Effects of Halogen, Chalcogen, Pnicogen, and Tetrel Bonds on IR and NMR Spectra



Abstract
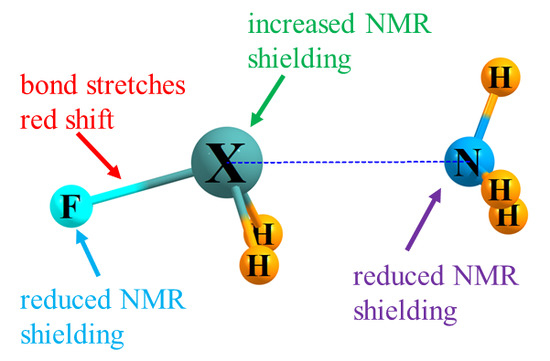
1. Introduction
2. Methods and Systems
3. Results
3.1. Energetics and IR Spectra
3.2. NMR Shielding
3.3. Atomic Charges
3.4. Electron Density Shifts
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Del Bene, J.E.; Alkorta, I.; Elguero, J. Exploring N…C tetrel and O…S chalcogen bonds in HN(CH)SX:OCS systems, for X = F, NC, Cl, CN, CCH, and H. Chem. Phys. Lett. 2019, 730, 466–471. [Google Scholar] [CrossRef]
- Gougoula, E.; Medcraft, C.; Alkorta, I.; Walker, N.R.; Legon, A.C. A chalcogen-bonded complex H3NS=C=S formed by ammonia and carbon disulfide characterised by chirped-pulse, broadband microwave spectroscopy. J. Chem. Phys. 2019, 150, 084307. [Google Scholar] [CrossRef] [PubMed]
- Alkorta, I.; Legon, A. An Ab Initio Investigation of the Geometries and Binding Strengths of Tetrel-, Pnictogen-, and Chalcogen-Bonded Complexes of CO2, N2O, and CS2 with Simple Lewis Bases: Some Generalizations. Molecules 2018, 23, 2250. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, S.J. Pnicogen and tetrel bonds—Tetrahedral Lewis acid centres. Struct. Chem. 2019, 30, 1141–1152. [Google Scholar] [CrossRef]
- Grabowski, S. Tetrel Bonds with π-Electrons Acting as Lewis Bases—Theoretical Results and Experimental Evidences. Molecules 2018, 23, 1183. [Google Scholar] [CrossRef] [PubMed]
- Alkorta, I.; Elguero, J.; Grabowski, S.J. Pnicogen and hydrogen bonds: Complexes between PH3X+ and PH2X systems. Phys. Chem. Chem. Phys. 2015, 17, 3261–3272. [Google Scholar] [CrossRef]
- Franconetti, A.; Quiñonero, D.; Frontera, A.; Resnati, G. Unexpected chalcogen bonds in tetravalent sulfur compounds. Phys. Chem. Chem. Phys. 2019, 21, 11313–11319. [Google Scholar] [CrossRef]
- Franconetti, A.; Frontera, A. Theoretical and Crystallographic Study of Lead(IV) Tetrel Bonding Interactions. Chem. Eur. J. 2019, 25, 6007–6013. [Google Scholar] [CrossRef]
- Frontera, A.; Bauzá, A. S⋯Sn Tetrel Bonds in the Activation of Peroxisome Proliferator-Activated Receptors (PPARs) by Organotin Molecules. Chem. Eur. J. 2018, 24, 16582–16587. [Google Scholar] [CrossRef]
- Murray, J.S.; Politzer, P. σ-Holes and Si···N intramolecular interactions. J. Mol. Model. 2019, 25, 101. [Google Scholar] [CrossRef]
- Clark, T.; Murray, J.S.; Politzer, P. A perspective on quantum mechanics and chemical concepts in describing noncovalent interactions. Phys. Chem. Chem. Phys. 2018, 20, 30076–30082. [Google Scholar] [CrossRef] [PubMed]
- Riley, K.E.; Tran, K.-A. Strength, character, and directionality of halogen bonds involving cationic halogen bond donors. Faraday Disc. 2017, 203, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Riley, K.E.; Vazquez, M.; Umemura, C.; Miller, C.; Tran, K.-A. Exploring the (Very Flat) Potential Energy Landscape of R–Br···π Interactions with Accurate CCSD(T) and SAPT Techniques. Chem. Eur. J. 2016, 22, 17690–17695. [Google Scholar] [CrossRef] [PubMed]
