
Development and Validation of an HPLC-ESI/MS/MS Method for the Determination of Amoxicillin, Its Major Metabolites, and Ampicillin Residues in Chicken Tissues

Abstract
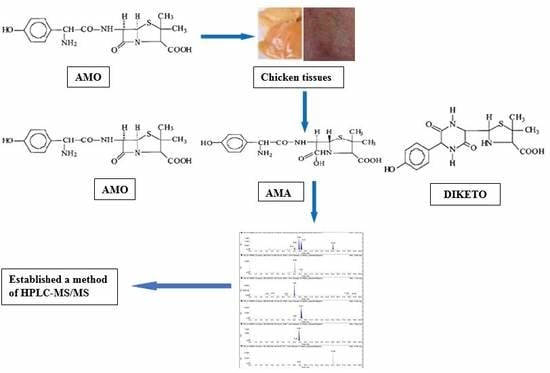
1. Introduction
2. Results and Discussion
2.1. Extraction Conditions
2.2. Mass Spectrometry
2.3. Chromatography
2.4. Method Validation
2.4.1. Specificity and Sensitivity
2.4.2. Linearity and Matrix Effects
2.4.3. Precision and Accuracy
2.4.4. CCα, CCβ, LOD, and LOQ
2.4.5. Stability
2.5. Comparison of HPLC- ESI/MS/MS and UPLC-MS/MS
2.6. Application of the Method
3. Materials and Methods
3.1. Ethics Statement
3.2. Experimental Animals
3.3. Apparatus
3.4. HPLC-MS/MS Instrumentation and Conditions
3.5. Sample Preparation
3.6. Method Validation
3.6.1. Specificity and Sensitivity
3.6.2. Linearity and Matrix Effects
3.6.3. Precision and Accuracy
3.6.4. CCα, CCβ, LOD, and LOQ
3.6.5. Stability Study
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- JECFA (Joint FAO/WHO Expert Committee on Food Additives). Residue Evaluation of Certain Veterinary Drugs (Seventy-Fifth Report of the Joint FAO/WHO Expert Committee on Food Additives) FAO JECFA Monographs 12; Food and Agriculture Organization of the United Nations: Rome, Italy, 2012. [Google Scholar]
- Chesa-Jimenez, J.; Peris, J.E.; Torres-Molina, F.; Granero, L. Low bioavailability of amoxicillin in rats as a consequence of presystemic degradation in the intestine. Antimicrob. Agents Chemother. 1994, 38, 842–847. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nagele, E.; Moritz, R. Structure elucidation of degradation products of the antibiotic amoxicillin with ion trap MS(n) and accurate mass determination by ESI TOF. J. Am. Soc. Mass Spectrom. 2005, 16, 1670–1676. [Google Scholar] [CrossRef] [PubMed]
- Anfossi, P.; Zaghini, A.; Grassigli, G.; Menotta, S.; Fedrizzi, G. Relative oral bioavailability of microgranulated amoxicillin in pigs. J. Vet. Pharmacol. Ther. 2002, 25, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Reyns, T.; Cherlet, M.; De Baere, S.; De Backer, P.; Croubels, S. Rapid method for the quantification of amoxicillin and its major metabolites in pig tissues by liquid chromatography-tandem mass spectrometry with emphasis on stability issues. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2008, 861, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Commission of the European Communities. List of exhibitors. Eur. Radiol. 1999, 9, 16–17. [Google Scholar] [CrossRef]
- Commission of the European Communities. Diario Oficial de las Comunidades Europeas No. 37/2010 L15/1. Off. J. Eur. Commun. L. 2009, 60, 16. [Google Scholar]
- Uddin, M.N.; Das, S.; Khan, S.H.; Shill, S.K.; Bhuiyan, H.R.; Karim, R. Simultaneous determination of amoxicillin and chloramphenicol and their drug interaction study by the validated UPLC method. J. Taibah Univ. Sci. 2016, 10, 755–765. [Google Scholar] [CrossRef]
- Batrawi, N.; Wahdan, S.; Al-Rimawi, F. A validated stability-indicating HPLC method for simultaneous determination of amoxicillin and enrofloxacin combination in an injectable suspension. Sci. Pharm. 2017, 85, 6. [Google Scholar] [CrossRef] [PubMed]
- Dousa, M.; Hosmanova, R. Rapid determination of amoxicillin in premixes by HPLC. J. Pharm. Biomed. Anal. 2005, 37, 373–377. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, F.A.; Nasr, J.J.M. Direct determination of ampicillin and amoxicillin residues in food samples after aqueous SDS extraction by micellar liquid chromatography with UV detection. Anal. Methods 2014, 6, 1523–1529. [Google Scholar] [CrossRef]
- Gamba, V.; Dusi, G. Liquid chromatography with fluorescence detection of amoxicillin and ampicillin in feeds using pre-column derivatization. Anal. Chim. Acta 2003, 483, 69–72. [Google Scholar] [CrossRef]
