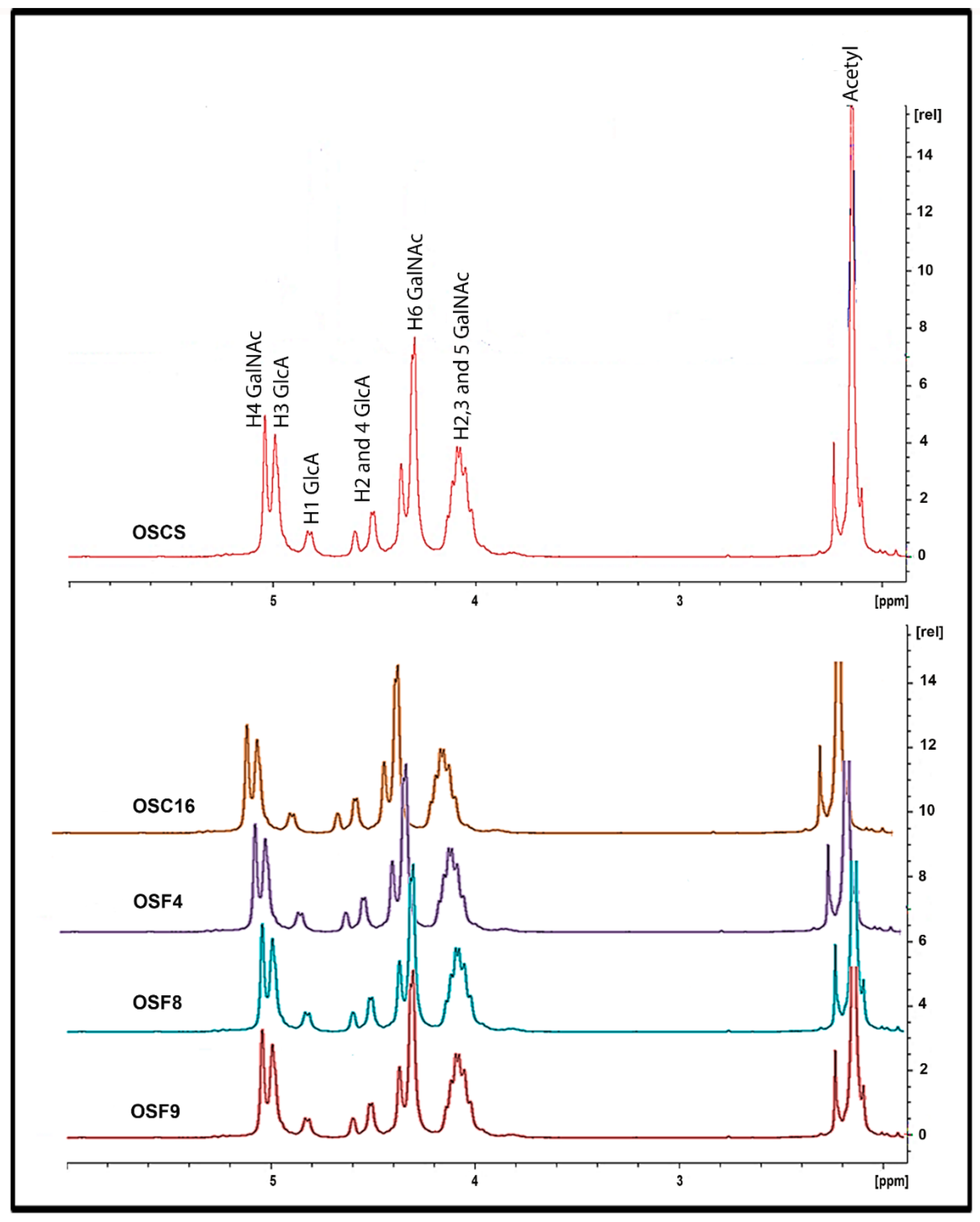
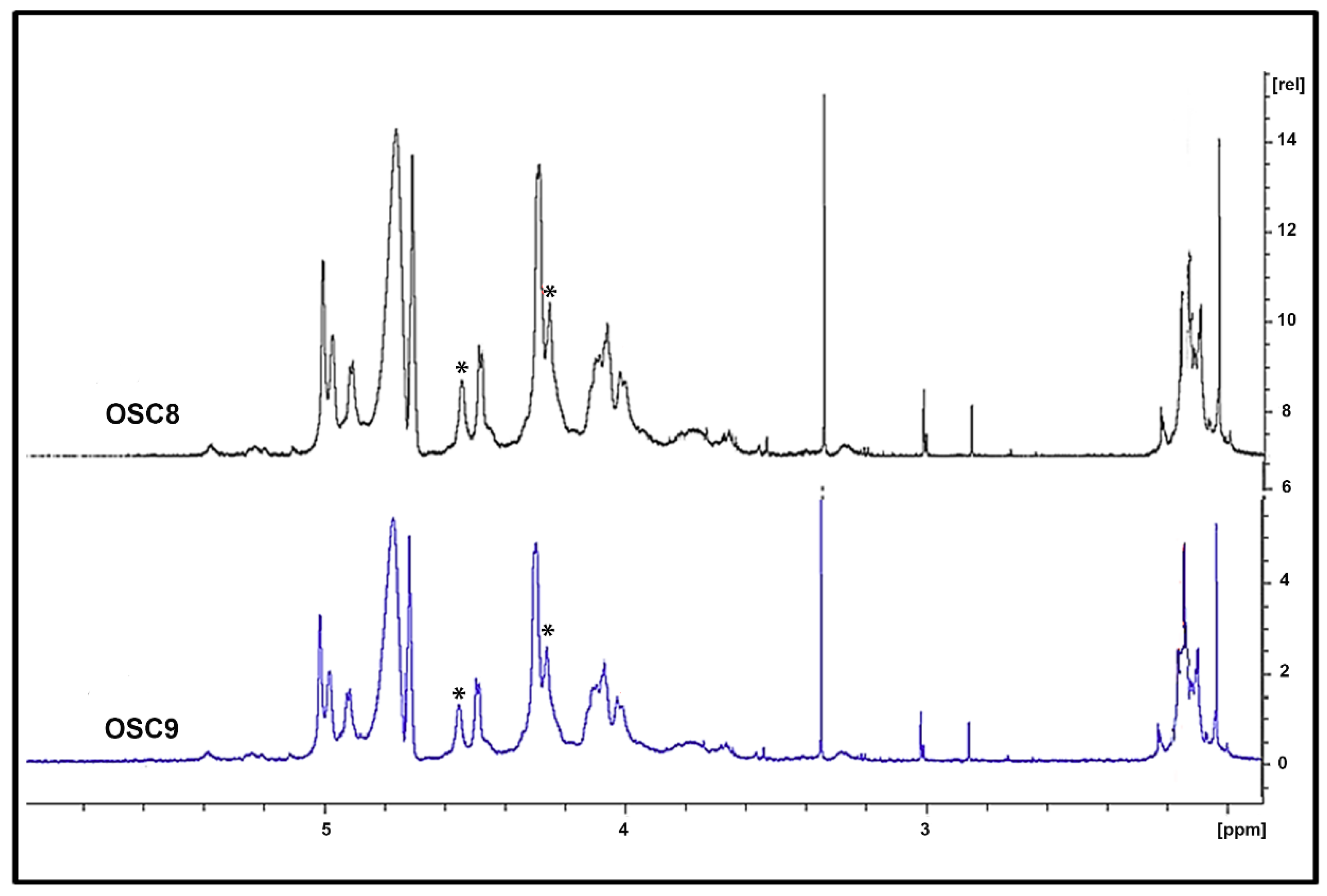
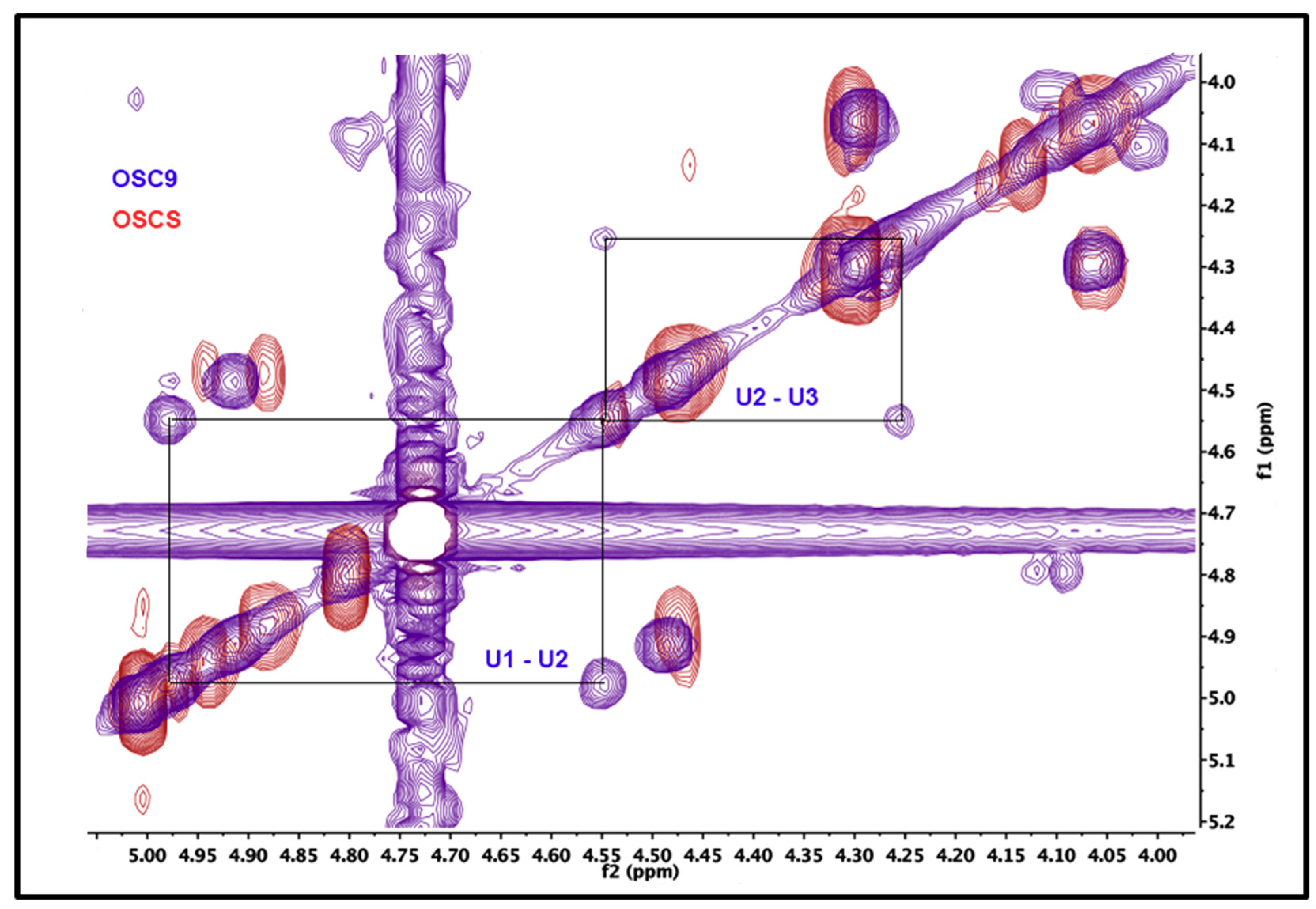
2.5. Structural Characterization of Heparin Contaminants by Nuclear Magnetic Resonance (NMR)
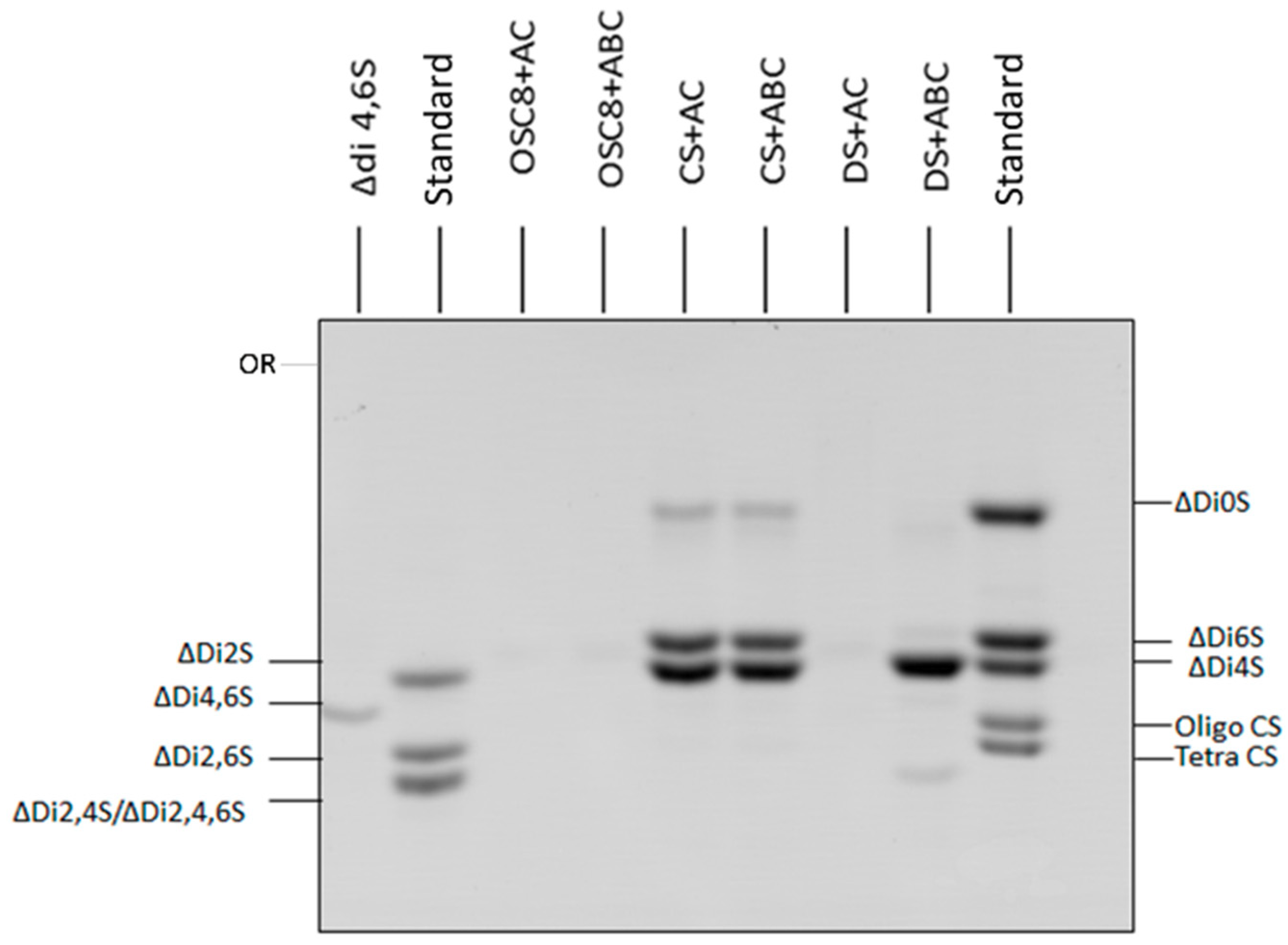
2.6. Exhaustive Degradation of OSC8 with Chondroitinases AC and ABC
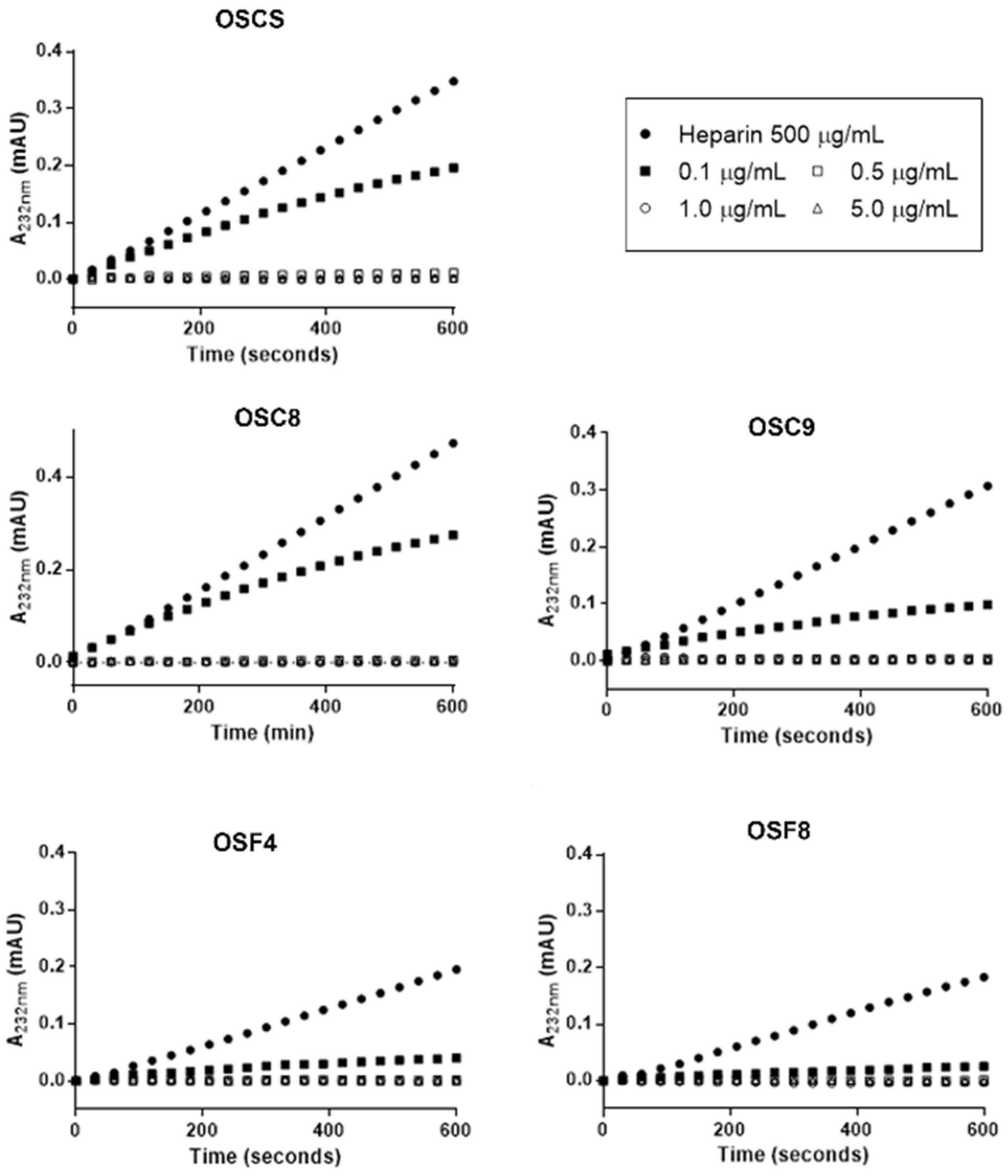
2.7. Effect of the Isolated Contaminants on Degradation of Heparin by Heparinase
2.8. APTT
2.9. Effects of the Contaminants on Endothelial Cells
2.10. Discussion
3. Materials and Methods
3.1. Heparin Samples and Standard OSCS
3.2. Agarose Gel Electrophoresis
3.3. Scanning UV Spectroscocy
3.4. Ion Exchange Chromatography for Isolation of the Sulfated Contaminants
3.5. Degradation with Different Glycosaminoglycan Lyases
3.6. Fluorophore Assisted Carbohydrate Electrophoresis (FACE)
3.7. Inhibitory Effect of OSCS on Heparinase Activity
3.8. Nuclear Magnetic Resonance (NMR) for Structural Characterization of the Contaminants in Heparin Preparations
3.9. APTT Assay
3.10. Endothelial Cells Culture
3.11. Cell Viability Assay
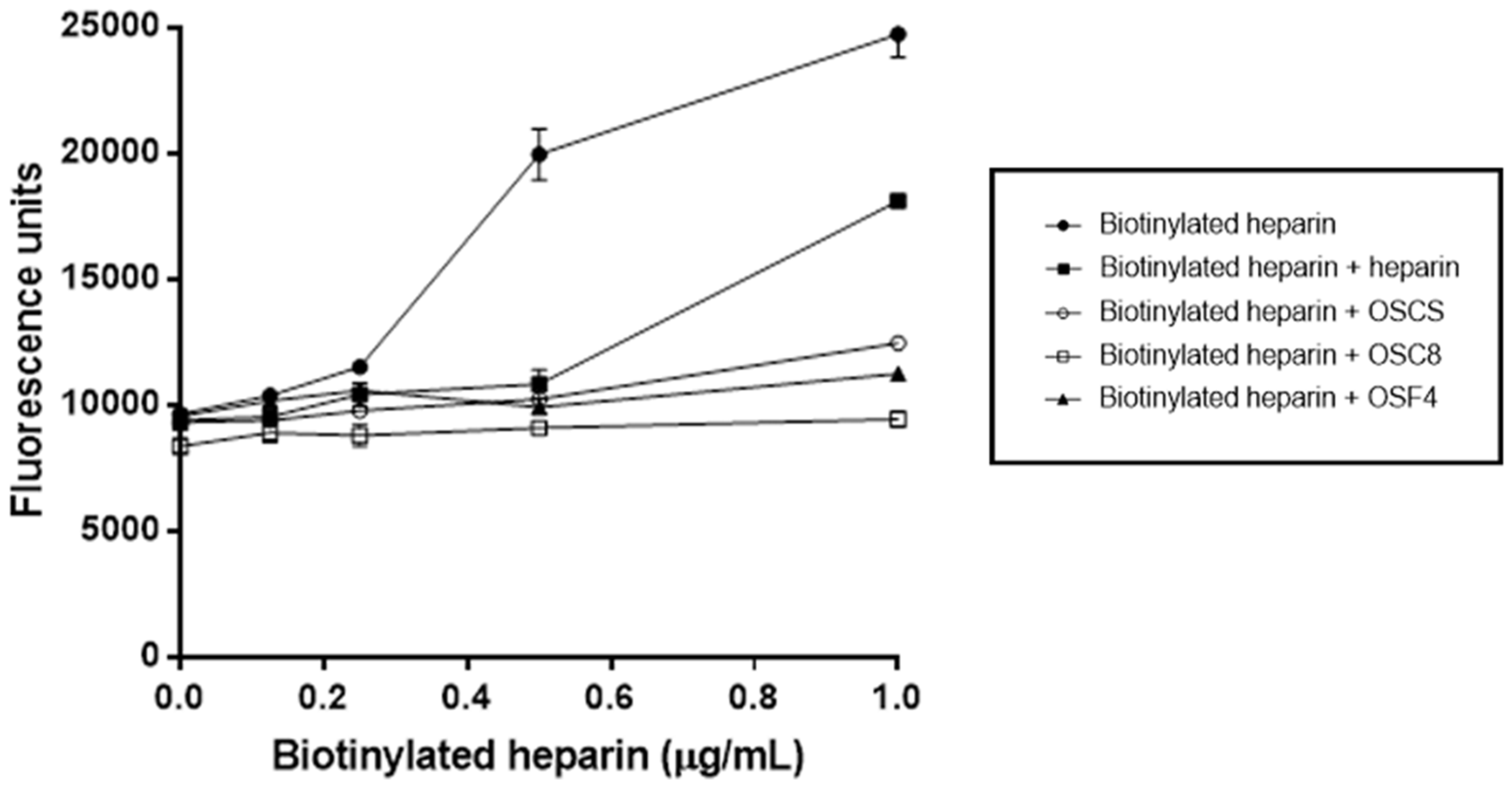
3.12. Competitive Binding Assay between Heparin and OSCSs
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Dietrich, C.P.; Nader, H.B.; McDuffie, N.N. Electrofocusing of heparin: Presence of 21 monomeric and dimeric molecular species in heparin preparations. An. Da Acad. Bras. De Cienc. 1975, 47, 301–309. [Google Scholar]
- Nader, H.B.; McDuffie, N.M.; Dietrich, C.P. Heparin fractionation by electrofocusing: Presence of 21 components of different molecular weights. Biochem. Biophys. Res. Commun. 1974, 57, 488–493. [Google Scholar] [CrossRef]
