
Cis/Trans Energetics in Epoxide, Thiirane, Aziridine and Phosphirane Containing Cyclopentanols: Effects of Intramolecular OH⋯O, S, N and P Contacts



Abstract
1. Introduction
2. Computational Methods
3. Results and Discussion
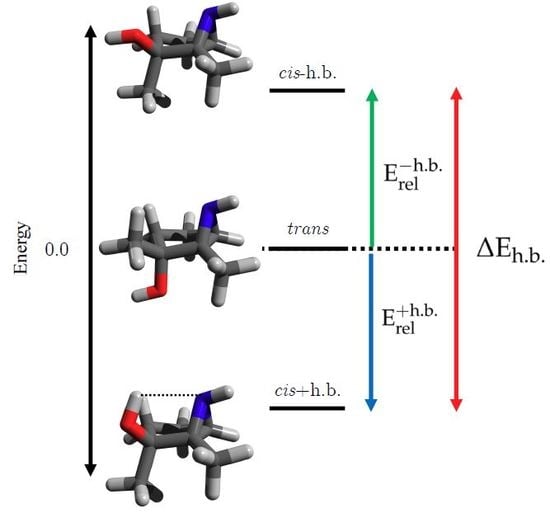
3.1. Energetics
3.2. Bond Lengths, Vibrational Frequencies and NMR Chemical Shielding Constants
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Shimazaki, M.; Hara, H.; Keisuke, S.; Tsuchihashi, G.I. On the use of epoxy alcohol-aldol rearrangement for stereoselective construction of quarternary carbon centers. Tetrahedron Lett. 1987, 28, 5891–5894. [Google Scholar] [CrossRef]
- Song, Z.L.; Fan, C.A.; Tu, Y.Q. Semipinacol Rearrangement in Natural Product Synthesis. Chem. Rev. 2011, 111, 7523–7556. [Google Scholar] [CrossRef] [PubMed]
- Parker, R.E.; Isaacs, N.S. Mechanisms Of Epoxide Reactions. Chem. Rev. 1959, 59, 737–799. [Google Scholar] [CrossRef]
- Maruoka, K.; Hasegawa, M.; Yamamoto, H.; Suzuki, K.; Shimazaki, M.; Tsuchihashi, G. Epoxy silyl ether rearrangements: A new, stereoselective approach to the synthesis of .beta.-hydroxy carbonyl compounds. J. Am. Chem. Soc. 1986, 108, 3827–3829. [Google Scholar] [CrossRef]
- Clarke, C.; Fleming, I.; Fortunak, J.M.; Gallagher, P.T.; Honan, M.C.; Mann, A.; Nubling, C.O.; Raithby, P.R.; Wolff, J. An approach to the synthesis of gelsemine: The intramolecular reaction of an allylsilane with an acyliminium ion for the synthesis of one of the quaternary centres. Tetrahedron 1988, 44, 3931–3944. [Google Scholar] [CrossRef]
- Jung, M.E.; Lee, W.S.; Sun, D. Synthesis of Four Diastereomeric 3,5-Dialkoxy-2,4-dimethylalkanals by a Simple Extension of the Non-Aldol Aldol Process to Bis(propionates). Org. Lett. 1999, 1, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.J.; Walsh, P.J. Asymmetric Addition of Alkylzinc Reagents to Cyclic Unsaturated Ketones and a Tandem Enantioselective Addition/Diastereoselective Epoxidation with Dioxygen. J. Am. Chem. Soc. 2003, 125, 9544–9545. [Google Scholar] [CrossRef] [PubMed]
- Suda, K.; Kikkawa, T.; Nakajima, S.i.; Takanami, T. Highly Regio- and Stereoselective Rearrangement of Epoxides to Aldehydes Catalyzed by High-Valent Metalloporphyrin Complex, Cr(TPP)OTf. J. Am. Chem. Soc. 2004, 126, 9554–9555. [Google Scholar] [CrossRef]
- Kita, Y.; Matsuda, S.; Inoguchi, R.; Ganesh, J.K.; Fujioka, H. Lewis Acid-Promoted Rearrangement of 2,3-Epoxy Alcohol Derivatives: Stereochemical Control and Selective Formation of Two Types of Chiral Quaternary Carbon Centers from the Single Carbon Skeleton. J. Org. Chem. 2006, 71, 5191–5197. [Google Scholar] [CrossRef]
- Tu, Y.Q.; Fan, C.A.; Ren, S.K.; Chan, A.S.C. Zinc bromide as catalyst for the stereoselective construction of quaternary carbon: Improved synthesis of diastereomerically enriched spirocyclic diols. J. Chem. Soc. Perkin Trans. 1 2000, 3791–3794. [Google Scholar] [CrossRef]