- Xie, K.; Jia, L.; Xu, D.; Guo, H.; Xie, X.; Huang, Y.; Chen, X.; Bao, W.; Dai, G.; Wang, J. Simultaneous determination of amoxicillin and ampicillin in eggs by reversed-phase high-performance liquid chromatography with fluorescence detection using pre-column derivatization. J. Chromatogr. Sci. 2012, 50, 620–624. [Google Scholar] [CrossRef] [PubMed]
- De Baere, S.; Cherlet, M.; Baert, K.; De Baere, P. Quantitative analysis of amoxycillin and its major metabolites in animal tissues by liquid chromatography combined with electrospray ionization tandem mass spectrometry. Anal. Chem. 2002, 74, 1393–1401. [Google Scholar] [CrossRef] [PubMed]
- Lugoboni, B.; Gazzotti, T.; Zironi, E.; Barbarossa, A.; Pagliuca, G. Development and validation of a liquid chromatography/tandem mass spectrometry method for quantitative determination of amoxicillin in bovine muscle. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2011, 879, 1980–1986. [Google Scholar] [CrossRef] [PubMed]
- Freitas, A.; Barbosa, J.; Ramos, F. Determination of amoxicillin stability in chicken meat by liquid chromatography–tandem mass spectrometry. Food Anal. Methods 2012, 5, 471–479. [Google Scholar] [CrossRef]
- Colin, P.; De Bock, L.; T’Jollyn, H.; Boussery, K.; Van Bocxlaer, J. Development and validation of a fast and uniform approach to quantify beta-lactam antibiotics in human plasma by solid phase extraction-liquid chromatography-electrospray-tandem mass spectrometry. Talanta 2013, 103, 285–293. [Google Scholar] [CrossRef]
- Sun, L.; Jia, L.; Xie, X.; Xie, K.; Wang, J.; Liu, J.; Cui, L.; Zhang, G.; Dai, G.; Wang, J. Quantitative analysis of amoxicillin, its major metabolites and ampicillin in eggs by liquid chromatography combined with electrospray ionization tandem mass spectrometry. Food Chem. 2016, 192, 313–318. [Google Scholar] [CrossRef]
- Jank, L.; Martins, M.T.; Arsand, J.B.; Motta, T.M.C.; Feijó, T.C.; dos Santos Castilhos, T.; Hoff, R.B.; Barreto, F.; Pizzolato, T.M. Liquid chromatography–tandem mass spectrometry multiclass method for 46 antibiotics residues in milk and meat: Development and validation. Food Anal. Methods 2017, 10, 2152–2164. [Google Scholar] [CrossRef]
- Liu, C.; Wang, H.; Jiang, Y.; Du, Z. Rapid and simultaneous determination of amoxicillin, penicillin G, and their major metabolites in bovine milk by ultra-high-performance liquid chromatography-tandem mass spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2011, 879, 533–540. [Google Scholar] [CrossRef]
- Liu, Y.; Zhu, K.; Wang, J.; Huang, X.; Wang, G.; Li, C.; Cao, J.; Ding, S. Simultaneous detection and comparative pharmacokinetics of amoxicillin, clavulanic acid and prednisolone in cows’ milk by UPLC-MS/MS. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2016, 1008, 74–80. [Google Scholar] [CrossRef]
- Wang, B.; Pang, M.; Xie, X.; Zhao, M.; Xie, K.; Zhang, Y.; Zhao, X.; Wang, Y.J.; Wang, R.; Wu, H.; et al. Quantitative analysis of amoxicillin, amoxicillin major metabolites, and ampicillin in chicken tissues via ultra-performance liquid chromatography-electrospray ionization tandem mass spectrometry. Food Anal. Methods 2017, 10, 3292–3305. [Google Scholar] [CrossRef]
- Directive, I.C. European Union Commission decision of 2002/657/EC implementing council directive 96/23/EC concerning the performance of analytical methods and the interpretation of results (2002/657/EC). Off. J. Eur. Commun. L. 2002, 221, 18–36. [Google Scholar]
- Food and Drug Administration; Center for Drug Evaluation and Research; Center for Veterinary Medicine; United States Department of Health and Human Services. Guidance for Industry: Bioanalytical Method Validation; Department of Health and Human Services: Washington, DC, USA, 2001.