- Perlin, A.S.; Mackie, D.M.; Dietrich, C.P. Evidence for a (1 leads to 4)-linked 4-O-(-L-idopyranosyluronic acid 2-sulfate)-(2-deoxy-2-sulfoamino-D-glucopyranosyl 6-sulfate) sequence in heparin. Long-range H-H coupling in 4-deoxy-hex-4-enopyranosides. Carbohydr. Res. 1971, 18, 185–194. [Google Scholar] [CrossRef]
- Lima, M.; Rudd, T.; Yates, E. New Applications of Heparin and Other Glycosaminoglycans. Molecules 2017, 22, 749. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Johnson, D.J.; Esmon, C.T.; Huntington, J.A. Structure of the antithrombin-thrombin-heparin ternary complex reveals the antithrombotic mechanism of heparin. Nat. Struct. Mol. Boil. 2004, 11, 857–862. [Google Scholar] [CrossRef] [PubMed]
- Baik, J.Y.; Dahodwala, H.; Oduah, E.; Talman, L.; Gemmill, T.R.; Gasimli, L.; Datta, P.; Yang, B.; Li, G.; Zhang, F.; et al. Optimization of bioprocess conditions improves production of a CHO cell-derived, bioengineered heparin. Biotechnol. J. 2015, 10, 1067–1081. [Google Scholar] [CrossRef]
- Nader, H.B.; Pinhal, M.A.; Bau, E.C.; Castro, R.A.; Medeiros, G.F.; Chavante, S.F.; Leite, E.L.; Trindade, E.S.; Shinjo, S.K.; Rocha, H.A.; et al. Development of new heparin-like compounds and other antithrombotic drugs and their interaction with vascular endothelial cells. Braz. J. Med Biol. Res. 2001, 34, 699–709. [Google Scholar] [CrossRef][Green Version]
- Oduah, E.I.; Linhardt, R.J.; Sharfstein, S.T. Heparin: Past, Present, and Future. Pharmaceuticals 2016, 9, 38. [Google Scholar] [CrossRef]