- Angeles, A.R.; Waters, S.P.; Danishefsky, S.J. Total Syntheses of (+)- and (−)-Peribysin E. J. Am. Chem. Soc. 2008, 130, 13765–13770. [Google Scholar] [CrossRef] [PubMed]
- Tanino, K.; Onuki, K.; Asano, K.; Miyashita, M.; Nakamura, T.; Takahashi, Y.; Kuwajima, I. Total Synthesis of Ingenol. J. Am. Chem. Soc. 2003, 125, 1498–1500. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.M.; Gu, P.; Tu, Y.Q.; Fan, C.A.; Zhang, Q. An Efficient Total Synthesis of (±)-Stemonamine. Org. Lett. 2008, 10, 1763–1766. [Google Scholar] [CrossRef] [PubMed]
- Fenster, M.D.B.; Dake, G.R. An Asymmetric Formal Synthesis of Fasicularin. Chem. Eur. J. 2005, 11, 639–649. [Google Scholar] [CrossRef] [PubMed]
- Epstein, O.L.; Cha, J.K. Rapid Access to the in,out-Tetracyclic Core of Ingenol. Angew. Chem. 2005, 44, 121–123. [Google Scholar] [CrossRef] [PubMed]
- Carr, J.M.; Snowden, T.S. Comparative reductive desymmetrization of 2,2-disubstituted-cycloalkane-1,3-diones. Tetrahedron 2008, 64, 2897–2905. [Google Scholar] [CrossRef]
- Wang, B.M.; Song, Z.L.; Fan, C.A.; Tu, Y.Q.; Shi, Y. Lewis Acid Promoted Highly Stereoselective Rearrangement of 2,3-Aziridino Alcohols: A New Efficient Approach to Amino Carbonyl Compounds. Org. Lett. 2002, 4, 363–366. [Google Scholar] [CrossRef]
- Tu, Y.Q.; Sun, L.D.; Wang, P.Z. Stereoselective Reductive Rearrangement of Hydroxy Epoxides: A New Method for Synthesis of 1,3-Diols1. J. Org. Chem. 1999, 64, 629–633. [Google Scholar] [CrossRef]
- Jung, M.E.; D’Amico, D.C. Enantiospecific synthesis of all four diastereomers of 2-methyl-3- [(trialkylsilyl)oxy]alkanals: Facile preparation of aldols by non-aldol chemistry. J. Am. Chem. Soc. 1993, 115, 12208–12209. [Google Scholar] [CrossRef]
- Zhu, Y.; Shu, L.; Tu, Y.; Shi, Y. Enantioselective Synthesis and Stereoselective Rearrangements of Enol Ester Epoxides. J. Org. Chem. 2001, 66, 1818–1826. [Google Scholar] [CrossRef]
- Abdo, Y.A.; Weeks, J.W.; Layfield, W.; Tremlett, W.M.; Graham, J.W.; Tabor, M.E.; Causey, S.E.; Carr, J.M.; Tschumper, G.S. Intramolecular Hydrogen Bonding in α-Epoxy Alcohols: A Conformational Analysis of 1,2-Dialkyl-2,3-epoxycyclopentanol Diastereomers. Chem. Lett. 2018, 47, 156–159. [Google Scholar] [CrossRef]
- De Ceglie, M.C.; Degennaro, L.; Falcicchio, A.; Luisi, R. Restricted rotations and stereodynamics of aziridine-2-methanol derivatives. Tetrahedron 2011, 67, 9382–9388. [Google Scholar] [CrossRef]
- Wang, M.C.; Wang, D.K.; Zhu, Y.; Liu, L.T.; Guo, Y.F. Enantiopure N-ferrocenylmethylaziridin-2-ylmethanols from l-serine: Synthesis, crystal structure and applications. Tetrahedron Asymmetry 2004, 15, 1289–1294. [Google Scholar] [CrossRef]
- Wang, M.C.; Wang, Y.H.; Li, G.W.; Sun, P.P.; Tian, J.X.; Lu, H.J. Applications of conformational design: Rational design of chiral ligands derived from a common chiral source for highly enantioselective preparations of (R)- and (S)-enantiomers of secondary alcohols. Tetrahedron Asymmetry 2011, 22, 761–768. [Google Scholar] [CrossRef]