- Straub, R.F.; Voyksner, R.D. Determination of penicillin G, ampicillin, amoxicillin, cloxacillin and cephapirin by high-performance liquid chromatography-electrospray mass spectrometry. J. Chromatogr. 1993, 647, 167–181. [Google Scholar] [CrossRef]
- De Baere, S.; Wassink, P.; Croubels, S.; Boever, S.D.; Baert, K.; Backer, P.D. Quantitative liquid chromatographic–mass spectrometric analysis of amoxycillin in broiler edible tissues. Anal. Chim. Acta 2005, 529, 221–227. [Google Scholar] [CrossRef]
- Franski, R.; Czerniel, J.; Kowalska, M.; Franska, M. Electrospray ionization collision-induced dissociation tandem mass spectrometry of amoxicillin and ampicillin and their degradation products. Rapid Commun. Mass Spectrom. 2014, 28, 713–722. [Google Scholar] [CrossRef] [PubMed]
- Tyczkowska, K.L.; Voyksner, R.D.; Straub, R.F.; Aronson, A.L. Simultaneous multiresidue analysis of beta-lactam antibiotics in bovine milk by liquid chromatography with ultraviolet detection and confirmation by electrospray mass spectrometry. J. AOAC Int. 1994, 77, 1122–1131. [Google Scholar] [PubMed]
- Bogialli, S.; Capitolino, V.; Curini, R.; Di Corcia, A.; Nazzari, M.; Sergi, M. Simple and rapid liquid chromatography−tandem mass spectrometry confirmatory assay for determining amoxicillin and ampicillin in bovine tissues and milk. J. Agric. Food Chem. 2004, 52, 3286–3291. [Google Scholar] [CrossRef] [PubMed]
- Larsen, E.H.; Ludwigsen, M.B. Determination of iodine in food-related certified reference materials using wet ashing and detection by inductively coupled plasma mass spectrometry. J. Anal. At. Spectrom. 1997, 12, 435–439. [Google Scholar] [CrossRef]
Sample Availability: Not available. |




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chicken Tissues | Extraction Agent | Recovery (%) (n = 6) | |||
|---|---|---|---|---|---|
| AMO | AMA | DIKETO | AMP | ||
| Muscle | 10 mL acetonitrile-water (80/20 v/v) | 85.14 ± 8.23 | 83.23 ± 5.81 | 90.32 ± 6.43 | 95.23 ± 9.13 |
| Liver | 86.18 ± 9.04 | 82.65 ± 6.31 | 95.42 ± 8.76 | 86.12 ± 8.54 | |
| Kidney | 83.06 ± 6.32 | 88.42 ± 7.86 | 93.02 ± 7.73 | 88.13 ± 10.32 | |
| Muscle | 12 mL acetonitrile-water (80/40 v/v) | 95.62 ± 10.32 | 92.49 ± 7.48 | 94.43 ± 6.98 | 99.32 ± 10.83 |
| Liver | 91.55 ± 11.19 | 81.21 ± 6.01 | 92.58 ± 10.08 | 85.08 ± 9.04 | |