- Van der Meer, J.Y.; Kellenbach, E.; van den Bos, L.J. From Farm to Pharma: An Overview of Industrial Heparin Manufacturing Methods. Molecules 2017, 22, 1025. [Google Scholar] [CrossRef]
- Guerrini, M.; Shriver, Z.; Bisio, A.; Naggi, A.; Casu, B.; Sasisekharan, R.; Torri, G. The tainted heparin story: an update. Thromb. Haemost. 2009, 102, 907–911. [Google Scholar] [CrossRef]
- Liverani, L.; Mascellani, G.; Spelta, F. Heparins: Process-related physico-chemical and compositional characteristics, fingerprints and impurities. Thromb. Haemost. 2009, 102, 846–853. [Google Scholar] [CrossRef]
- Sassaki, G.L.; Riter, D.S.; Santana Filho, A.P.; Guerrini, M.; Lima, M.A.; Cosentino, C.; Souza, L.M.; Cipriani, T.R.; Rudd, T.R.; Nader, H.B.; et al. A robust method to quantify low molecular weight contaminants in heparin: detection of tris(2-n-butoxyethyl) phosphate. Analyst 2011, 136, 2330–2338. [Google Scholar] [CrossRef]
- Guerrini, M.; Beccati, D.; Shriver, Z.; Naggi, A.; Viswanathan, K.; Bisio, A.; Capila, I.; Lansing, J.C.; Guglieri, S.; Fraser, B.; et al. Oversulfated chondroitin sulfate is a contaminant in heparin associated with adverse clinical events. Nat. Biotechnol. 2008, 26, 669–675. [Google Scholar] [CrossRef]
- Ramacciotti, E.; Wahi, R.; Messmore, H.L. Contaminated heparin preparations, severe adverse events and the contact system. Clin. Appl. Thromb. Hemost. 2008, 14, 489–491. [Google Scholar] [CrossRef]