- Wojtulewski, S.; J Grabowski, S. Different donors and acceptors for intramolecular hydrogen bonds. Chem. Phys. Lett. 2003, 378, 388–394. [Google Scholar] [CrossRef]
- Grabowski, S. An estimation of strength of intramolecular hydrogen bonds—Ab initio and AIM studies. J. Mol. Struct. 2001, 562, 137–143. [Google Scholar] [CrossRef]
- Froimowicz, P.; Zhang, K.; Ishida, H. Intramolecular Hydrogen Bonding in Benzoxazines: When Structural Design Becomes Functional. Chem. Eur. J. 2016, 22, 2691–2707. [Google Scholar] [CrossRef]
- Grabowski, S.J. Ab Initio Calculations on Conventional and Unconventional Hydrogen BondsStudy of the Hydrogen Bond Strength. J. Phys. Chem. A 2001, 105, 10739–10746. [Google Scholar] [CrossRef]
- Howard, D.L.; Kjaergaard, H.G. Hydrogen bonding to divalent sulfur. Phys. Chem. Chem. Phys. 2008, 10, 4113–4118. [Google Scholar] [CrossRef]
- Hansen, A.S.; Du, L.; Kjaergaard, H.G. Positively Charged Phosphorus as a Hydrogen Bond Acceptor. J. Phys. Chem. Lett. 2014, 5, 4225–4231. [Google Scholar] [CrossRef]
- Lane, J.R.; Hansen, A.S.; Mackeprang, K.; Kjaergaard, H.G. Kinetic Energy Density as a Predictor of Hydrogen- Bonded OH-Stretching Frequencies. J. Phys. Chem. A 2017, 121, 3452–3460. [Google Scholar] [CrossRef] [PubMed]
- Møller, K.H.; Hansen, A.S.; Kjaergaard, H.G. Gas Phase Detection of the NH-P Hydrogen Bond and Importance of Secondary Interactions. J. Phys. Chem. A 2015, 119, 10988–10998. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Tang, S.; Hansen, A.; Frandsen, B.; Maroun, Z.; Kjærgaard, H. Subtle differences in the hydrogen bonding of alcohol to divalent oxygen and sulfur. Chem. Phys. Lett. 2017, 667, 146–153. [Google Scholar] [CrossRef]
- Rosenberg, R.E. The Strength of Hydrogen Bonds between Fluoro-Organics and Alcohols, a Theoretical Study. J. Phys. Chem. A 2018, 122, 4521–4529. [Google Scholar] [CrossRef] [PubMed]
- Howard, J.C.; Gray, J.L.; Hardwick, A.J.; Nguyen, L.T.; Tschumper, G.S. Getting down to the fundamentals of hydrogen bonding: Anharmonic vibrational frequencies of the hetero and homogeneous dimers of HF and H2O from ab initio electronic structure computations. J. Chem. Theory Comput. 2014, 10, 5426–5435. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.N.; Tschumper, G.S. Hydrogen Bonding in the Mixed HF/HCl Dimer: Is It Better to Give or Receive? J. Comput. Chem. 2018, 39, 839–843. [Google Scholar] [CrossRef] [PubMed]
- Sexton, T.M.; Howard, J.C.; Tschumper, G.S. Dissociation Energy of the H2O⋯HF Dimer. J. Phys. Chem. A 2018, 122, 4902–4908. [Google Scholar] [CrossRef]
- Dreux, K.M.; Tschumper, G.S. Examination of the structures, energetics, and vibrational frequencies of small sulfur-containing prototypical dimers, (H2S)2 and H2O/H2S. J. Comput. Chem. 2019, 40, 229–236. [Google Scholar] [CrossRef]