| Kidney | 94.36 ± 9.56 | 93.29 ± 11.83 | 98.39 ± 10.33 | 87.32 ± 10.81 | |
| Muscle | 20 mL acetonitrile-water (90/10 v/v) | 80.31 ± 9.48 | 80.98 ± 7.83 | 85.76 ± 7.43 | 90.65 ± 10.05 |
| Liver | 82.38 ± 10.54 | 78.65 ± 8.93 | 88.34 ± 8.93 | 80.98 ± 10.38 | |
| Kidney | 79.54 ± 7.89 | 83.43 ± 8.76 | 89.87 ± 9.98 | 83.78 ± 9.36 | |
| Retention Time (min) | Analyte | Precursor Ions (m/z) | Product Ions (m/z) | Declustering Potential (V) | Collision Energy (eV) |
|---|---|---|---|---|---|
| 8.06 | AMO | 366.4 | 114.0 * 208.0 160.0 | 50 | 29 19 29 |
| 7.95 | AMA | 384.4 | 323.1 * 189.0 160.0 | 45 | 19 29 34 |
| 9.25 | DIKETO | 366.4 | 160.1 * 114.1 207.1 | 52 | 22 52 18 |
| 8.92 | AMP | 350.4 | 106.1 * 192.1 160.1 | 50 | 22 23 18 |
| 15.12 | PV | 351.5 | 160.1 * 114.1 192.2 | 50 | 19 46 15 |
| Matrix | Analyte | Linearity | Determination Coefficient (r2) | Linearity Range (μg/kg) | LOD (μg/kg) | LOQ (μg/kg) |
|---|---|---|---|---|---|---|
| Muscle | AMO | y = 0.6622x + 0.0080 | 0.9999 | 2.08~2000 | 0.52 | 2.08 |
| AMA | y = 0.4318x − 0.0086 | 0.9999 | 4.10~10000 | 1.04 | 4.10 | |
| DIKETO | y = 1.8081x + 0.2443 | 0.9989 | 0.45~2000 | 0.15 | 0.45 | |
| AMP | y = 2.5286x + 0.2507 | 0.9968 | 0.30~1000 | 0.10 | 0.30 | |
| Liver | AMO | y = 0.6474x + 0.0575 | 0.9998 | 3.60~2000 | 0.85 | 3.60 |
| AMA | y = 0.5496x − 0.0139 | 0.9999 | 6.40~5000 | 1.65 | 6.40 | |
| DIKETO | y = 2.3948x + 0.0657 | 0.9999 | 0.90~2000 | 0.30 | 0.90 | |
| AMP | y = 3.3512x + 0.0286 | 0.9999 | 0.60~1000 | 0.20 | 0.60 | |
| Kidney | AMO | y = 0.6128x + 0.0124 | 0.9999 | 4.50~2000 | 1.20 | 4.50 |
| AMA | y = 0.4512x + 0.0127 | 0.9997 | 8.50~5000 | 2.20 | 8.50 | |
| DIKETO | y = 1.9630x + 0.1911 | 0.9994 | 1.38~2000 | 0.46 | 1.38 | |
| AMP | y = 2.8912x + 0.0601 | 0.9999 | 0.90~1000 | 0.30 | 0.90 |
| Analyte | Matrix | Muscle | Liver | Kidney | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Added Levels (μg/kg) | 25 | 50 | 100 | 25 | 50 | 100 | 25 | 50 | 100 | |
| AMO | Matrix effect (%) | −39 | −22 | −34 | −30 | −28 | −32 | −43 | −32 | −38 |
| AMA | −42 | −34 | −38 | −36 | −35 | −39 | −41 | −39 | −43 | |
| DIKETO | +25 | +42 | +20 | +28 | +38 | +25 | +18 | +33 | +26 | |
| AMP | +21 | +45 | +22 | +19 | +34 | +26 | +25 | +40 | +28 | |
