Primaquine and Chloroquine Fumardiamides as Promising Antiplasmodial Agents

 ,
,  and
and
Abstract
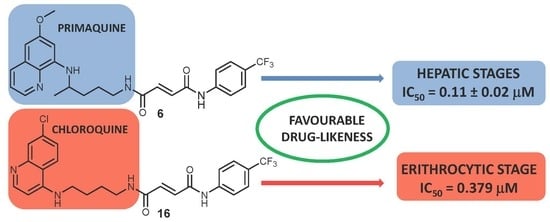
1. Introduction
2. Results and Discussion
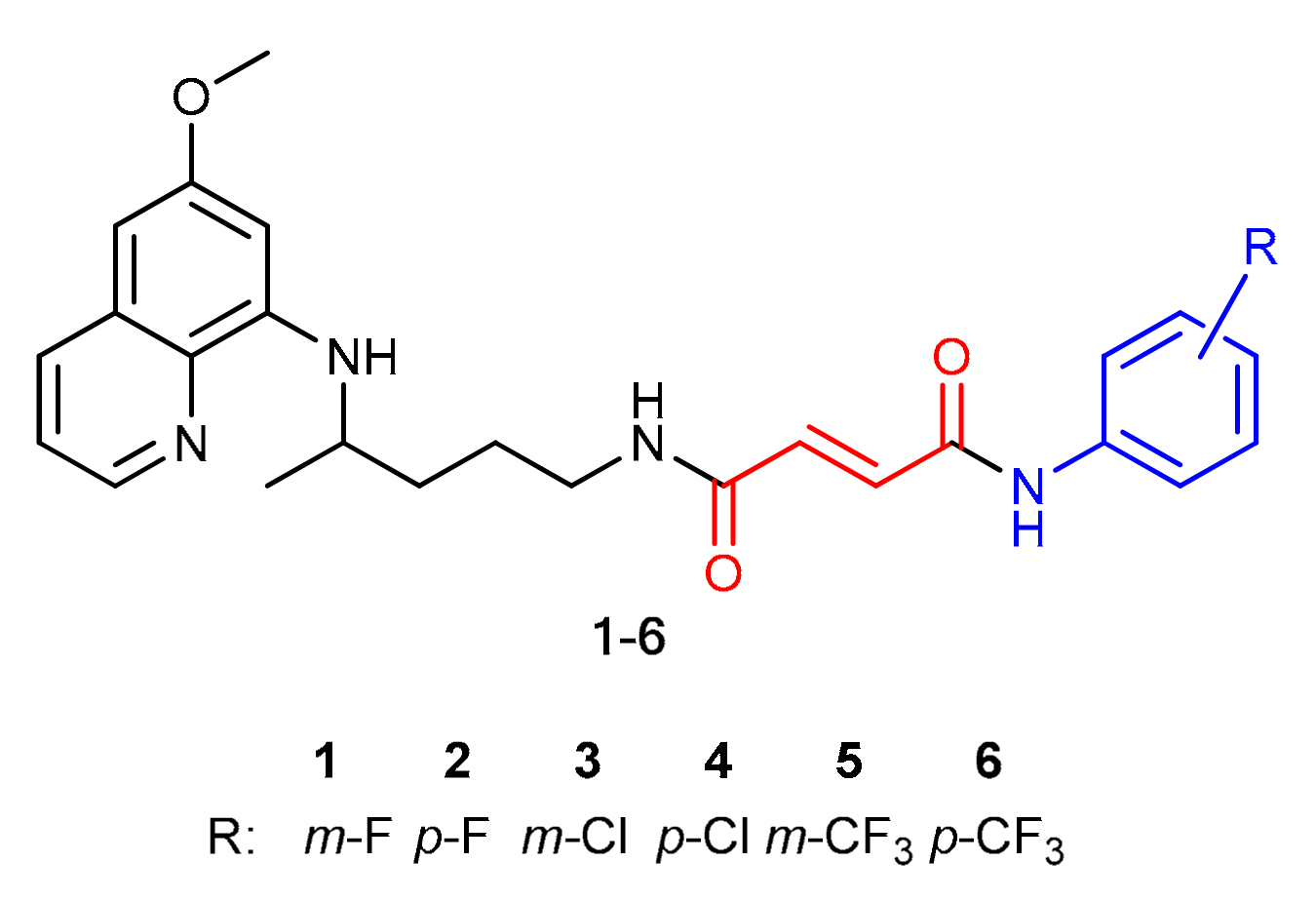
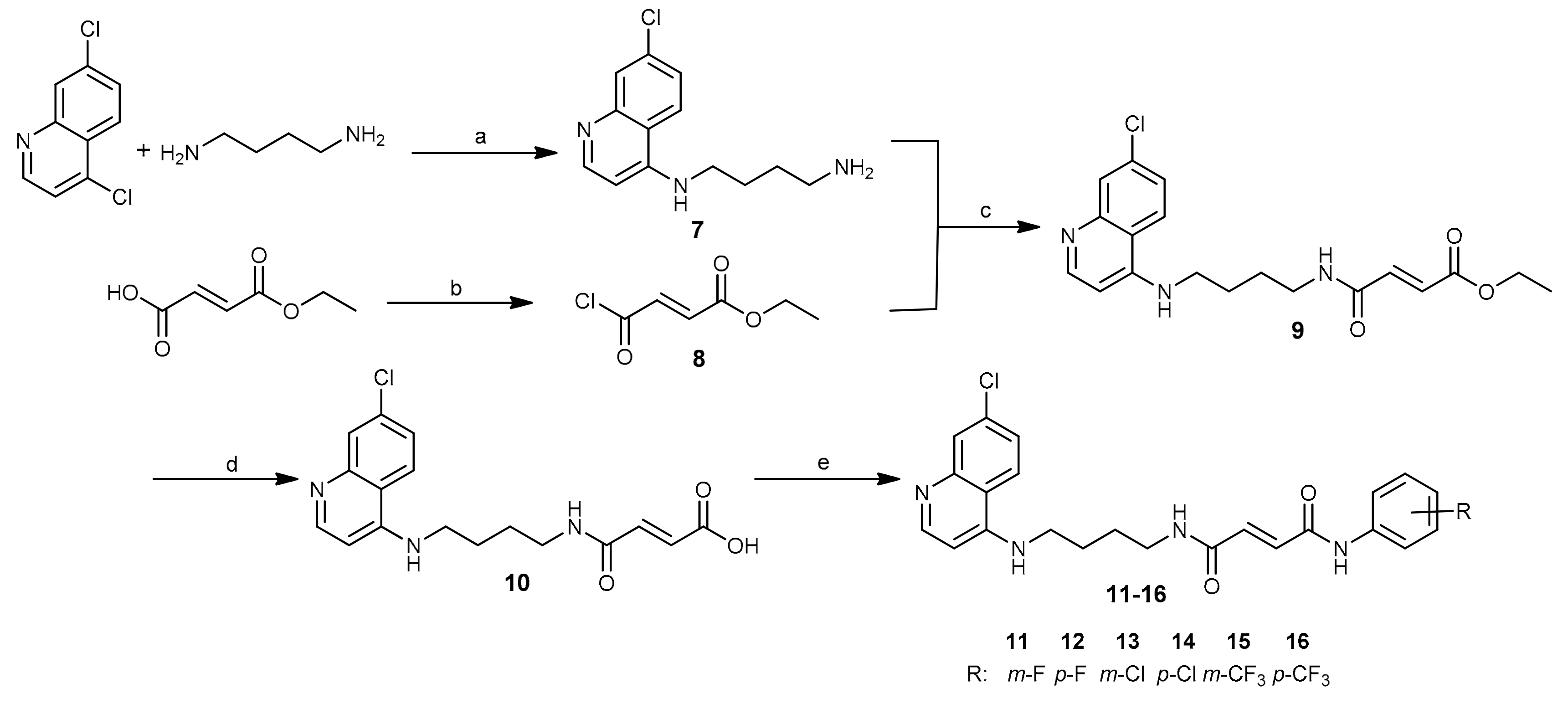
2.1. Chemistry
2.2. Antiplasmodial Activity
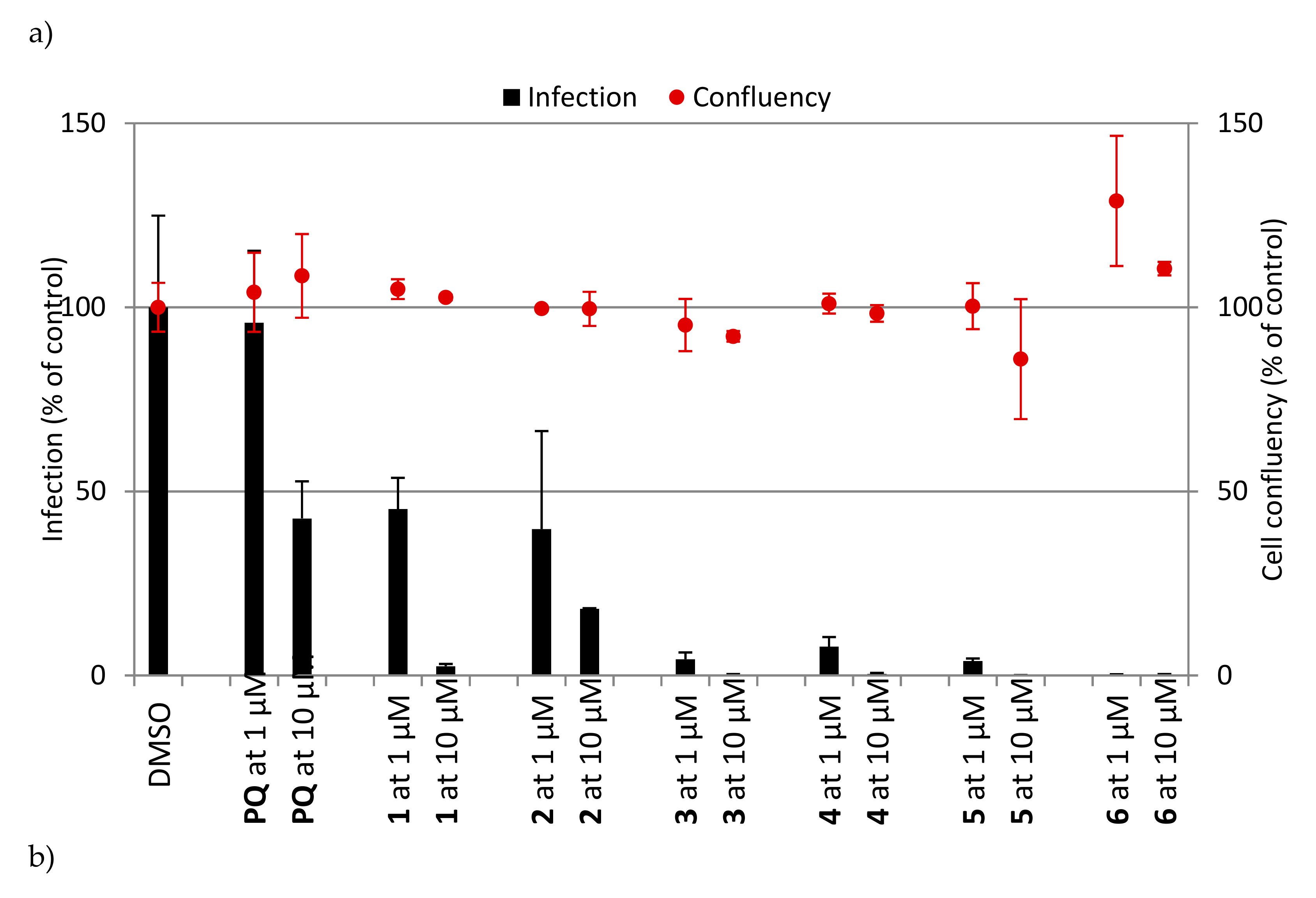
2.2.1. Erythrocytic Stages
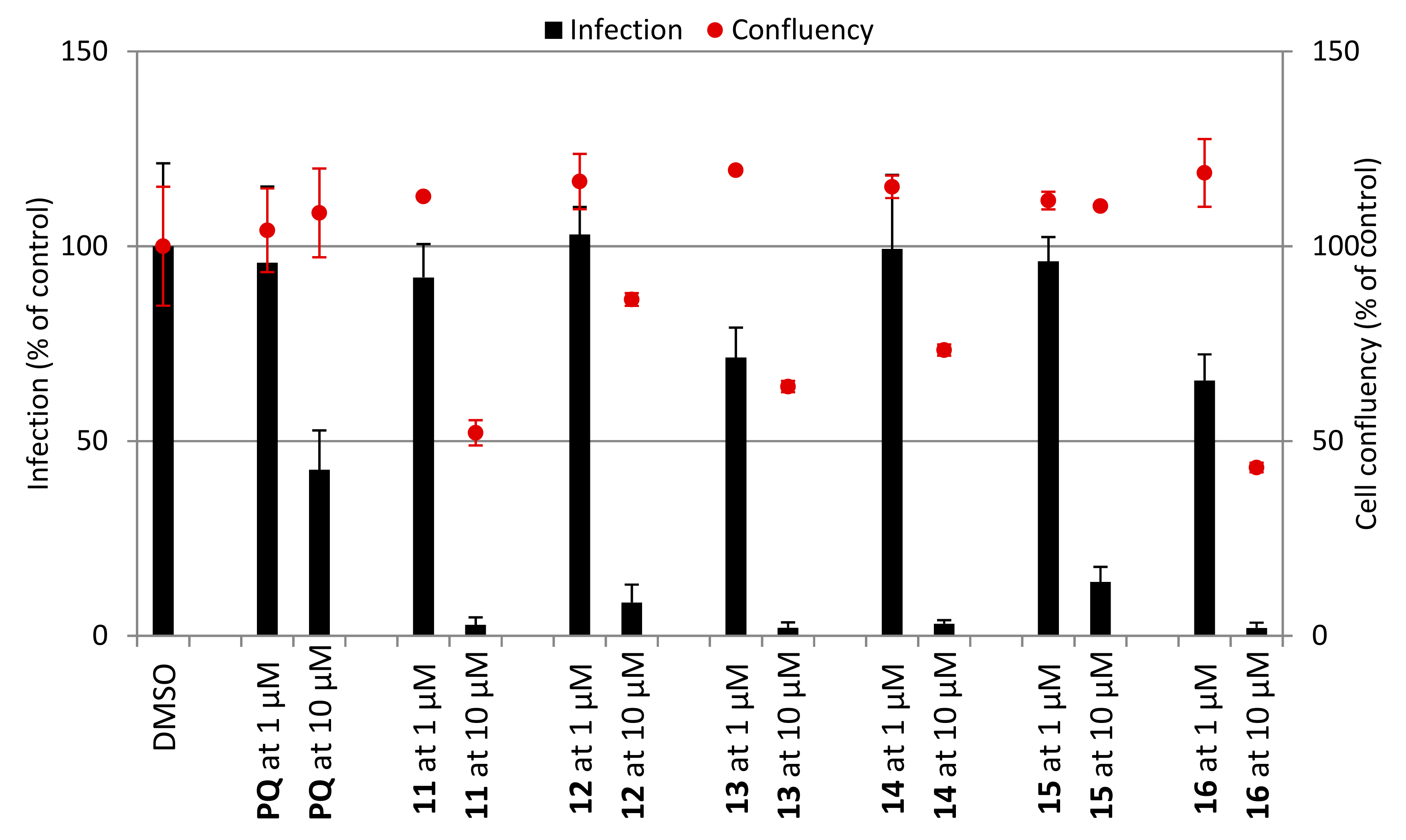
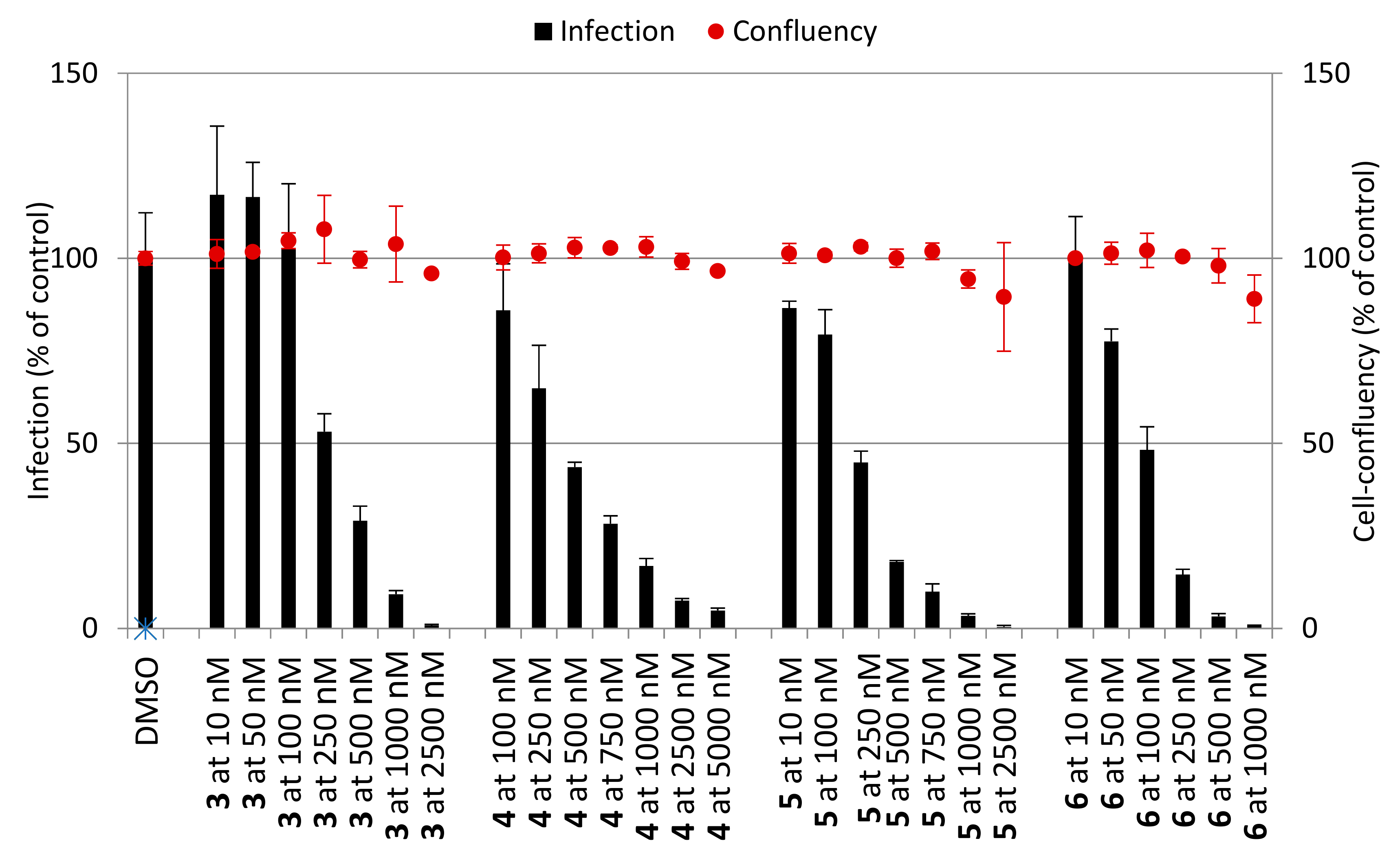
2.2.2. Hepatic Stages
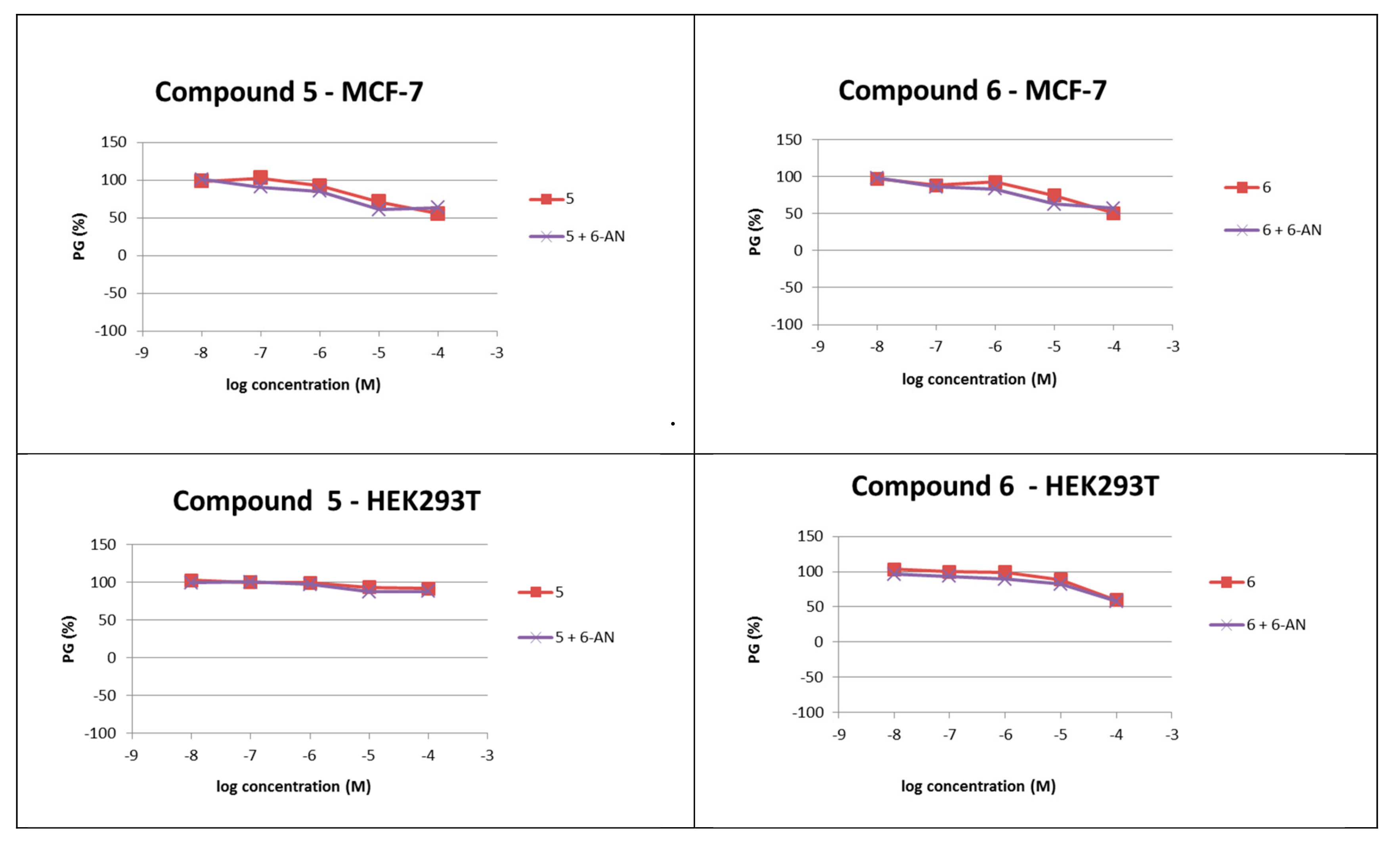
2.2.3. Cytotoxicity Assay in Human Cell Lines
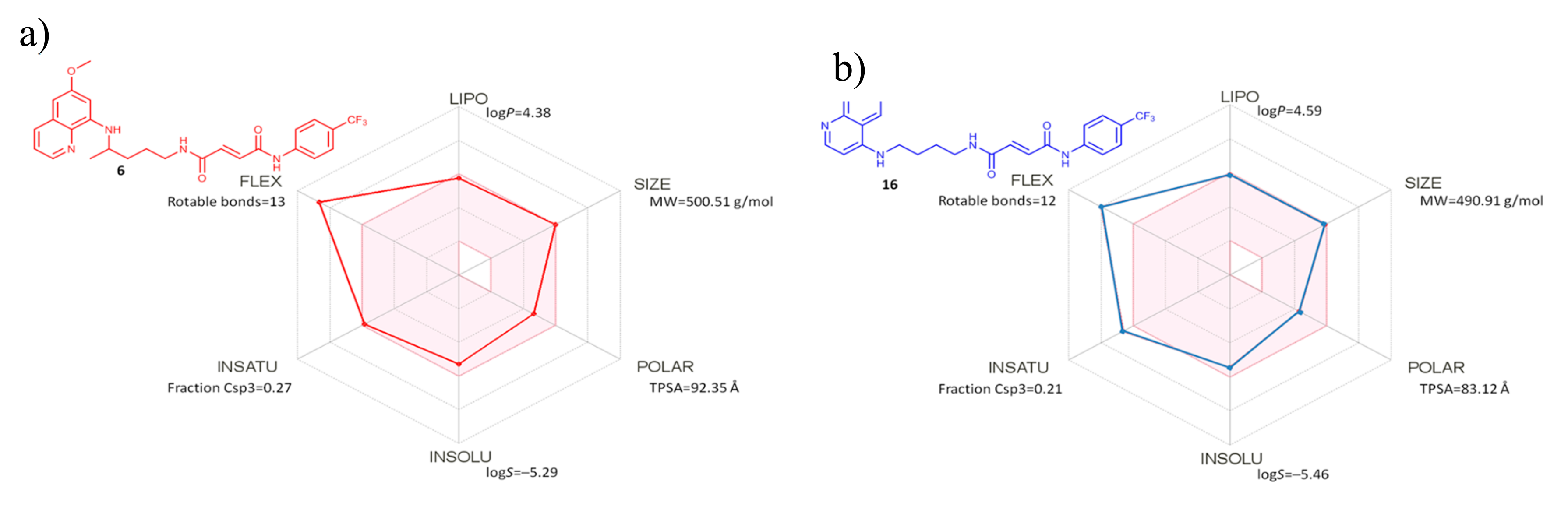
2.2.4. Evaluation of Drug-Like Properties
3. Materials and Methods
3.1. Chemistry
3.1.1. Materials and Methods
3.1.2. Procedures for the Synthesis of Compounds 7–10.
3.1.3. General Procedure for the Synthesis of Fumardiamides 11–16
3.2. In Vitro Drug Sensitivity Assay Against Erythrocytic Stages of P. falciparum
3.3. In Vitro Activity Against P. berghei Hepatic Stages
3.4. Cytotoxicity Assay in Human Cell Lines