- De Oliveira, P.R.; Rittner, R. The relevant effect of an intramolecular hydrogen bond on the conformational equilibrium of cis-3-methoxycyclohexanol compared to trans-3-methoxycyclohexanol and cis-1,3-dimethoxycyclohexane. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2005, 61, 1737–1745. [Google Scholar] [CrossRef]
- Kuhn, B.; Mohr, P.; Stahl, M. Intramolecular Hydrogen Bonding in Medicinal Chemistry. J. Med. Chem. 2010, 53, 2601–2611. [Google Scholar] [CrossRef]
- Takeda, T.; Suzuki, Y.; Kawamata, J.; Noro, S.I.; Nakamura, T.; Akutagawa, T. The emergent intramolecular hydrogen bonding effect on the electronic structures of organic electron acceptors. Phys. Chem. Chem. Phys. 2017, 19, 23905–23909. [Google Scholar] [CrossRef] [PubMed]
- Karas, L.J.; Batista, P.R.; Viesser, R.V.; Tormena, C.F.; Rittner, R.; de Oliveira, P.R. Trends of intramolecular hydrogen bonding in substituted alcohols: A deeper investigation. Phys. Chem. Chem. Phys. 2017, 19, 16904–16913. [Google Scholar] [CrossRef] [PubMed]
- Bhadra, M.; Lee, J.Y.C.; Cowley, R.E.; Kim, S.; Siegler, M.A.; Solomon, E.I.; Karlin, K.D. Intramolecular Hydrogen Bonding Enhances Stability and Reactivity of Mononuclear Cupric Superoxide Complexes. J. Am. Chem. Soc. 2018, 140, 9042–9045. [Google Scholar] [CrossRef] [PubMed]
- Pan, M.; Zhao, Y.; Zeng, X.; Zou, J. Efficient Absorption of CO2 by Introduction of Intramolecular Hydrogen Bonding in Chiral Amino Acid Ionic Liquids. Energy Fuels 2018, 32, 6130–6135. [Google Scholar] [CrossRef]
- Lakshmipriya, A.; Chaudhary, M.; Mogurampelly, S.; Klein, M.L.; Suryaprakash, N. Intramolecular Hydrogen Bonding Appetency for Conformational Penchants in Oxalohydrazide Fluoro Derivatives: NMR, MD, QTAIM, and NCI Studies. J. Phys. Chem. A 2018, 122, 2703–2713. [Google Scholar] [CrossRef]
- Hubbard, T.A.; Brown, A.J.; Bell, I.A.W.; Cockroft, S.L. The Limit of Intramolecular H-Bonding. J. Am. Chem. Soc. 2016, 138, 15114–15117. [Google Scholar] [CrossRef] [PubMed]
- Giordanetto, F.; Tyrchan, C.; Ulander, J. Intramolecular Hydrogen Bond Expectations in Medicinal Chemistry. Med. Chem. Lett. 2017, 8, 139–142. [Google Scholar] [CrossRef]
- Lane, J.R.; Schroder, S.D.; Saunders, G.C.; Kjaergaard, H.G. Intramolecular Hydrogen Bonding in Substituted Aminoalcohols. J. Phys. Chem. A 2016, 120, 6371–6378. [Google Scholar] [CrossRef]
- Nagy, P.I. Theoretical Studies of the Solvent Effect on the Conformation of the HO-C-C-X (X = F, NH2, NO2) Moiety with Competing Intra- and Intermolecular Hydrogen Bonds. J. Phys. Chem. A 2012, 116, 7726–7741. [Google Scholar] [CrossRef]
- Abraham, M.H.; Abraham, R.J.; Aliev, A.E.; Tormena, C.F. Is there an intramolecular hydrogen bond in 2-halophenols? A theoretical and spectroscopic investigation. Phys. Chem. Chem. Phys. 2015, 17, 25151–25159. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Kendall, R.A.; Dunning, T.H.; Harrison, R.J. Electron affinities of the first row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 1992, 96, 6796–6806. [Google Scholar] [CrossRef]