- Scheiner, S. The pnicogen bond: Its relation to hydrogen, halogen, and other noncovalent bonds. Acc. Chem. Res. 2013, 46, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Zierkiewicz, W.; Michalczyk, M.; Wysokiński, R.; Scheiner, S. On the ability of pnicogen atoms to engage in both σ and π-hole complexes. Heterodimers of ZF2C6H5 (Z = P, As, Sb, Bi) and NH3. J. Mol. Model. 2019, 25, 152. [Google Scholar] [CrossRef]
- Zierkiewicz, W.; Michalczyk, M.; Wysokiński, R.; Scheiner, S. Dual Geometry Schemes in Tetrel Bonds: Complexes between TF4 (T = Si, Ge, Sn) and Pyridine Derivatives. Molecules 2019, 24, 376. [Google Scholar] [CrossRef]
- Zierkiewicz, W.; Fanfrlík, J.; Michalczyk, M.; Michalska, D.; Hobza, P. S⋯N chalcogen bonded complexes of carbon disulfide with diazines. Theoretical study. Chem. Phys. 2018, 500, 37–44. [Google Scholar] [CrossRef]
- Dong, W.; Niu, B.; Liu, S.; Cheng, J.; Liu, S.; Li, Q. Comparison of σ-/π-Hole Tetrel Bonds between TH3F/F2TO and H2CX (X = O, S, Se). ChemPhysChem 2019, 20, 627–635. [Google Scholar]
- Dong, W.; Wang, Y.; Cheng, J.; Yang, X.; Li, Q. Competition between σ-hole pnicogen bond and π-hole tetrel bond in complexes of CF2=CFZH2 (Z = P, As, and Sb). Mol. Phys. 2019, 117, 251–259. [Google Scholar] [CrossRef]
- Dong, W.; Li, Q.; Scheiner, S. Comparative Strengths of Tetrel, Pnicogen, Chalcogen, and Halogen Bonds and Contributing Factors. Molecules 2018, 23, 1681. [Google Scholar] [CrossRef]
- Stasyuk, O.A.; Sedlak, R.; Guerra, C.F.; Hobza, P. Comparison of the DFT-SAPT and Canonical EDA Schemes for the Energy Decomposition of Various Types of Noncovalent Interactions. J. Chem. Theory Comput. 2018, 14, 3440–3450. [Google Scholar] [CrossRef] [PubMed]
- Sedlak, R.; Eyrilmez, S.M.; Hobza, P.; Nachtigallova, D. The role of the s-holes in stability of non-bonded chalcogenide⋯benzene interactions: The ground and excited states. Phys. Chem. Chem. Phys. 2018, 20, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Esrafili, M.; Mousavian, P. Strong Tetrel Bonds: Theoretical Aspects and Experimental Evidence. Molecules 2018, 23, 2642. [Google Scholar] [CrossRef] [PubMed]
- Scheiner, S.; Adhikari, U. Abilities of different electron donors (D) to engage in a P⋯D noncovalent interaction. J. Phys. Chem. A 2011, 115, 11101–11110. [Google Scholar] [CrossRef] [PubMed]
- Esrafili, M.D.; Mousavian, P.; Mohammadian-Sabet, F. Tuning of pnicogen and chalcogen bonds by an aerogen-bonding interaction: A comparative ab initio study. Mol. Phys. 2019, 117, 58–66. [Google Scholar] [CrossRef]
- Scilabra, P.; Terraneo, G.; Resnati, G. The Chalcogen Bond in Crystalline Solids: A World Parallel to Halogen Bond. Acc. Chem. Res. 2019, 52, 1313–1324. [Google Scholar] [CrossRef]