- Blossom, D.B.; Kallen, A.J.; Patel, P.R.; Elward, A.; Robinson, L.; Gao, G.; Langer, R.; Perkins, K.M.; Jaeger, J.L.; Kurkjian, K.M.; et al. Outbreak of adverse reactions associated with contaminated heparin. N. Engl. J. Med. 2008, 359, 2674–2684. [Google Scholar] [CrossRef]
- Lima, M.A.; Rudd, T.R.; de Farias, E.H.; Ebner, L.F.; Gesteira, T.F.; de Souza, L.M.; Mendes, A.; Cordula, C.R.; Martins, J.R.; Hoppensteadt, D.; et al. A new approach for heparin standardization: combination of scanning UV spectroscopy, nuclear magnetic resonance and principal component analysis. PLoS ONE 2011, 6, e15970. [Google Scholar] [CrossRef]
- Kishimoto, T.K.; Viswanathan, K.; Ganguly, T.; Elankumaran, S.; Smith, S.; Pelzer, K.; Lansing, J.C.; Sriranganathan, N.; Zhao, G.; Galcheva-Gargova, Z.; et al. Contaminated heparin associated with adverse clinical events and activation of the contact system. N. Engl. J. Med. 2008, 358, 2457–2467. [Google Scholar] [CrossRef]
- Zhang, Z.; Weiwer, M.; Li, B.; Kemp, M.M.; Daman, T.H.; Linhardt, R.J. Oversulfated chondroitin sulfate: impact of a heparin impurity, associated with adverse clinical events, on low-molecular-weight heparin preparation. J. Med. Chem. 2008, 51, 5498–5501. [Google Scholar] [CrossRef]
- Guerrini, M.; Zhang, Z.; Shriver, Z.; Naggi, A.; Masuko, S.; Langer, R.; Casu, B.; Linhardt, R.J.; Torri, G.; Sasisekharan, R. Orthogonal analytical approaches to detect potential contaminants in heparin. Proc. Natl. Acad. Sci. USA 2009, 106, 16956–16961. [Google Scholar] [CrossRef]
- Calabro, A.; Midura, R.; Wang, A.; West, L.; Plaas, A.; Hascall, V.C. Fluorophore-assisted carbohydrate electrophoresis (FACE) of glycosaminoglycans. Osteoarthr. Cartil. 2001, 9 (Suppl. A), S16–S22. [Google Scholar] [CrossRef]
