
Enantioselective Protonation of Radical Anion Intermediates in Photoallylation and Photoreduction Reactions of 3,3-Diaryl-1,1-dicyano-2-methylprop-1-ene with Allyltrimethylsilane


Abstract

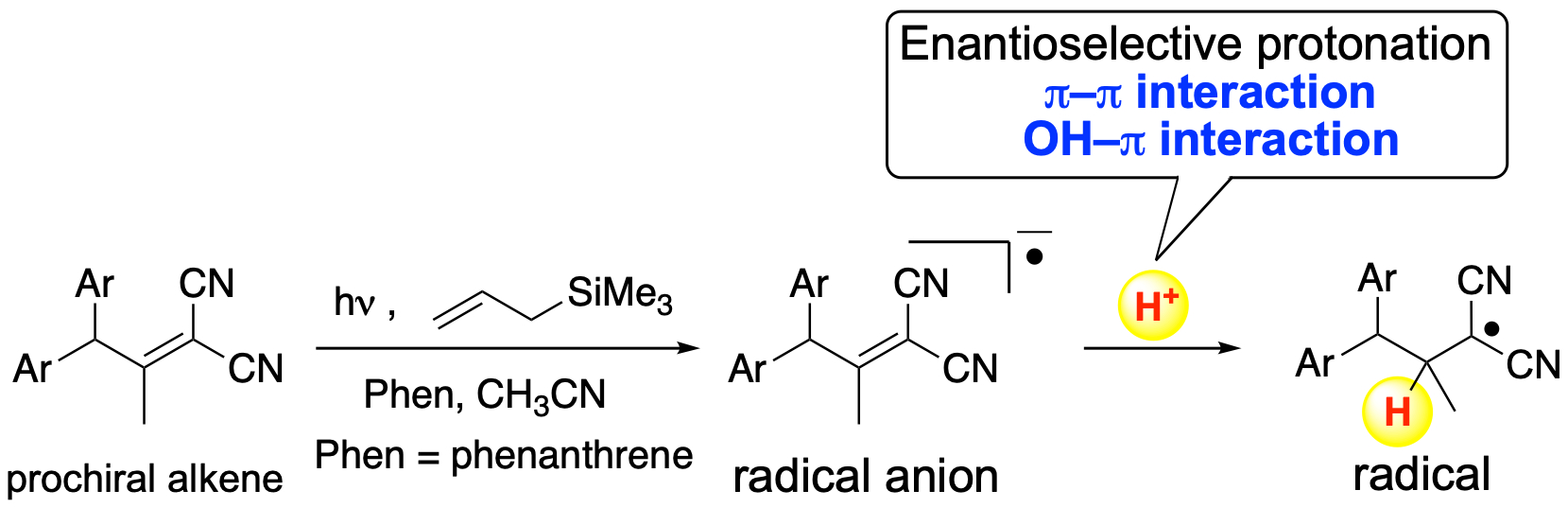
1. Introduction
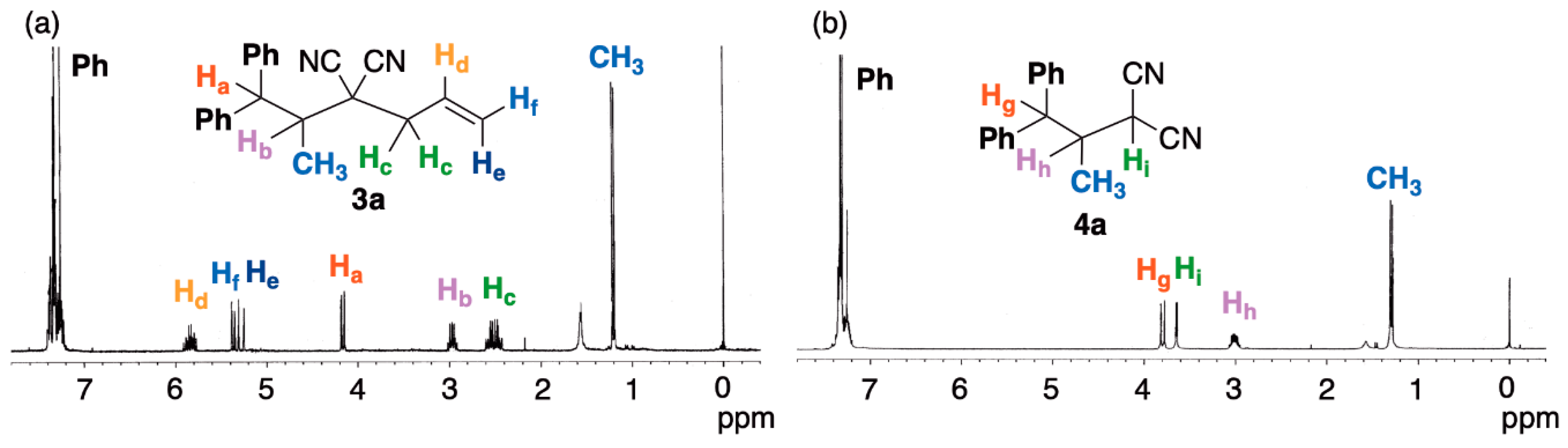
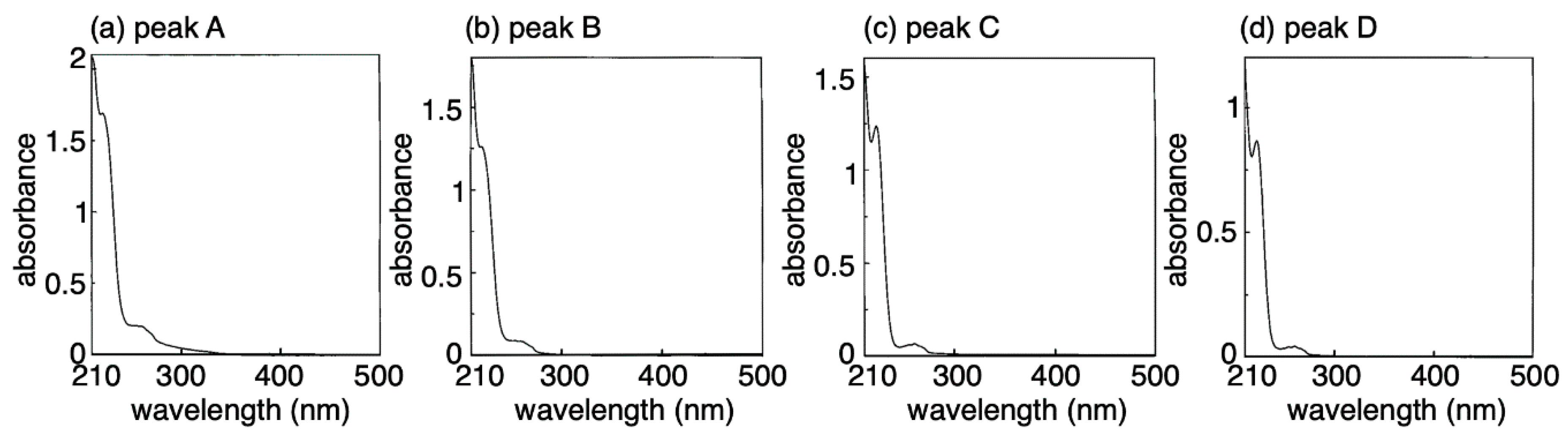
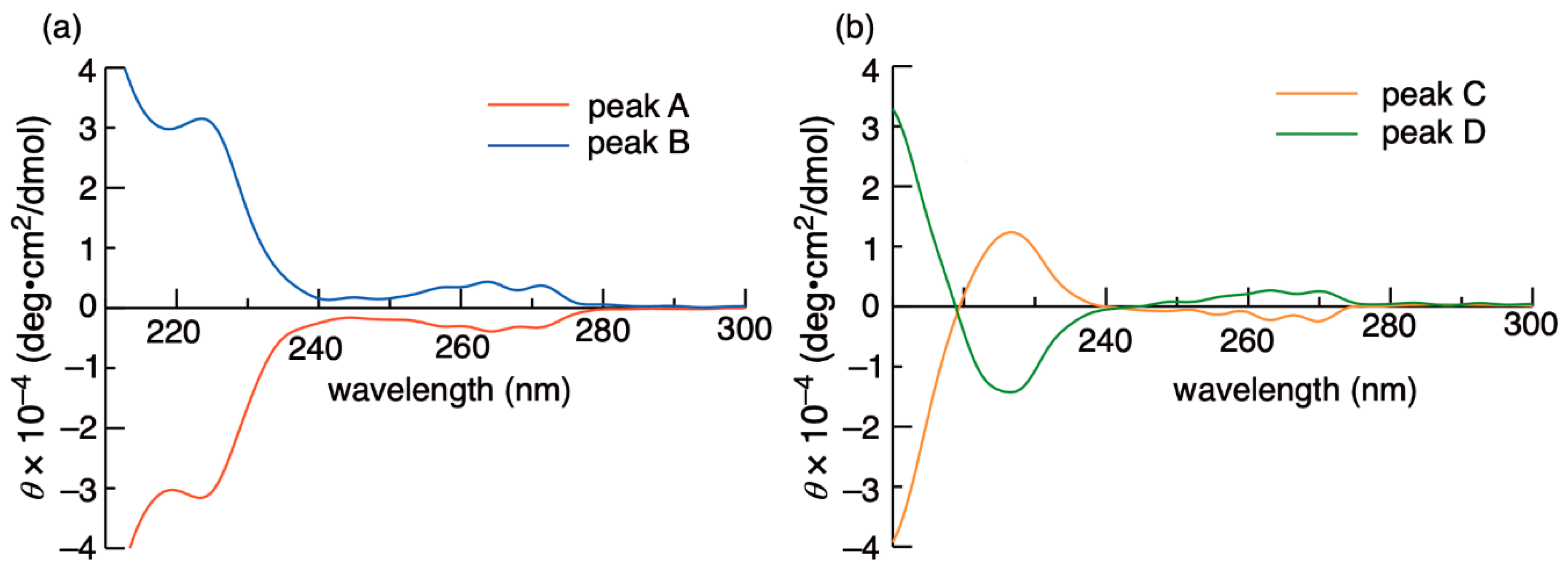
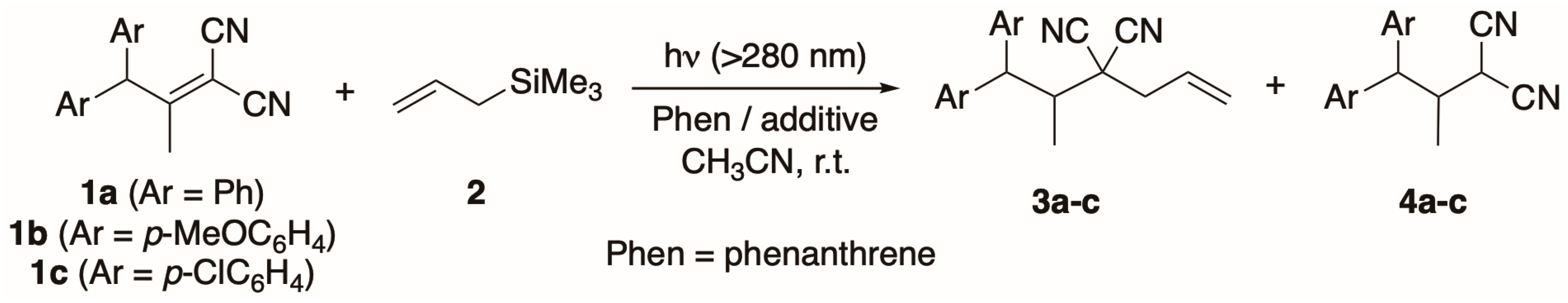
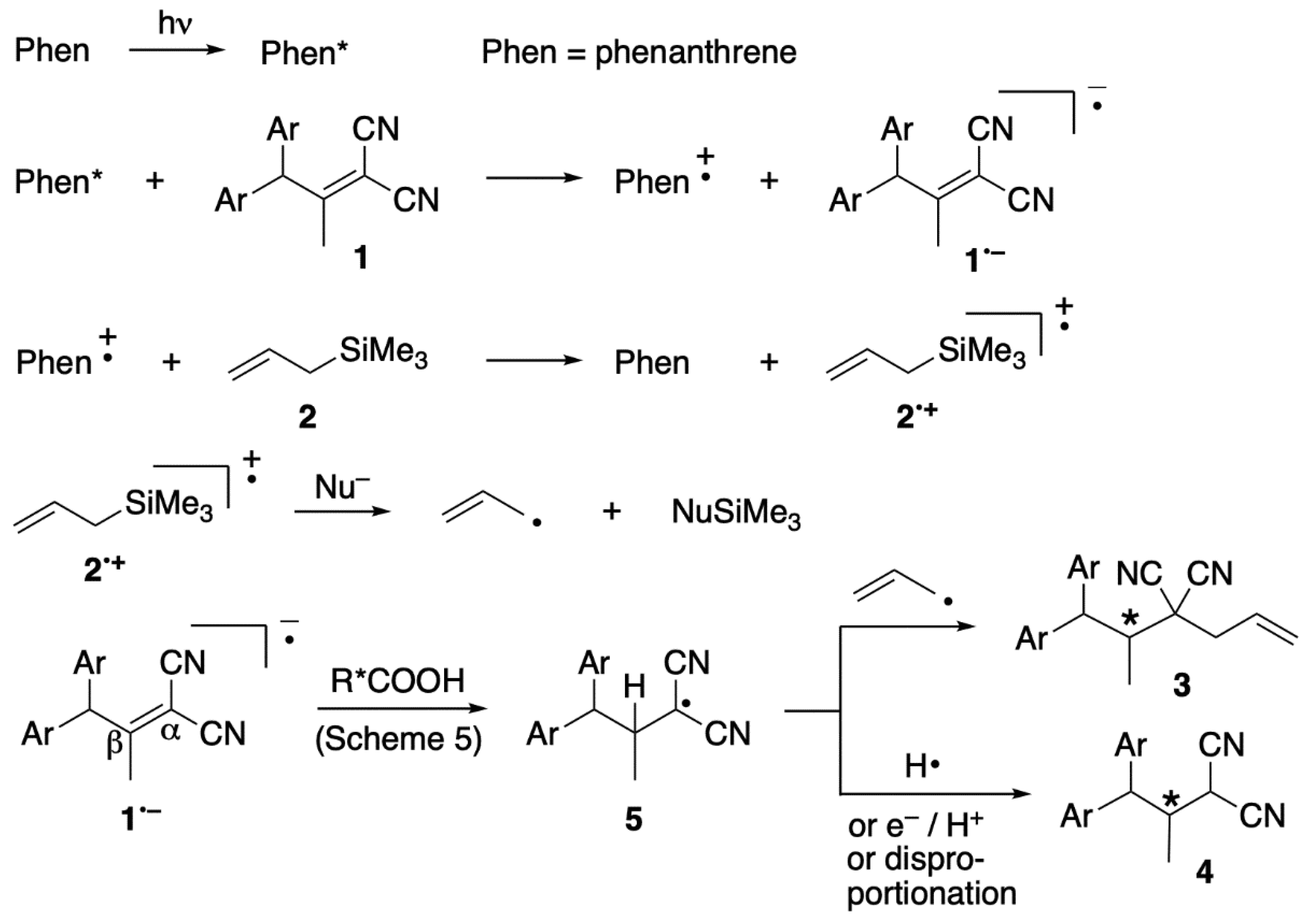
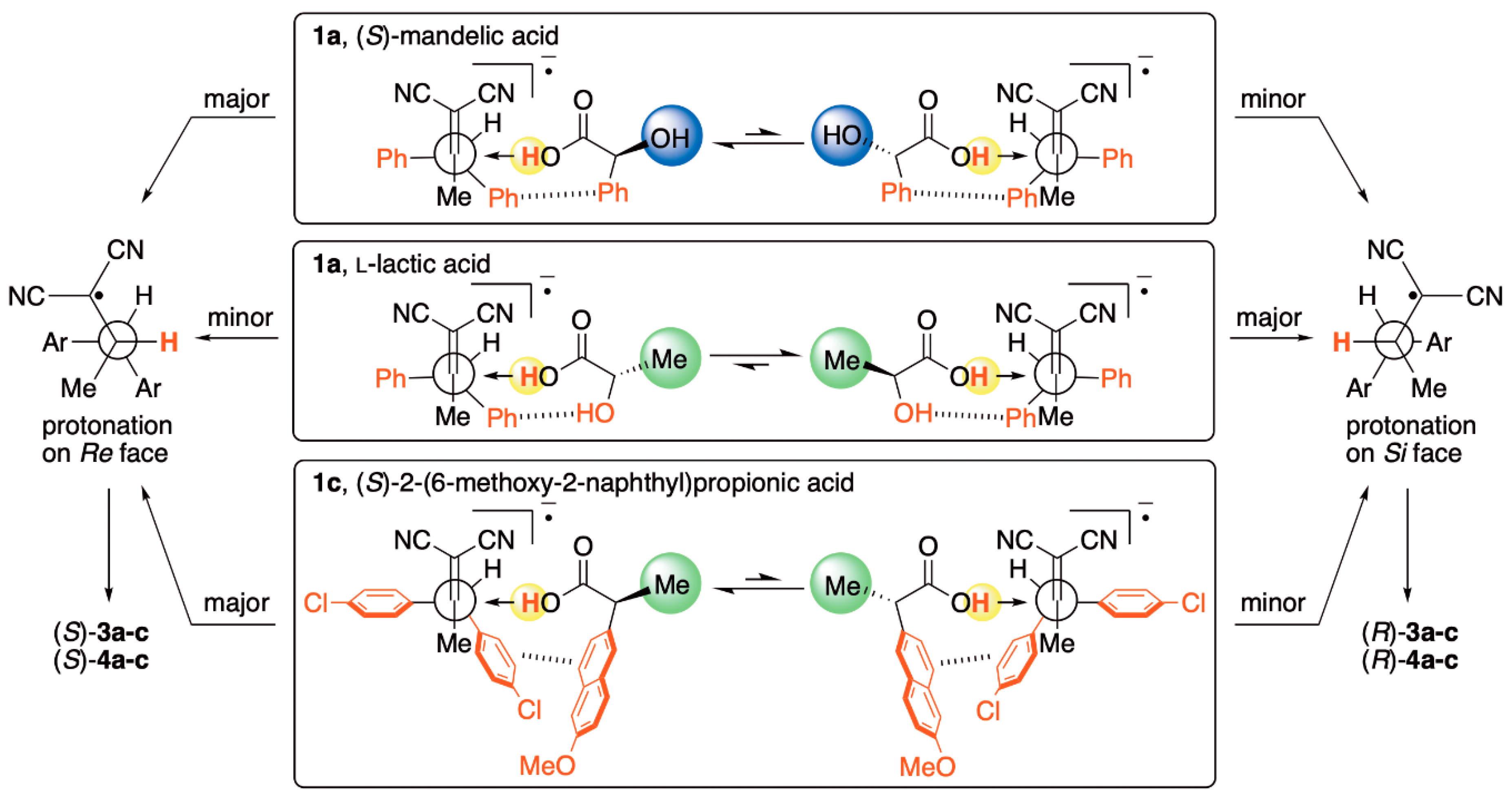
2. Results and Discussion
3. Experimental Section
3.1. Materials and Equipments
3.2. Preparation of 1a
3.3. Preparation of 1b
3.4. Preparation of 1c
3.5. General Procedure for Photoreactions
3.6. Resolution of Enantiomers
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Neunteufel, R.A.; Arnold, D.R. Radical Ions in Photochemistry. I. The 1, 1-Diphenylethylene Cation Radical. J. Am. Chem. Soc. 1973, 95, 4080–4081. [Google Scholar] [CrossRef]
- Maroulis, A.J.; Shigemitsu, Y.; Arnold, D.R. Radical Ions in Photochemistry. 5. Photosensitized (Electron Transfer) Cyanation of Olefins. J. Am. Chem. Soc. 1978, 100, 535–541. [Google Scholar] [CrossRef]
- Yasuda, M.; Yamashita, T.; Matsumoto, T.; Shima, K.; Pac, C. Synthetic Application of Photochemical Electron Transfer to Direct Amination of Arenes by Ammonia and Primary Amines. J. Org. Chem. 1985, 50, 3667–3669. [Google Scholar] [CrossRef]