- Kuwano, S.; Suzuki, T.; Yamanaka, M.; Tsutsumi, R.; Arai, T. Catalysis Based on C−I···π Halogen Bonds: Electrophilic Activation of 2-Alkenylindoles by Cationic Halogen-Bond Donors for [4 + 2] Cycloadditions. Angew. Chem. Int. Ed. 2019, 58, 10220–10224. [Google Scholar] [CrossRef]
- Scheiner, S. Comparison of halide receptors based on H, halogen, chalcogen, pnicogen, and tetrel bonds. Faraday Disc. 2017, 203, 213–226. [Google Scholar] [CrossRef]
- Dreger, A.; Wonner, P.; Engelage, E.; Walter, S.M.; Stoll, R.; Huber, S.M. A halogen-bonding-catalysed Nazarov cyclisation reaction. Chem. Commun. 2019, 55, 8262–8265. [Google Scholar] [CrossRef]
- Trievel, R.C.; Scheiner, S. Crystallographic and Computational Characterization of Methyl Tetrel Bonding in S-Adenosylmethionine-Dependent Methyltransferases. Molecules 2018, 23, 2965. [Google Scholar] [CrossRef]
- Vanderkooy, A.; Gupta, A.K.; Földes, T.; Lindblad, S.; Orthaber, A.; Pápai, I.; Erdélyi, M. Halogen Bonding Helicates Encompassing Iodonium Cations. Angew. Chem. Int. Ed. 2019, 58, 9012–9016. [Google Scholar] [CrossRef]
- Pan, F.; Chen, Y.; Li, S.; Jiang, M.; Rissanen, K. Iodine Clathrated: A Solid-State Analogue of the Iodine–Starch Complex. Chem. Eur. J. 2019, 25, 7485–7488. [Google Scholar] [CrossRef]
- Perera, M.D.; Aakeröy, C.B. Organocatalysis by a multidentate halogen-bond donor: An alternative to hydrogen-bond based catalysis. New J. Chem. 2019, 43, 8311–8314. [Google Scholar] [CrossRef]
- Scheiner, S. Differential Binding of Tetrel-Bonding Bipodal Receptors to Monatomic and Polyatomic Anions. Molecules 2019, 24, 227. [Google Scholar] [CrossRef]
- Scheiner, S. Assembly of Effective Halide Receptors from Components. Comparing Hydrogen, Halogen, and Tetrel Bonds. J. Phys. Chem. A 2017, 121, 3606–3615. [Google Scholar] [CrossRef]
- Del Bene, J.E.; Alkorta, I.; Elguero, J. Anionic complexes of F− and Cl− with substituted methanes: Hydrogen, halogen, and tetrel bonds. Chem. Phys. Lett. 2016, 655–656, 115–119. [Google Scholar] [CrossRef]
- Scheiner, S. Can two trivalent N atoms engage in a direct N⋯N noncovalent interaction? Chem. Phys. Lett. 2011, 514, 32–35. [Google Scholar] [CrossRef]
- Southern, S.A.; Bryce, D.L. NMR Investigations of Noncovalent Carbon Tetrel Bonds. Computational Assessment and Initial Experimental Observation. J. Phys. Chem. A 2015, 119, 11891–11899. [Google Scholar] [CrossRef]
- Scheiner, S. Comparison of CH···O, SH···O, Chalcogen, and Tetrel Bonds Formed by Neutral and Cationic Sulfur-Containing Compounds. J. Phys. Chem. A 2015, 119, 9189–9199. [Google Scholar] [CrossRef]
- Vincent de Paul, N.N.; Scheiner, S. Comparison of p-hole tetrel bonding with s-hole halogen bonds in complexes of XCN (X = F, Cl, Br, I) and NH3. Phys. Chem. Chem. Phys. 2016, 18, 3581–3590. [Google Scholar] [CrossRef]