- Dunning, T.H. Gaussian Basis Sets for Use in Correlated Molecular Calculations. I. The Atoms Boron through Neon and Hydrogen. J. Chem. Phys. 1989, 90, 1007. [Google Scholar] [CrossRef]
- Woon, D.E.; Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. III. The atoms aluminum through argon. J. Chem. Phys. 1993, 98, 1358–1371. [Google Scholar] [CrossRef]
- Dunning, T.H.; Peterson, K.A.; Wilson, A.K. Gaussian basis sets for use in correlated molecular calculations. X. The atoms aluminum through argon revisited. J. Chem. Phys. 2001, 114, 9244–9253. [Google Scholar] [CrossRef]
- Helgaker, T.; Jaszuński, M.; Ruud, K. Ab Initio Methods for the Calculation of NMR Shielding and Indirect Spin-Spin Coupling Constants. Chem. Rev. 1999, 99, 293–352. [Google Scholar] [CrossRef] [PubMed]
- Ditchfield, R. Self-consistent perturbation theory of diamagnetism. Mol. Phys. 1974, 27, 789–807. [Google Scholar] [CrossRef]
- Mardirossian, N.; Head-Gordon, M. How Accurate Are the Minnesota Density Functionals for Noncovalent Interactions, Isomerization Energies, Thermochemistry, and Barrier Heights Involving Molecules Composed of Main-Group Elements? J. Chem. Theory Comput. 2016, 12, 4303–4325. [Google Scholar] [CrossRef]
- Kozuch, S.; Bachrach, S.M.; Martin, J.M. Conformational Equilibria in Butane-1,4-diol: A Benchmark of a Prototypical System with Strong Intramolecular H-bonds. J. Phys. Chem. A 2014, 118, 293–303. [Google Scholar] [CrossRef]
- Møller, C.; Plesset, M.S. Note on an Approximation Treatment for Many-Electron Systems. Phys. Rev. 1934, 46, 618–622. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision E.01; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Werner, H.J.; Knowles, P.J.; Knizia, G.; Manby, F.R.; Schütz, M. Molpro: A general purpose quantum chemistry program package. Comput. Mol. Sci. 2012, 2, 242–253. [Google Scholar] [CrossRef]
- Werner, H.J.; Knowles, P.J.; Knizia, G.; Manby, F.R.; Schütz, M.; Celani, P.; Györffy, W.; Kats, D.; Korona, T.; Lindh, R.; et al. MOLPRO, version 2015.1; A Package of ab Initio Programs; Ab Initio Software: Lexington, MA, USA, 2015. [Google Scholar]
- Alkorta, I.; Rozas, I.; Elguero, J. Non-conventional hydrogen bonds. Chem. Soc. Rev. 1998, 27, 163–170. [Google Scholar] [CrossRef]
- Tang, T.H.; Deretey, E.; Knak Jensen, S.J.; Csizmadia, I.G. Hydrogen bonds: Relation between lengths and electron densities at bond critical points. Eur. Phys. J. D 2006, 37, 217–222. [Google Scholar] [CrossRef]
- Mata, I.; Alkorta, I.; Molins, E.; Espinosa, E. Universal Features of the Electron Density Distribution in Hydrogen-Bonding Regions: A Comprehensive Study Involving HX (X = H, C, N, O, F, S, Cl) Interactions. Chem. Eur. J. 2010, 16, 2442–2452. [Google Scholar] [CrossRef] [PubMed]
- Nowroozi, A.; Raissi, H.; Hajiabadi, H.; Jahani, P.M. Reinvestigation of intramolecular hydrogen bond in malonaldehyde derivatives: An ab initio, AIM and NBO study. Int. J. Quantum Chem. 2011, 111, 3040–3047. [Google Scholar] [CrossRef]