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- World Health Organization. World Malaria Report 2018; World Health Organization: Geneva, Switzerland, 2018. [Google Scholar]
- World Health Organization. Global Technical Strategy for Malaria 2016–2030; World Health Organization: Geneva, Switzerland, 2015. [Google Scholar]
- Prudêncio, M.; Rodriguez, A.; Mota, M.M. The silent path to thousands of merozoites: The Plasmodium liver stage. Nat. Rev. Microbiol. 2006, 4, 849–856. [Google Scholar] [CrossRef] [PubMed]
- Campo, B.; Vandal, O.; Wesche, D.L.; Burrows, J.N. Killing the hypnozoite drug discovery approaches to prevent relapse in Plasmodium vivax. Pathog. Glob. Health 2015, 109, 107–122. [Google Scholar] [CrossRef] [PubMed]
- White, N.J. Determinants of relapse periodicity in Plasmodium vivax malaria. Malar J. 2011, 10, 297. [Google Scholar] [CrossRef] [PubMed]
- Vale, N.; Moreira, R.; Gomes, P. Primaquine revisited six decades after its discovery. Eur. J. Med. Chem. 2009, 44, 937–953. [Google Scholar] [CrossRef] [PubMed]
- Pybus, B.S.; Marcsisin, S.R.; Jin, X.; Deye, G.; Sousa, J.C.; Li, Q.; Caridha, D.; Zeng, Q.; Reichard, G.A.; Ockenhouse, C.; et al. The metabolism of primaquine to its active metabolite is dependent on CYP 2D6. Malar. J. 2013, 12, 212. [Google Scholar] [CrossRef]
- Leslie, T.; Rab, M.A.; Ahmadzai, H.; Durrani, N.; Fayaz, M.; Kolaczinski, J.; Rowland, M. Compliance with 14-day primaquine therapy for radical cure of vivax malaria a randomized placebo-controlled trial comparing unsupervised with supervised treatment. Trans. R. Soc. Trop. Med. Hyg. 2004, 98, 168–173. [Google Scholar] [CrossRef]
- Rajapakse, S.; Rodrigo, C.; Fernando, S.D. Tafenoquine for preventing relapse in people with Plasmodium vivax malaria. Cochrane Database Syst. Rev. 2015, 29, CD010458. [Google Scholar] [CrossRef]
- Lacerda, M.V.G.; Llanos-Cuentas, A.; Krudsood, S.; Lon, C.; Saunders, D.L.; Mohammed, R.; Yilma, D.; Pereira, D.; Espino, F.E.J.; Mia, R.Z.; et al. Single-dose tafenoquine to prevent relapse of Plasmodium vivax malaria. N. Engl. J. Med. 2019, 380, 215–228. [Google Scholar] [CrossRef]