- Liu, M.; Li, Q.; Scheiner, S. Comparison of tetrel bonds in neutral and protonated complexes of pyridineTF3 and furanTF3 (T = C, Si, and Ge) with NH3. Phys. Chem. Chem. Phys. 2017, 19, 5550–5559. [Google Scholar] [CrossRef]
- Scheiner, S. Systematic Elucidation of Factors That Influence the Strength of Tetrel Bonds. J. Phys. Chem. A 2017, 121, 5561–5568. [Google Scholar] [CrossRef]
- Hadzi, D.; Bratos, S. Vibrational Spectroscopy of the hydrogen bond. In The Hydrogen Bond. Recent Developments in Theory and Experiments; Schuster, P., Zundel, G., Sandorfy, C., Eds.; North-Holland Publishing Co.: Amsterdam, The Netherlands, 1976; Volume 2, pp. 565–611. [Google Scholar]
- Gilli, G.; Gilli, P. The Nature of the Hydrogen Bond; Oxford University Press: Oxford, UK, 2009; p. 313. [Google Scholar]
- Schuster, P.; Zundel, G.; Sandorfy, C. The Hydrogen Bond. Recent Developments in Theory and Experiments; North-Holland Publishing Co.: Amsterdam, The Netherlands, 1976. [Google Scholar]
- Scheiner, S. Hydrogen Bonding: A Theoretical Perspective; Oxford University Press: New York, NY, USA, 1997; p. 375. [Google Scholar]
- Hobza, P.; Havlas, Z. Blue-shifting hydrogen bonds. Chem. Rev. 2000, 100, 4253–4264. [Google Scholar] [CrossRef]
- Masunov, A.; Dannenberg, J.J.; Contreras, R.H. C-H bond-shortening upon hydrogen bond formation: Influence of an electric field. J. Phys. Chem. A 2001, 105, 4737–4740. [Google Scholar] [CrossRef]
- Gu, Y.; Kar, T.; Scheiner, S. Fundamental properties of the CH⋯O interaction: Is it a true hydrogen bond? J. Am. Chem. Soc. 1999, 121, 9411–9422. [Google Scholar] [CrossRef]
- Qian, W.; Krimm, S. C-H⋯O and O-H⋯O hydrogen bonding in formic acid dimer structures: A QM/MM study confirms the common origin of their different spectroscopic behavior. J. Phys. Chem. A 2002, 106, 11663–11671. [Google Scholar] [CrossRef]
- Jabłoński, M. Red and blue shifted hydridic bonds. J. Comput. Chem. 2014, 35, 1739–1747. [Google Scholar] [CrossRef]
- Gu, Y.; Kar, T.; Scheiner, S. Comparison of the CH⋯N and CH⋯O interactions involving substituted alkanes. J. Mol. Struct. 2000, 552, 17–31. [Google Scholar] [CrossRef]
- Bene, J.E.D.; Alkorta, I.; Elguero, J. Properties of cationic pnicogen-bonded complexes F4-nHnP+: N-base with H–P···N linear and n = 1–4. Mol. Phys. 2016, 114, 102–117. [Google Scholar] [CrossRef]
- Ellington, T.L.; Reves, P.L.; Simms, B.L.; Wilson, J.L.; Watkins, D.L.; Tschumper, G.S.; Hammer, N.I. Quantifying the Effects of Halogen Bonding by Haloaromatic Donors on the Acceptor Pyrimidine. ChemPhysChem 2017, 18, 1267–1273. [Google Scholar] [CrossRef]
- Esrafili, M.D.; Vakili, M. The effect of hydrogen-bonding cooperativity on the strength and properties of σ-hole interactions: An ab initio study. Mol. Phys. 2017, 115, 913–924. [Google Scholar] [CrossRef]