- Contreras-Garcia, J.; Johnson, E.R.; Keinan, S.; Chaudret, R.; Piquemal, J.P.; Beratan, D.N.; Yang, W. NCIPLOT: A Program for Plotting Noncovalent Interaction Regions. J. Chem. Theor. Comp. 2011, 7, 625–632. [Google Scholar] [CrossRef]
- Thomsen, D.L.; Axson, J.L.; Schroder, S.D.; Lane, J.R.; Vaida, V.; Kjaergaard, H.G. Intramolecular Interactions in 2-Aminoethanol and 3-Aminopropanol. J. Phys. Chem. A 2013, 117, 10260–10273. [Google Scholar] [CrossRef]
- Rusinska-Roszak, D.; Sowinski, G. Estimation of the Intramolecular OH...OC Hydrogen Bond Energy via the Molecular Tailoring Approach. Part I: Aliphatic Structures. J. Chem. Inf. Model. 2014, 54, 1963–1977. [Google Scholar] [CrossRef]
- Afonin, A.V.; Vashchenko, A.V.; Sigalov, M.V. Estimating the energy of intramolecular hydrogen bonds from 1H NMR and QTAIM calculations. Org. Biomol. Chem. 2016, 14, 11199–11211. [Google Scholar] [CrossRef]
- Quiquempoix, L.; Bogdan, E.; Wells, N.J.; Le Questel, J.Y.; Graton, J.; Linclau, B. A Study of Intramolecular Hydrogen Bonding in Levoglucosan Derivatives. Molecules 2017, 22, 518. [Google Scholar] [CrossRef]
- Raissi, H.; Yoosean, M.; Mollania, F.; Farzad, F.; Nowroozi, A.R.; loghmaninejad, D. Ab initio and DFT computational studies on molecular conformations and strength of the intramolecular hydrogen bond in different conformers of 3-amino-2-iminomethyl acryl aldehyde. Comput. Theor. Chem. 2011, 966, 299–305. [Google Scholar] [CrossRef]
- Otto, K.E.; Xue, Z.; Zielke, P.; Suhm, M.A. The Raman spectrum of isolated water clusters. Phys. Chem. Chem. Phys. 2014, 16, 9849–9858. [Google Scholar] [CrossRef] [PubMed]
- Strickler, S.J. Molecular spectra and molecular structure. Volume 3, electronic spectra and electronic structure of polyatomic molecules (Herzberg, Gerhard). J. Chem. Educ. 1967, 44, A760. [Google Scholar] [CrossRef]
- Iogansen, A. Direct proportionality of the hydrogen bonding energy and the intensification of the stretching XH vibration in infrared spectra. Spectrochim. Acta A 1999, 55, 1585–1612. [Google Scholar] [CrossRef]
- Das, P.; Das, P.K.; Arunan, E. Conformational Stability and Intramolecular Hydrogen Bonding in 1,2-Ethanediol and 1,4-Butanediol. J. Phys. Chem. A 2015, 119, 3710–3720. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Hedberg, K. Conformational analysis. 13. 2-Fluoroethanol. An investigation of the molecular structure and conformational composition at 20, 156, and 240 °C. Estimate of the anti-gauche energy difference. J. Am. Chem. Soc. 1989, 111, 6909–6913. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| A | M | R | M06-2X | MP2 | CCSD(T)//MP2 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| O | Me | Me | |||||||||
| O | Et | Me | |||||||||
| O | Me | Et | |||||||||
| S | Me | Me | |||||||||
| S | Et | Me | |||||||||
| S | Me | Et | |||||||||
| N | Me | Me | |||||||||
| N | Et | Me | |||||||||