- Llanos-Cuentas, A.; Lacerda, M.V.G.; Hien, T.T.; Vélez, I.D.; Namaik-Larp, C.; Chu, S.S.; Villegas, M.F.; Val, F.; Monteiro, W.M.; Brito, M.A.M.; et al. Tafenoquine versus primaquine to prevent relapse of Plasmodium vivax malaria. N Engl. J. Med. 2019, 380, 229–241. [Google Scholar] [CrossRef]
- Llanos-Cuentas, A.; Lacerda, M.V.G.; Rueangweerayut, R.; Krudsood, S.; Gupta, S.K.; Kochar, S.K.; Arthur, P.; Chuenchom, N.; Möhrle, J.J.; Duparc, S.; et al. Tafenoquine plus chloroquine for the treatment and relapse prevention of Plasmodium vivax malaria (DETECTIVE): A multicentre, double-blind, randomised, phase 2b dose-selection study. Lancet 2014, 383, 1049–1058. [Google Scholar] [CrossRef]
- Pavić, K.; Perković, I.; Cindrić, M.; Pranjić, M.; Martin-Kleiner, I.; Kralj, M.; Schols, D.; Hadjipavlou-Litina, D.; Katsori, A.-M.; Zorc, B. Novel semicarbazides and ureas of primaquine with bulky aryl or hydroxyalkyl substituents, Synthesis, cytostatic and antioxidative activity. Eur. J. Med. Chem. 2014, 86, 502–514. [Google Scholar]