- Klett, M.W.; Johnson, R.P. Photogeneration of Triphenyl C3 Radical Cations: Deprotonation and Nucleophilic Addition as Competitive Pathways. J. Am. Chem. Soc. 1985, 107, 6615–6620. [Google Scholar] [CrossRef]
- Kavarnos, G.J.; Turro, N.J. Photosensitization by Reversible Electron Transfer: Theories, Experimental Evidence, and Examples. Chem. Rev. 1986, 86, 401–449. [Google Scholar] [CrossRef]
- Yasuda, M.; Yamashita, T.; Shima, K.; Pac, C. Direct Photoamination of Arenes with Ammonia and Primary Amines in the Presence of Electron Acceptors. J. Org. Chem. 1987, 52, 753–759. [Google Scholar] [CrossRef]
- Mattay, J. Charge Transfer and Radical Ions in Photochemistry. Angew. Chem. Int. Ed. Engl. 1987, 26, 825–845. [Google Scholar] [CrossRef]
- Hasegawa, E.; Xu, W.; Mariano, P.S.; Yoon, U.C.; Kim, J.U. Electron-Transfer-Induced Photoadditions of the Silyl Amine Et2NCH2TMS to α, β-Unsaturated Cyclohexenones. Dual Reaction Pathways Based on Ion-Pair-Selective Cation-Radical Chemistry. J. Am. Chem. Soc. 1988, 110, 8099–8111. [Google Scholar] [CrossRef]
- Yoon, U.C.; Mariano, P.S. Mechanistic and Synthetic Aspects of Amine-Enone Single Electron Transfer Photochemistry. Acc. Chem. Res. 1992, 25, 233–240. [Google Scholar] [CrossRef]
- Mangion, D.; Arnold, D.R. Photochemical Nucleophile-Olefin Combination, Aromatic Substitution Reaction. Its Synthetic Development and Mechanistic Exploration. Acc. Chem. Res. 2002, 35, 297–304. [Google Scholar] [CrossRef]