| N | Me | Et | |||||||||
| P | Me | Me | |||||||||
| P | Et | Me | |||||||||
| P | Me | Et | |||||||||
| A | M | R | |||||
|---|---|---|---|---|---|---|---|
| O | Me | Me | 3874 | 3872 | 3843 | ||
| O | Et | Me | 3880 | 3881 | 3841 | ||
| O | Me | Et | 3870 | 3872 | 3843 | ||
| S | Me | Me | 3873 | 3874 | 3830 | ||
| S | Et | Me | 3878 | 3882 | 3827 | ||
| S | Me | Et | 3869 | 3874 | 3828 | ||
| N | Me | Me | 3877 | 3874 | 3831 | ||
| N | Et | Me | 3883 | 3884 | 3832 | ||
| N | Me | Et | 3867 | 3873 | 3826 | ||
| P | Me | Me | 3871 | 3871 | 3837 | ||
| P | Et | Me | 3877 | 3878 | 3836 | ||
| P | Me | Et | 3868 | 3872 | 3836 |
| A | M | R | |||||
|---|---|---|---|---|---|---|---|
| O | Me | Me | 0.9609 | 0.9605 | 0.9633 | ||
| O | Et | Me | 0.9606 | 0.9605 | 0.9636 | ||
| O | Me | Et | 0.9612 | 0.9610 | 0.9635 | ||
| S | Me | Me | 0.9611 | 0.9610 | 0.9640 | ||
| S | Et | Me | 0.9607 | 0.9605 | 0.9641 | ||
| S | Me | Et | 0.9613 | 0.9610 | 0.9641 | ||
| N | Me | Me | 0.9608 | 0.9609 | 0.9642 | ||
| N | Et | Me | 0.9604 | 0.9602 | 0.9641 | ||
| N | Me | Et | 0.9611 | 0.9609 | 0.9644 | ||
| P | Me | Me | 0.9612 | 0.9611 | 0.9633 | ||
| P | Et | Me | 0.9608 | 0.9607 | 0.9633 | ||
| P | Me | Et | 0.9614 | 0.9611 | 0.9634 |
| A | M | R | |||||
|---|---|---|---|---|---|---|---|
| O | Me | Me | 31.90 | 31.42 | 31.01 | ||
| O | Et | Me | 31.12 | 30.86 | 30.72 | ||
| O | Me | Et | 31.89 | 31.47 | 30.88 | ||
| S | Me | Me | 31.84 | 31.35 | 30.84 | ||
| S | Et | Me | 31.14 | 30.62 | 30.71 | ||
| S | Me | Et | 31.80 | 31.34 | 30.72 | ||
| N | Me | Me | 31.90 | 31.51 | 30.57 | ||
| N | Et | Me | 31.22 | 30.90 | 30.54 | ||
| N | Me | Et | 31.96 | 31.59 | 30.40 | ||
| P | Me | Me | 31.96 | 31.42 | 31.11 | ||
| P | Et | Me | 31.27 | 30.62 | 31.12 | ||
| P | Me | Et | 31.94 | 31.49 | 31.01 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smith, B.E.; Carr, J.M.; Tschumper, G.S. Cis/Trans Energetics in Epoxide, Thiirane, Aziridine and Phosphirane Containing Cyclopentanols: Effects of Intramolecular OH⋯O, S, N and P Contacts. Molecules 2019, 24, 2523. https://doi.org/10.3390/molecules24142523
Smith BE, Carr JM, Tschumper GS. Cis/Trans Energetics in Epoxide, Thiirane, Aziridine and Phosphirane Containing Cyclopentanols: Effects of Intramolecular OH⋯O, S, N and P Contacts. Molecules. 2019; 24(14):2523. https://doi.org/10.3390/molecules24142523
Chicago/Turabian StyleSmith, Ben E., Jeremy M. Carr, and Gregory S. Tschumper. 2019. "Cis/Trans Energetics in Epoxide, Thiirane, Aziridine and Phosphirane Containing Cyclopentanols: Effects of Intramolecular OH⋯O, S, N and P Contacts" Molecules 24, no. 14: 2523. https://doi.org/10.3390/molecules24142523
APA StyleSmith, B. E., Carr, J. M., & Tschumper, G. S. (2019). Cis/Trans Energetics in Epoxide, Thiirane, Aziridine and Phosphirane Containing Cyclopentanols: Effects of Intramolecular OH⋯O, S, N and P Contacts. Molecules, 24(14), 2523. https://doi.org/10.3390/molecules24142523