- Cordula, C.R.; Lima, M.A.; Shinjo, S.K.; Gesteira, T.F.; Pol-Fachin, L.; Coulson-Thomas, V.J.; Verli, H.; Yates, E.A.; Rudd, T.R.; Pinhal, M.A.; et al. On the catalytic mechanism of polysaccharide lyases: evidence of His and Tyr involvement in heparin lysis by heparinase I and the role of Ca2+. Mol. BioSystems 2014, 10, 54–64. [Google Scholar] [CrossRef]
- Aich, U.; Shriver, Z.; Tharakaraman, K.; Raman, R.; Sasisekharan, R. Competitive inhibition of heparinase by persulfonated glycosaminoglycans: A tool to detect heparin contamination. Anal. Chem. 2011, 83, 7815–7822. [Google Scholar] [CrossRef][Green Version]
- Cavalcante, R.S.; Brito, A.S.; Palhares, L.; Lima, M.A.; Cavalheiro, R.P.; Nader, H.B.; Sassaki, G.L.; Chavante, S.F. 2,3-Di-O-sulfo glucuronic acid: An unmodified and unusual residue in a highly sulfated chondroitin sulfate from Litopenaeus vannamei. Carbohydr. Polym. 2018, 183, 192–200. [Google Scholar] [CrossRef]
- Kinoshita-Toyoda, A.; Yamada, S.; Haslam, S.M.; Khoo, K.H.; Sugiura, M.; Morris, H.R.; Dell, A.; Sugahara, K. Structural determination of five novel tetrasaccharides containing 3-O-sulfated D-glucuronic acid and two rare oligosaccharides containing a beta-D-glucose branch isolated from squid cartilage chondroitin sulfate E. Biochemistry 2004, 43, 11063–11074. [Google Scholar] [CrossRef]
- Maruyama, T.; Toida, T.; Imanari, T.; Yu, G.; Linhardt, R.J. Conformational changes and anticoagulant activity of chondroitin sulfate following its O-sulfonation. Carbohydr. Res. 1998, 306, 35–43. [Google Scholar] [CrossRef]