- Ohashi, M.; Nakatani, K.; Maeda, H.; Mizuno, K. Photochemical Monoalkylation of Propanedinitrile by Electron-Rich Alkenes. Org. Lett. 2008, 10, 2741–2743. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, M.; Nakatani, K.; Maeda, H.; Mizuno, K. Photoinduced Tandem Three-Component Coupling of Propanedinitrile, 2,5-Dimethylhexa-2,4-diene, and Cyanoarenes. J. Org. Chem. 2008, 73, 8348–8351. [Google Scholar] [CrossRef] [PubMed]
- Yoshimi, Y. Photoinduced electron transfer-promoted decarboxylative radical reactions of aliphatic carboxylic acids by organic photoredox system. J. Photochem. Photobiol. A Chem. 2017, 342, 116–130. [Google Scholar] [CrossRef]
- Maeda, H.; Takayama, H.; Segi, M. Photoinduced three-component coupling reactions of electron deficient alkenes, dienes and active methylene compounds. Photochem. Photobiol. Sci. 2018, 17, 1118–1126. [Google Scholar] [CrossRef] [PubMed]
- Pandey, G.; Reddy, G.D.; Chakrabarti, D. Stereoselectivity in the photoinduced electron transfer (PET) promoted intramolecular cyclisations of 1-alkenyl-2-silyl-piperidines and -pyrrolidines: Rapid construction of 1-azabicyclo [m.n.0] alkenes and stereoselective synthesis of (±)-isoretronecanol and (±)-epilupinine. J. Chem. Soc. Perkin Trans. 1 1996, 219–224. [Google Scholar] [CrossRef]
- Heinemann, C.; Demuth, M. Short Biomimetic Synthesis of a Steroid by Photoinduced Electron Transfer and Remote Asymmetric Induction. J. Am. Chem. Soc. 1999, 121, 4894–4895. [Google Scholar] [CrossRef]
- Asaoka, S.; Kitazawa, T.; Wada, T.; Inoue, Y. Enantiodifferentiating Anti-Markovnikov Photoaddition of Alcohols to 1,1-Diphenylalkenes Sensitized by Chiral Naphthalenecarboxylates. J. Am. Chem. Soc. 1999, 121, 8486–8498. [Google Scholar] [CrossRef]