- Gholipour, A.; Farhadi, S.; Neyband, R.S. Theoretical investigation of the nature and strength of simultaneous interactions of π–π stacking and halogen bond including NMR, SAPT, AIM and NBO analysis. Struct. Chem. 2016, 27, 1543–1551. [Google Scholar] [CrossRef]
- Cormanich, R.A.; Rittner, R.; O’Hagan, D.; Bühl, M. Inter-and intramolecular CF···C=O interactions on aliphatic and cyclohexane carbonyl derivatives. J. Comput. Chem. 2016, 37, 25–33. [Google Scholar] [CrossRef]
- Viger-Gravel, J.; Leclerc, S.; Korobkov, I.; Bryce, D.L. Correlation between 13C chemical shifts and the halogen bonding environment in a series of solid para-diiodotetrafluorobenzene complexes. CrystEngComm 2013, 15, 3168–3177. [Google Scholar] [CrossRef]
- Alkorta, I.; Sánchez-Sanz, G.; Elguero, J.; Del Bene, J.E. Influence of hydrogen bonds on the P···P pnicogen bond. J. Chem. Theory Comput. 2012, 8, 2320–2327. [Google Scholar] [CrossRef]
- Ma, N.; Zhang, Y.; Ji, B.; Tian, A.; Wang, W. Structural competition between halogen bonds and lone-pair···p interactions in solution. ChemPhysChem 2012, 13, 1411–1414. [Google Scholar] [CrossRef]
- Jooneghani, S.G.N.; Gholipour, A. Mutual cooperation of π-π stacking and pnicogen bond interactions of substituted monomeric Lawesson’s reagent and pyridine rings: Theoretical insight into Pyr||X-PhPS2⊥pyr complexes. Chem. Phys. Lett. 2019, 721, 91–98. [Google Scholar] [CrossRef]
- Watson, B.; Grounds, O.; Borley, W.; Rosokha, S.V. Resolving the halogen vs. hydrogen bonding dichotomy in solutions: Intermolecular complexes of trihalomethanes with halide and pseudohalide anions. Phys. Chem. Chem. Phys. 2018, 20, 21999–22007. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Dunning, T.H.J. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Woon, D.E.; Dunning, T.H., Jr. Gaussian basis sets for use in correlated molecular calculations. V. Core-valence basis sets for boron through neon. J. Chem. Phys. 1995, 103, 4572–4585. [Google Scholar] [CrossRef]
- Feller, D. The role of databases in support of computational chemistry calculations. J. Comput. Chem. 1996, 17, 1571–1586. [Google Scholar] [CrossRef]
- Schuchardt, K.L.; Didier, B.T.; Elsethagen, T.; Sun, L.; Gurumoorthi, V.; Chase, J.; Li, J.; Windus, T.L. Basis Set Exchange: A Community Database for Computational Sciences. J. Chem. Infor. Model. 2007, 47, 1045–1052. [Google Scholar] [CrossRef]
- Ang, S.J.; Ser, C.T.; Wong, M.W. Modeling halogen bonding with planewave density functional theory: Accuracy and challenges. J. Comput. Chem. 2019, 40, 1829–1835. [Google Scholar] [CrossRef]
- Orlova, A.P.; Jasien, P.G. Halogen bonding in self-assembling systems: A comparison of intra-and interchain binding energies. Comput. Theor. Chem. 2018, 1139, 63–69. [Google Scholar] [CrossRef]