- Li, B.; Suwan, J.; Martin, J.G.; Zhang, F.; Zhang, Z.; Hoppensteadt, D.; Clark, M.; Fareed, J.; Linhardt, R.J. Oversulfated chondroitin sulfate interaction with heparin-binding proteins: new insights into adverse reactions from contaminated heparins. Biochem. Pharmacol. 2009, 78, 292–300. [Google Scholar] [CrossRef]
- Gray, A.; Litinas, E.; Jeske, W.; Fareed, J.; Hoppensteadt, D. Interactions of oversulfated chondroitin sulfate (OSCS) from different sources with unfractionated heparin. Clin. Appl. Thromb. Hemost. 2012, 18, 166–173. [Google Scholar] [CrossRef]
- Boucas, R.I.; Trindade, E.S.; Tersariol, I.L.; Dietrich, C.P.; Nader, H.B. Development of an enzyme-linked immunosorbent assay (ELISA)-like fluorescence assay to investigate the interactions of glycosaminoglycans to cells. Anal. Chim. Acta 2008, 618, 218–226. [Google Scholar] [CrossRef]
- Szajek, A.Y.; Chess, E.; Johansen, K.; Gratzl, G.; Gray, E.; Keire, D.; Linhardt, R.J.; Liu, J.; Morris, T.; Mulloy, B.; et al. The US regulatory and pharmacopeia response to the global heparin contamination crisis. Nat. Biotechnol. 2016, 34, 625–630. [Google Scholar] [CrossRef]
- Viskov, C.; Bouley, E.; Hubert, P.; Martinez, C.; Herman, F.; Jeske, W.; Hoppensteadt, D.; Walenga, J.M.; Fareed, J. Isolation and characterization of contaminants in recalled unfractionated heparin and low-molecular-weight heparin. Clin. Appl. Thromb. Hemost. 2009, 15, 395–401. [Google Scholar] [CrossRef]