- Bertrand, S.; Hoffmann, N.; Humbel, S.; Pete, J.P. Diastereoselective Tandem Addition-Cyclization Reactions of Unsaturated Tertiary Amines Initiated by Photochemical Electron Transfer (PET). J. Org. Chem. 2000, 65, 8690–8703. [Google Scholar] [CrossRef]
- Asaoka, S.; Wada, T.; Inoue, Y. Microenvironmental Polarity Control of Electron-Transfer Photochirogenesis. Enantiodifferentiating Polar Addition of 1, 1-Diphenyl-1-alkenes Photosensitized by Saccharide Naphthalenecarboxylates. J. Am. Chem. Soc. 2003, 125, 3008–3027. [Google Scholar] [CrossRef]
- Bauer, A.; Westkämper, F.; Grimme, S.; Bach, T. Catalytic enantioselective reactions driven by photoinduced electron transfer. Nature 2005, 436, 1139–1140. [Google Scholar] [CrossRef]
- Ma, N.; Shi, W.; Zhang, R.; Zhu, Z.; Jiang, Z. Study on improved diastereoselectivity in photo-induced electron transfer pinacol coupling reactions of substituted acetophenones. Tetrahedron Lett. 2011, 52, 718–720. [Google Scholar] [CrossRef]
- Mizuno, K.; Ikeda, M.; Otsuji, Y. Dual Regioselectivity in the Photoallylation of Electron-Deficient Alkenes by Allylic Silanes. Chem. Lett. 1988, 17, 1507–1510. [Google Scholar] [CrossRef]
- Hayamizu, T.; Ikeda, M.; Maeda, H.; Mizuno, K. Photoallylation and Photoreduction of Cyclohexylidenepropanedinitrile by Use of Allyltrimethylsilane via Photoinduced Electron Transfer: Control of the Product Ratio Depending on pKa Values of Additives. Org. Lett. 2001, 3, 1277–1280. [Google Scholar] [CrossRef]