- Forni, A.; Pieraccini, S.; Franchini, D.; Sironi, M. Assessment of DFT Functionals for QTAIM Topological Analysis of Halogen Bonds with Benzene. J. Phys. Chem. A 2016, 120, 9071–9080. [Google Scholar] [CrossRef]
- Bauzá, A.; García-Llinás, X.; Frontera, A. Charge-assisted triel bonding interactions in solid state chemistry: A combined computational and crystallographic study. Chem. Phys. Lett. 2016, 666, 73–78. [Google Scholar] [CrossRef]
- Esrafili, M.D.; Vessally, E. A theoretical evidence for cooperative enhancement in aerogen-bonding interactions: Open-chain clusters of KrOF2 and XeOF2. Chem. Phys. Lett. 2016, 662, 80–85. [Google Scholar] [CrossRef]
- Mardirossian, N.; Head-Gordon, M. How Accurate Are the Minnesota Density Functionals for Noncovalent Interactions, Isomerization Energies, Thermochemistry, and Barrier Heights Involving Molecules Composed of Main-Group Elements? J. Chem. Theory Comput. 2016, 12, 4303–4325. [Google Scholar] [CrossRef]
- Liao, M.S.; Lu, Y.; Scheiner, S. Performance assessment of density-functional methods for study of charge-transfer complexes. J. Comput. Chem. 2003, 24, 623–631. [Google Scholar] [CrossRef]
- Latajka, Z.; Scheiner, S. Primary and secondary basis set superposition error at the SCF and MP2 levels: H3N--Li+ and H2O--Li+. J. Chem. Phys. 1987, 87, 1194–1204. [Google Scholar] [CrossRef]
- Boys, S.F.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Ditchfield, R. GIAO studies of magnetic shielding in FHF- and HF. Chem. Phys. Lett. 1976, 40, 53–56. [Google Scholar] [CrossRef]
- Alkorta, I.; Elguero, J. Ab initio hybrid DFT-GIAO calculations of the shielding produced by carbon-carbon bonds and aromatic rings in 1H NMR spectroscopy. New J. Chem. 1998, 22, 381–385. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Murray, J.S.; Resnati, G.; Politzer, P. Close contacts and noncovalent interactions in crystals. Faraday Disc. 2017, 203, 113–130. [Google Scholar] [CrossRef]
- Adhikari, U.; Scheiner, S. The S⋯N noncovalent interaction: Comparison with hydrogen and halogen bonds. Chem. Phys. Lett. 2011, 514, 36–39. [Google Scholar] [CrossRef]
- Adhikari, U.; Scheiner, S. Sensitivity of pnicogen, chalcogen, halogen and H-bonds to angular distortions. Chem. Phys. Lett. 2012, 532, 31–35. [Google Scholar] [CrossRef]
- Scheiner, S. Detailed comparison of the pnicogen bond with chalcogen, halogen and hydrogen bonds. Int. J. Quantum Chem. 2013, 113, 1609–1620. [Google Scholar] [CrossRef]
- Horn, P.R.; Mao, Y.; Head-Gordon, M. Probing non-covalent interactions with a second generation energy decomposition analysis using absolutely localized molecular orbitals. Phys. Chem. Chem. Phys. 2016, 18, 23067–23079. [Google Scholar] [CrossRef]
- Schütz, M.; Brdarski, S.; Widmark, P.-O.; Lindh, R.; Karlström, G. The water dimer interaction energy: Convergence to the basis set limit at the correlated level. J. Chem. Phys. 1997, 107, 4597–4605. [Google Scholar] [CrossRef]