- Bianchini, P.; Nader, H.B.; Takahashi, H.K.; Osima, B.; Straus, A.H.; Dietrich, C.P. Fractionation and identification of heparin and other acidic mucopolysaccharides by a new discontinuous electrophoretic method. J. Chromatogr. A 1980, 196, 455–462. [Google Scholar] [CrossRef]
- Dietrich, C.P.; Silva, M.E.; Michelacci, Y.M. Sequential degradation of heparin in Flavobacterium heparinum. Purification and properties of five enzymes involved in heparin degradation. J. Boil. Chem. 1973, 248, 6408–6415. [Google Scholar]
- Nader, H.B.; Porcionatto, M.A.; Tersariol, I.L.; Pinhal, M.A.; Oliveira, F.W.; Moraes, C.T.; Dietrich, C.P. Purification and substrate specificity of heparitinase I and heparitinase II from Flavobacterium heparinum. Analyses of the heparin and heparan sulfate degradation products by 13C NMR spectroscopy. J. Boil. Chem. 1990, 265, 16807–16813. [Google Scholar]
- Nader, H.B.; Kobayashi, E.Y.; Chavante, S.F.; Tersariol, I.L.; Castro, R.A.; Shinjo, S.K.; Naggi, A.; Torri, G.; Casu, B.; Dietrich, C.P. New insights on the specificity of heparin and heparan sulfate lyases from Flavobacterium heparinum revealed by the use of synthetic derivatives of K5 polysaccharide from E. coli and 2-O-desulfated heparin. Glycoconj. J. 1999, 16, 265–270. [Google Scholar] [CrossRef]
- Calabro, A.; Benavides, M.; Tammi, M.; Hascall, V.C.; Midura, R.J. Microanalysis of enzyme digests of hyaluronan and chondroitin/dermatan sulfate by fluorophore-assisted carbohydrate electrophoresis (FACE). Glycobiology 2000, 10, 273–281. [Google Scholar] [CrossRef]
- Heparin Sodium Stage 2 Monograph. Pharmacopeia Forum. Interim Revision Announcement. 2019, Volume 35, pp. 1–4. Available online: https://www.uspnf.com/sites/default/files/usp_pdf/EN/USPNF/key-issues/heparinSodiumMonograph.pdf (accessed on 1 June 2019).