- Mizuno, K.; Hayamizu, T.; Maeda, H. Regio- and stereoselective functionalization of electron-deficient alkenes by organosilicon compounds via photoinduced electron transfer. Pure Appl. Chem. 2003, 75, 1049–1054. [Google Scholar] [CrossRef]
- Hayamizu, T.; Maeda, H.; Ikeda, M.; Mizuno, K. Highly stereoselective carbon-functionalization of electron-deficient arylalkenes by use of organosilicon compounds via photoinduced electron transfer. Tetrahedron Lett. 2001, 42, 2361–2364. [Google Scholar] [CrossRef]
- Hayamizu, T.; Maeda, H.; Mizuno, K. Diastereoselective Protonation on Radical Anions of Electron-Deficient Alkenes via Photoinduced Electron Transfer. J. Org. Chem. 2004, 69, 4997–5004. [Google Scholar] [CrossRef] [PubMed]
- Maeda, H.; Nishitsuji, N.; Mizuno, K. Diastereoselective protonation on a radical anion in the photoallylation and photoreduction of 1, 1-dicyano-2-methyl-3-phenyl-1-butene by allyltrimethylsilane. Res. Chem. Intermed. 2010, 36, 577–585. [Google Scholar] [CrossRef]
- Dinnocenzo, J.P.; Farid, S.; Goodman, J.L.; Gould, I.R.; Todd, W.P.; Mattes, S.L. Nucleophile-Assisted Cleavage of Silane Cation Radicals. J. Am. Chem. Soc. 1989, 111, 8973–8975. [Google Scholar] [CrossRef]
- Dockery, K.P.; Dinnocenzo, J.P.; Farid, S.; Goodman, J.L.; Gould, I.R.; Todd, W.P. Nucleophile-Assisted Cleavage of Benzyltrialkylsilane Cation Radicals. J. Am. Chem. Soc. 1997, 119, 1876–1883. [Google Scholar] [CrossRef]