- Perković, I.; Antunović, M.; Marijanović, I.; Pavić, K.; Ester, K.; Kralj, M.; Vlainić, J.; Kosalec, I.; Schols, D.; Hadjipavlou-Litina, D.; et al. Novel urea and bis-urea primaquine derivatives with hydroxyphenyl and halogenphenyl substituents: Synthesis and biological evaluation. Eur. J. Med. Chem. 2016, 124, 622–636. [Google Scholar] [CrossRef]
- Pavić, K.; Perković, I.; Gilja, P.; Kozlina, F.; Ester, K.; Kralj, M.; Schols, D.; Hadjipavlou-Litina, D.; Pontiki, E.; Zorc, B. Design, synthesis and biological evaluation of novel primaquine-cinnamic acid conjugates of amide and acylsemicarbazide type. Molecules 2016, 21, 1629. [Google Scholar] [CrossRef]
- Pavić, K.; Perković, I.; Pospíšilová, Š.; Machado, M.; Fontinha, D.; Prudêncio, M.; Jampilek, J.; Coffey, A.; Endersen, L.; Rimac, H.; et al. Primaquine hybrids as promising antimycobacterial and antimalarial agents. Eur. J. Med. Chem. 2018, 143, 769–779. [Google Scholar] [CrossRef]
- Vlainić, J.; Kosalec, I.; Pavić, K.; Hadjipavlou-Litina, D.; Pontiki, E.; Zorc, B. Insights into biological activity of ureidoamides with primaquine and amino acid moieties. J. Enzym. Inhib. Med. Chem. 2018, 33, 376–382. [Google Scholar] [CrossRef]