- Alcívar León, C.D.; Echeverría, G.A.; Piro, O.E.; Ulic, S.E.; Jios, J.L.; Burgos Paci, M.; Argüello, G.A. The role of halogen C–X1⋯X2–C contact on the preferred conformation of 2-perhalomethylchromones in solid state. Chem. Phys. 2016, 472, 142–155. [Google Scholar] [CrossRef]
- Chaudhary, P.; Goettel, J.T.; Mercier, H.P.A.; Sowlati-Hashjin, S.; Hazendonk, P.; Gerken, M. Lewis Acid Behavior of SF4: Synthesis, Characterization, and Computational Study of Adducts of SF4 with Pyridine and Pyridine Derivatives. Chem. Eur. J. 2015, 21, 6247–6256. [Google Scholar] [CrossRef]
- Hauchecorne, D.; van der Veken, B.J.; Herrebout, W.A.; Hansen, P.E. A 19F NMR study of C–I⋯p halogen bonding. Chem. Phys. 2011, 381, 5–10. [Google Scholar] [CrossRef]
- Pinheiro, P.D.S.M.; Rodrigues, D.A.; Alves, M.A.; Tinoco, L.W.; Ferreira, G.B.; de Sant’Anna, C.M.R.; Fraga, C.A.M. Theoretical and experimental characterization of 1,4-N⋯S s-hole intramolecular interactions in bioactive N-acylhydrazone derivatives. New J. Chem. 2018, 42, 497–505. [Google Scholar] [CrossRef]
- Del Bene, J.E.; Alkorta, I.; Sanchez-Sanz, G.; Elguero, J. Structures, energies, bonding, and NMR properties of pnicogen complexes H2XP:NXH2 (X = H, CH3, NH2, OH, F, Cl). J. Phys. Chem. A 2011, 115, 13724–13731. [Google Scholar] [CrossRef]
- Mokrai, R.; Barrett, J.; Apperley, D.C.; Batsanov, A.S.; Benkő, Z.; Heift, D. Weak Pnictogen Bond with Bismuth: Experimental Evidence Based on Bi–P Through-Space Coupling. Chem. Eur. J. 2019, 25, 4017–4024. [Google Scholar] [CrossRef]
- Szell, P.M.J.; Cavallo, G.; Terraneo, G.; Metrangolo, P.; Gabidullin, B.; Bryce, D.L. Comparing the Halogen Bond to the Hydrogen Bond by Solid-State NMR Spectroscopy: Anion Coordinated Dimers from 2-and 3-Iodoethynylpyridine Salts. Chem. Eur. J. 2018, 24, 11364–11376. [Google Scholar] [CrossRef]
- Cerreia Vioglio, P.; Catalano, L.; Vasylyeva, V.; Nervi, C.; Chierotti, M.R.; Resnati, G.; Gobetto, R.; Metrangolo, P. Natural Abundance 15N and 13C Solid-State NMR Chemical Shifts: High Sensitivity Probes of the Halogen Bond Geometry. Chem. Eur. J. 2016, 22, 16819–16828. [Google Scholar] [CrossRef]
- Thangavadivale, V.; Aguiar, P.M.; Jasim, N.A.; Pike, S.J.; Smith, D.A.; Whitwood, A.C.; Brammer, L.; Perutz, R.N. Self-complementary nickel halides enable multifaceted comparisons of intermolecular halogen bonds: Fluoride ligands vs. other halides. Chem. Sci. 2018, 9, 3767–3781. [Google Scholar] [CrossRef]
- Masoodi, H.R.; Bagheri, S.; Ranjbar, M. Theoretical study of cooperativity between hydrogen bond‒hydrogen bond, halogen bond–halogen bond and hydrogen bond–halogen bond in ternary FX…diazine…XF (X = H and Cl) complexes. Mol. Phys. 2016, 114, 3464–3474. [Google Scholar] [CrossRef]