- de Lijser, H.J.P.; Snelgrove, D.W.; Dinnocenzo, J.P. Nucleophilic Substitutions on Silane Cation Radicals: Stepwise or Concerted? J. Am. Chem. Soc. 2001, 123, 9698–9699. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Nishii, S.; Ibuka, T. Acyclic Stereocontrol via an Electron-Transfer Process. Remarkable Stereochemical Difference between One- and Two-Electron Events. J. Am. Chem. Soc. 1988, 110, 617–618. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Nishii, S.; Ibuka, T. Diastereofacial Selection in the Conjugate Reduction of γ-Alkyl-α, β-Unsaturated Carbonyl Derivatives. Stereocontrol Dictated by Aromatic Ring-Pd Interaction. J. Chem. Soc. Perkin Trans. 1 1989, 1703–1705. [Google Scholar] [CrossRef]
- Paddon-Row, M.N.; Rondan, N.G.; Houk, K.N. Staggered Models for Asymmetric Induction: Attack Trajectories and Conformations of Allylic Bonds from ab Initio Transition Structures of Addition Reactions. J. Am. Chem. Soc. 1982, 104, 7162–7166. [Google Scholar] [CrossRef]
- Texier-Boullet, F.; Foucaud, A. Knoevenagel condensation catalysed by aluminum oxide. Tetrahedron Lett. 1982, 23, 4927–4928. [Google Scholar] [CrossRef]
- Kozik, B.; Wilamowski, J.; Góra, M.; Sepiol, J.J. Synthesis of α-arylnaphthalenes from diphenylacetaldehydes and 1, 1-diphenylacetones. Tetrahedron 2008, 64, 6452–6460. [Google Scholar] [CrossRef]