- Levatić, J.; Pavić, K.; Perković, I.; Uzelac, L.; Ester, K.; Kralj, M.; Kaiser, M.; Rottmann, M.; Supek, F.; Zorc, B. Machine learning prioritizes synthesis of primaquine ureidoamides with high antimalarial activity and attenuated cytotoxicity. Eur. J. Med. Chem. 2018, 146, 651–667. [Google Scholar] [CrossRef]
- Beus, M.; Rajić, Z.; Maysinger, D.; Mlinarić, Z.; Antunović, M.; Marijanović, I.; Fontinha, D.; Prudêncio, M.; Held, J.; Olgen, S.; et al. SAHA-primaquine hybrids (sahaquines) as potential anticancer and antimalarial compounds. Chem. Open 2018, 7, 624–638. [Google Scholar]
- Rajić, Z.; Beus, M.; Michnova, H.; Vlainić, J.; Persoons, L.; Kosalec, I.; Jampilek, J.; Schols, D.; Keser, T.; Zorc, B. Asymmetric primaquine and halogenaniline fumardiamides as novel biologically active Michael acceptors. Molecules 2018, 23, 1724. [Google Scholar] [CrossRef]
- Pérez, B.; Teixeira, C.; Albuquerque, I.S.; Gut, J.; Rosenthal, P.J.; Prudêncio, M.; Gomes, P. Primacins, N-cinnamoyl-primaquine conjugates, with improved liver-stage antimalarial activity. Med. Chem. Commun. 2012, 3, 1170–1172. [Google Scholar] [CrossRef]
- Chemicalize, 2017; ChemAxon Ltd.: Budapest, Hungary, 2017; Available online: https://chemicalize.com (accessed on 15 April 2019).
- Baell, J.B.; Holloway, G.A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef]
- Swiss ADME Programs, Swiss Institute of Bioinformatics, Lausanne, Switzerland. Available online: http://www.swissadme.ch (accessed on 10 April 2019).
- Held, J.; Gebru, T.; Kalesse, M.; Jansen, R.; Gerth, K.; Müller, R.; Mordmüller, B. Antimalarial activity of the myxobacterial macrolide chlorotonil A. Antimicrob. Agents Chemother. 2014, 58, 6378–6384. [Google Scholar] [CrossRef]
- Noedl, H.; Bronnert, J.; Yingyuen, K.; Attlmayr, B.; Kollaritsch, H.; Fukuda, M. Simple histidine-rich protein 2 double-site sandwich enzyme-linked immunosorbent assay for use in malaria drug sensitivity testing. Antimicrob. Agents Chemother. 2005, 49, 3575–3577. [Google Scholar] [CrossRef]
- R Core Team. A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018; Available online: https://www.R-project.org/ (accessed on 25 March 2019).