| Analyte | Spiked Concentration (μg/kg) | Recovery (%) (n = 6) | RSD (%) (n = 6) | Intra-day RSD (%) (n = 6) | Inter-day RSD (%) (n = 18) |
|---|---|---|---|---|---|
| AMO | 2.08 | 81.25 ± 10.62 | 13.07 | 6.98 | 10.23 |
| 25.00 | 106.32 ± 3.11 | 2.93 | 5.11 | 6.32 | |
| 50.00 a | 96.24 ± 9.72 | 10.10 | 11.09 | 12.06 | |
| 100.00 | 90.83 ± 7.88 | 8.68 | 10.08 | 12.66 | |
| AMA | 4.10 | 79.62 ± 9.65 | 12.12 | 11.74 | 14.96 |
| 25.00 | 90.49 ± 6.49 | 7.18 | 8.65 | 10.39 | |
| 50.00 | 94.77 ± 8.24 | 8.69 | 11.00 | 13.67 | |
| 100.00 | 93.19 ± 9.19 | 9.86 | 12.07 | 15.39 | |
| DIKETO | 0.45 | 82.53 ± 10.23 | 12.40 | 13.25 | 14.68 |
| 25.00 | 103.46 ± 5.54 | 5.36 | 4.20 | 11.42 | |
| 50.00 | 95.22 ± 7.77 | 8.16 | 9.53 | 10.35 | |
| 100 | 104.50 ± 9.67 | 9.25 | 13.73 | 15.33 | |
| AMP | 0.30 | 85.69 ± 10.28 | 12.00 | 14.85 | 14.58 |
| 25.00 | 107.62 ± 6.02 | 5.59 | 2.95 | 3.21 | |
| 50.00 a | 107.33 ± 11.07 | 10.31 | 11.32 | 9.73 | |
| 100.00 | 104.75 ± 10.10 | 9.64 | 14.16 | 15.00 |
| Analyte | Spiked Concentration (μg/kg) | Recovery (%) (n = 6) | RSD (%) (n = 6) | Intra-day RSD (%) (n = 6) | Inter-day RSD (%) (n = 18) |
|---|---|---|---|---|---|
| AMO | 3.60 | 75.20 ± 11.83 | 15.73 | 16.54 | 18.56 |
| 25.00 | 97.24 ± 12.72 | 13.08 | 9.93 | 8.97 | |
| 50.00 a | 93.43 ± 12.82 | 13.72 | 5.89 | 7.32 | |
| 100.00 | 92.93 ± 10.19 | 10.97 | 7.93 | 7.64 | |
| AMA | 6.40 | 80.55 ± 13.20 | 16.39 | 15.62 | 16.33 |
| 25.00 | 97.70 ± 11.26 | 11.53 | 11.44 | 10.42 | |
| 50.00 | 83.09 ± 5.90 | 7.10 | 3.09 | 6.41 | |
| 100.00 | 90.29 ± 13.34 | 14.77 | 5.01 | 8.27 | |
| DIKETO | 0.90 | 84.60 ± 12.10 | 14.30 | 14.98 | 16.25 |
| 25.00 | 101.01 ± 14.76 | 14.62 | 7.55 | 11.84 | |
| 50.00 | 93.52 ± 10.49 | 11.21 | 9.24 | 10.45 | |
| 100.00 | 99.66 ± 7.88 | 7.91 | 7.11 | 7.60 | |
| AMP | 0.60 | 79.65 ± 10.88 | 13.66 | 14.05 | 16.54 |
| 25.00 | 99.48 ± 6.03 | 6.06 | 9.07 | 14.30 | |
| 50.00 a | 86.09 ± 8.10 | 9.40 | 8.79 | 10.77 | |
| 100.00 | 100.00 ± 10.91 | 10.91 | 8.15 | 8.47 |
| Analyte | Spiked Concentration (μg/kg) | Recovery (%) (n = 6) | RSD (%) (n = 6) | Intra-day RSD (%) (n = 6) | Inter-day RSD (%) (n = 18) |
|---|---|---|---|---|---|
| AMO | 4.50 | 83.24 ± 11.25 | 13.52 | 13.87 | 14.23 |
| 25.00 | 92.42 ± 10.74 | 11.62 | 6.36 | 11.95 | |
| 50.00 a | 95.10 ± 9.31 | 9.79 | 10.58 | 9.29 | |
| 100.00 | 102.87 ± 10.01 | 9.73 | 4.18 | 9.18 | |