- Henze, H.R.; Leslie, W.B. Synthesis of 5-Benzohydryl-5-Substituted Hydantoins. J. Org. Chem. 1950, 15, 901–907. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Substrate | Additive | Irradiation Time/h | Yields/% | |
|---|---|---|---|---|---|
| 3 | 4 | ||||
| 1 | 1a (Ar = Ph) | none | 4 | 0 b | 53 b |
| 2 | 1a (Ar = Ph) | acetic acid c | 4 | 34 b | 31 b |
| 3 | 1b (Ar = p-MeOC6H4) | none | 24 | 0 d | 44 d |
| 4 | 1b (Ar = p-MeOC6H4) | acetic acid c | 24 | 20 d | 46 d |
| 5 | 1c (p-ClC6H4) | none | 2 | 0 d | 36 d |
| 6 | 1c (p-ClC6H4) | acetic acid c | 2 | 33 d | 47 d |
| Entry | Substrate | Additive | Irradiation Time/h | Yields/% (ee/%) | ||
|---|---|---|---|---|---|---|
| 3 | 4 | |||||
| 1 | 1a (Ar = Ph) | none | 4 | 0 b | 53 b | |
| 2 | 1a (Ar = Ph) |  | acetic acidc | 4 | 34 b | 31 b |
| 3 | 1a (Ar = Ph) |  | (R)-mandelic acid | 4 | 34 b (+1.5 d,e) | 27 b (+4.1 d,e) |
| 4 | 1a (Ar = Ph) |  | (S)-mandelic acid | 4 | 22 b (−3.4 d,f) | 26 b (−4.8 d,f) |
| 5 | 1a (Ar = Ph) |  | l-lactic acid | 4 | 28b (+0.6 d,e) | 39 b (+3.2 d,e) |
| 6 | 1a (Ar = Ph) |  | dibenzoyl l-tartaric acid | 4 | 31 b (−2.0 d,f) | 16 b (−3.6 d,f) |
| 7 | 1b (Ar = p-MeOC6H4) | none | 24 | 0 g | 44 g | |
| 8 | 1b (Ar = p-MeOC6H4) | | acetic acid c | 24 | 20 g | 46 g |
| 9 | 1b (Ar = p-MeOC6H4) | | (R)-mandelic acid | 24 | 54 g (nd h) | 28g (nd h) |
| 10 | 1b (Ar = p-MeOC6H4) | | (S)-mandelic acid | 24 | 13 g (nd h) | 48g (nd h) |
| 11 | 1c (p-ClC6H4) | none | 2 | 0 g | 36 g | |
| 12 | 1c (p-ClC6H4) | | acetic acid c | 2 | 33 g | 47 g |
| 13 | 1c (p-ClC6H4) | | (R)-mandelic acid | 2 | 27 g (+2.0 i,j) | 55 g (+2.0 i,j) |