- Machado, M.; Sanches-Vaz, M.; Cruz, J.P.; Mendes, A.M.; Prudêncio, M. Inhibition of Plasmodium hepatic infection by antiretroviral compounds. Front. Cell. Infect. Microbiol. 2017, 7, 1–9. [Google Scholar] [CrossRef]
- Ploemen, I.H.J.; Prudêncio, M.; Douradinha, B.G.; Ramesar, J.; Fonager, J.; Gemert, G.J.; Luty, A.J.F.; Hermsen, C.C.; Sauerwein, R.W.; Baptista, F.G.; et al. Visualisation and quantitative analysis of the rodent malaria liver stage by real time imaging. PLoS ONE 2009, 4, e7881. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compd. | Structure | IC50 1 (µM) Pf3D7 | IC50 (µM) PfDd2 | IC50 (µM) P. bergei |
|---|---|---|---|---|
| 1 |  | 13.77 | 20 | n.d. 2 |
| 2 |  | 7.74 | >13 | n.d. |
| 3 |  | >13 | 13.58 | 0.27 ± 0.06 3 |
| 4 |  | >13 | >13 | 0.39 ± 0.01 |
| 5 |  | 13.91 | 10.67 | 0.27 ± 0.08 |
| 6 |  | 12.96 | 18.43 | 0.11 ± 0.02 |
| 11 |  | 0.144 | 7.024 | n.d. |
| 12 |  | 0.067 | 1.879 | n.d. |
| 13 |  | 0.083 | 6.961 | n.d. |
| 14 |  | 0.035 | 0.89 | n.d. |
| 15 |  | 0.194 | 3.217 | n.d. |
| 16 |  | 0.035 | 0.379 | n.d. |
| PQ 4 | 1.994 | 1.816 | 8.4 ± 3.4 | |
| CQ 5 | 0.0037 | 0.241 | n.d. |
| Compd. | IC50 1(µM) MCF-7 | IC50 2 (µM) HEK293T |
|---|---|---|
| 5 | ≥100 | ≥100 |
| 6 | ≥100 | ≥100 |
| 6-AN + 5 | ≥100 | ≥100 |
| 6-AN + 6 | ≥100 | ≥100 |
| 6-AN | 2 ± 0.3 | 10 ± 7 |
| Compd. | Molecular Formula | Number of Atoms | MW | log P | HBD | HBA | Lipinski Score 1 | MR (cm3/mol) | TPSA (Å2) |
|---|---|---|---|---|---|---|---|---|---|
| 11 | C23H22ClFN4O2 | 53 | 440.90 | 3.57 | 3 | 4 | 4 | 122.34 | 83.12 |
| 12 | C23H22ClFN4O2 | 53 | 440.90 | 3.57 | 3 | 4 | 4 | 122.34 | 83.12 |
| 13 | C23H22Cl2N4O2 | 53 | 457.36 | 4.03 | 3 | 4 | 4 | 126.93 | 83.12 |
| 14 | C23H22Cl2N4O2 | 53 | 457.36 | 4.03 | 3 | 4 | 4 | 126.93 | 83.12 |
| 15 | C24H22ClF3N4O2 | 56 | 490.91 | 4.30 | 3 | 4 | 4 | 128.10 | 83.12 |
| 16 | C24H22ClF3N4O2 | 56 | 490.91 | 4.30 | 3 | 4 | 4 | 128.10 | 83.12 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beus, M.; Fontinha, D.; Held, J.; Rajić, Z.; Uzelac, L.; Kralj, M.; Prudêncio, M.; Zorc, B. Primaquine and Chloroquine Fumardiamides as Promising Antiplasmodial Agents. Molecules 2019, 24, 2812. https://doi.org/10.3390/molecules24152812
Beus M, Fontinha D, Held J, Rajić Z, Uzelac L, Kralj M, Prudêncio M, Zorc B. Primaquine and Chloroquine Fumardiamides as Promising Antiplasmodial Agents. Molecules. 2019; 24(15):2812. https://doi.org/10.3390/molecules24152812
Chicago/Turabian StyleBeus, Maja, Diana Fontinha, Jana Held, Zrinka Rajić, Lidija Uzelac, Marijeta Kralj, Miguel Prudêncio, and Branka Zorc. 2019. "Primaquine and Chloroquine Fumardiamides as Promising Antiplasmodial Agents" Molecules 24, no. 15: 2812. https://doi.org/10.3390/molecules24152812
APA StyleBeus, M., Fontinha, D., Held, J., Rajić, Z., Uzelac, L., Kralj, M., Prudêncio, M., & Zorc, B. (2019). Primaquine and Chloroquine Fumardiamides as Promising Antiplasmodial Agents. Molecules, 24(15), 2812. https://doi.org/10.3390/molecules24152812