Sample Availability: No compounds were synthesized. |



{kind=link}
{kind=link}
{kind=link}
| −Eint, kcal/mol | E(2), kcal/mol | Δr (AF), Å | Δν (AF), cm−1 | I Ratio a | |
|---|---|---|---|---|---|
| halogen | |||||
| FCl | 12.01 | 39.3 | 0.060 | −174.9 | 7.7 |
| FBr | 16.28 | 51.3 | 0.061 | −107.1 | 5.0 |
| FI | 19.17 | 43.6 | 0.055 | −48.6 | 2.9 |
| chalcogen | |||||
| FSH | 9.12 | 21.0 | 0.034 | −85.8 | 3.0 |
| FSeH | 12.25 | 30.9 | 0.042 | −70.3 | 2.6 |
| FTeH | 16.14 | 20.9 b | 0.042 | −44.8 | 1.9 |
| pnicogen | |||||
| FPH2 | 7.39 | 13.9 | 0.022 | −58.1 | 1.9 |
| FAsH2 | 8.98 | 16.3 | 0.029 | −47.3 | 1.8 |
| FSbH2 | 11.75 | 17.2 | 0.034 | −42.1 | 1.5 |
| tetrel | |||||
| FSiH3 | 9.01 | 18.1 | 0.027 | −74.7 | 3.4 |
| FGeH3 | 8.50 | 12.7 | 0.025 | −55.2 | 1.6 |
| FSnH3 | 12.18 | 13.3 | 0.030 | −54.9 | 1.2 |
| A | F | H a | N | |
|---|---|---|---|---|
| halogen | ||||
| FCl | 478.2 | −272.7 | - | −20.2 |
| FBr | 1674.9 | −391.2 | - | −17.6 |
| FI | 72.3 | −421.7 | - | −6.9 |
| chalcogen | ||||
| FSH | 264.0 | −184.1 | 3.8 | −11.5 |
| FSeH a | 847.5 | −234.0 | 4.9 | −12.5 |
| FTeH | 34.8 | −233.4 | 5.0 | −8.3 |
| pnicogen | ||||
| FPH2 | 68.2 | −92.0 | 1.2 | −10.6 |
| FAsH2 | 165.9 | −88.6 | 1.1 | −8.7 |
| FSbH2 | 5.0 | −88.3 | 1.0 | −12.4 |
| tetrel | ||||
| FSiH3 | 46.3 | −70.0 | 0.4 | −26.6 |
| FGeH3 | 70.7 | −54.8 | 0.3 | −16.3 |
| FSnH3 | −0.3 | −59.7 | 0.3 | −21.3 |
| A | F | H a | N | |
|---|---|---|---|---|
| halogen | ||||
| FCl | −0.076 | −0.087 | - | 0.067 |
| FBr | −0.084 | −0.099 | - | 0.066 |
| FI | −0.067 | −0.086 | - | 0.031 |
| chalcogen | ||||
| FSH | −0.044 | −0.050 | 0.005 | 0.019 |
| FSeH a | −0.058 | −0.061 | 0.004 | 0.026 |
| FTeH | −0.053 | −0.056 | 0.001 | 0.007 |
| pnicogen | ||||
| FPH2 | −0.035 | −0.029 | 0.003 | 0.003 |
| FAsH2 | −0.035 | −0.034 | 0.002 | 0.001 |
| FSbH2 | −0.036 | −0.035 | −0.001 | −0.007 |
| tetrel | ||||
| FSiH3 | −0.040 | −0.028 | −0.005 | 0.005 |
| FGeH3 | −0.016 | −0.027 | −0.005 | −0.004 |
| FSnH3 | −0.012 | −0.029 | −0.010 | −0.011 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, J.; Scheiner, S. Effects of Halogen, Chalcogen, Pnicogen, and Tetrel Bonds on IR and NMR Spectra. Molecules 2019, 24, 2822. https://doi.org/10.3390/molecules24152822
Lu J, Scheiner S. Effects of Halogen, Chalcogen, Pnicogen, and Tetrel Bonds on IR and NMR Spectra. Molecules. 2019; 24(15):2822. https://doi.org/10.3390/molecules24152822
Chicago/Turabian StyleLu, Jia, and Steve Scheiner. 2019. "Effects of Halogen, Chalcogen, Pnicogen, and Tetrel Bonds on IR and NMR Spectra" Molecules 24, no. 15: 2822. https://doi.org/10.3390/molecules24152822
APA StyleLu, J., & Scheiner, S. (2019). Effects of Halogen, Chalcogen, Pnicogen, and Tetrel Bonds on IR and NMR Spectra. Molecules, 24(15), 2822. https://doi.org/10.3390/molecules24152822