| 14 | 1c (p-ClC6H4) | | (S)-mandelic acid | 2 | 24 g (−2.6 i,k) | 61 g (−0.6 i,k) |
| 15 | 1c (p-ClC6H4) |  | (S)-2-(6-methoxy-2-naphthyl)propionic acid | 2 | 0 g | 68g (−3.5 i,k) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maeda, H.; Iida, M.; Ogawa, D.; Mizuno, K. Enantioselective Protonation of Radical Anion Intermediates in Photoallylation and Photoreduction Reactions of 3,3-Diaryl-1,1-dicyano-2-methylprop-1-ene with Allyltrimethylsilane. Molecules 2019, 24, 2677. https://doi.org/10.3390/molecules24142677
Maeda H, Iida M, Ogawa D, Mizuno K. Enantioselective Protonation of Radical Anion Intermediates in Photoallylation and Photoreduction Reactions of 3,3-Diaryl-1,1-dicyano-2-methylprop-1-ene with Allyltrimethylsilane. Molecules. 2019; 24(14):2677. https://doi.org/10.3390/molecules24142677
Chicago/Turabian StyleMaeda, Hajime, Masayuki Iida, Daisuke Ogawa, and Kazuhiko Mizuno. 2019. "Enantioselective Protonation of Radical Anion Intermediates in Photoallylation and Photoreduction Reactions of 3,3-Diaryl-1,1-dicyano-2-methylprop-1-ene with Allyltrimethylsilane" Molecules 24, no. 14: 2677. https://doi.org/10.3390/molecules24142677
APA StyleMaeda, H., Iida, M., Ogawa, D., & Mizuno, K. (2019). Enantioselective Protonation of Radical Anion Intermediates in Photoallylation and Photoreduction Reactions of 3,3-Diaryl-1,1-dicyano-2-methylprop-1-ene with Allyltrimethylsilane. Molecules, 24(14), 2677. https://doi.org/10.3390/molecules24142677