- Buonassisi, V.; Venter, J.C. Hormone and neurotransmitter receptors in an established vascular endothelial cell line. Proc. Natl. Acad. Sci. USA 1976, 73, 1612–1616. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |










© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mendes, A.; Meneghetti, M.C.Z.; Palladino, M.V.; Justo, G.Z.; Sassaki, G.L.; Fareed, J.; Lima, M.A.; Nader, H.B. Crude Heparin Preparations Unveil the Presence of Structurally Diverse Oversulfated Contaminants. Molecules 2019, 24, 2988. https://doi.org/10.3390/molecules24162988
Mendes A, Meneghetti MCZ, Palladino MV, Justo GZ, Sassaki GL, Fareed J, Lima MA, Nader HB. Crude Heparin Preparations Unveil the Presence of Structurally Diverse Oversulfated Contaminants. Molecules. 2019; 24(16):2988. https://doi.org/10.3390/molecules24162988
Chicago/Turabian StyleMendes, Aline, Maria C. Z. Meneghetti, Marcelly Valle Palladino, Giselle Zenker Justo, Guilherme L. Sassaki, Jawed Fareed, Marcelo A. Lima, and Helena B. Nader. 2019. "Crude Heparin Preparations Unveil the Presence of Structurally Diverse Oversulfated Contaminants" Molecules 24, no. 16: 2988. https://doi.org/10.3390/molecules24162988
APA StyleMendes, A., Meneghetti, M. C. Z., Palladino, M. V., Justo, G. Z., Sassaki, G. L., Fareed, J., Lima, M. A., & Nader, H. B. (2019). Crude Heparin Preparations Unveil the Presence of Structurally Diverse Oversulfated Contaminants. Molecules, 24(16), 2988. https://doi.org/10.3390/molecules24162988