| AMA | 8.50 | 85.67 ± 11.98 | 13.98 | 14.52 | 15.30 |
| 25.00 | 95.09 ± 10.18 | 10.70 | 5.96 | 13.66 | |
| 50.00 | 95.80 ± 12.71 | 13.26 | 5.22 | 10.32 | |
| 100.00 | 103.56 ± 9.52 | 9.19 | 3.52 | 6.63 | |
| DIKETO | 1.38 | 79.85 ± 10.65 | 13.34 | 14.66 | 12.68 |
| 25.00 | 99.91 ± 9.03 | 9.04 | 6.98 | 9.96 | |
| 50.00 | 100.57 ± 9.19 | 9.14 | 8.24 | 6.38 | |
| 100.00 | 101.40 ± 8.66 | 8.54 | 5.84 | 7.28 | |
| AMP | 0.90 | 80.50 ± 11.20 | 13.91 | 14.80 | 13.95 |
| 25.00 | 101.33 ± 7.36 | 7.27 | 7.15 | 7.76 | |
| 50.00 a | 89.95 ± 10.82 | 12.03 | 6.12 | 10.47 | |
| 100.00 | 101.04 ± 9.28 | 9.19 | 5.89 | 7.45 |
| Method | Recovery (%) (n = 6) | RSDmax (%) (n = 6) | LOD (μg/kg) | LOQ (μg/kg) | CCα (μg/kg) | CCβ (μg/kg) |
|---|---|---|---|---|---|---|
| UPLC-MS/MS | 72.05–108.13 | 16.35% | 0.01–1.36 | 0.05–5.44 | 52.62–57.26 | 55.23–64.51 |
| HPLC-ESI/MS/MS | 75.20–107.62 | 16.39% | 0.10–2.20 | 0.30–8.50 | 57.71–61.25 | 65.41–72.50 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, L.; Wang, B.; Diao, Z.; Zhao, M.; Xie, K.; Zhang, P.; Wang, X.; Zhang, T.; Wang, J. Development and Validation of an HPLC-ESI/MS/MS Method for the Determination of Amoxicillin, Its Major Metabolites, and Ampicillin Residues in Chicken Tissues. Molecules 2019, 24, 2652. https://doi.org/10.3390/molecules24142652
Chen L, Wang B, Diao Z, Zhao M, Xie K, Zhang P, Wang X, Zhang T, Wang J. Development and Validation of an HPLC-ESI/MS/MS Method for the Determination of Amoxicillin, Its Major Metabolites, and Ampicillin Residues in Chicken Tissues. Molecules. 2019; 24(14):2652. https://doi.org/10.3390/molecules24142652
Chicago/Turabian StyleChen, Lan, Bo Wang, Zhixiang Diao, Min Zhao, Kaizhou Xie, Peiyang Zhang, Xutang Wang, Tao Zhang, and Jinyu Wang. 2019. "Development and Validation of an HPLC-ESI/MS/MS Method for the Determination of Amoxicillin, Its Major Metabolites, and Ampicillin Residues in Chicken Tissues" Molecules 24, no. 14: 2652. https://doi.org/10.3390/molecules24142652
APA StyleChen, L., Wang, B., Diao, Z., Zhao, M., Xie, K., Zhang, P., Wang, X., Zhang, T., & Wang, J. (2019). Development and Validation of an HPLC-ESI/MS/MS Method for the Determination of Amoxicillin, Its Major Metabolites, and Ampicillin Residues in Chicken Tissues. Molecules, 24(14), 2652. https://doi.org/10.3390/molecules24142652