At-Line Monitoring of the Extraction Process of Rosmarini Folium via Wet Chemical Assays, UHPLC Analysis, and Newly Developed Near-Infrared Spectroscopic Analysis Methods

 , ,
, ,  ,
, 
Abstract
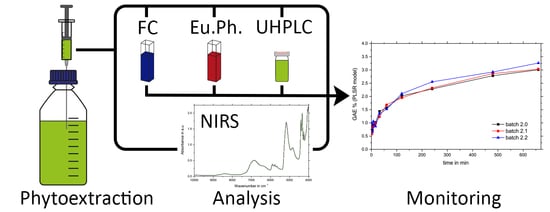
1. Introduction
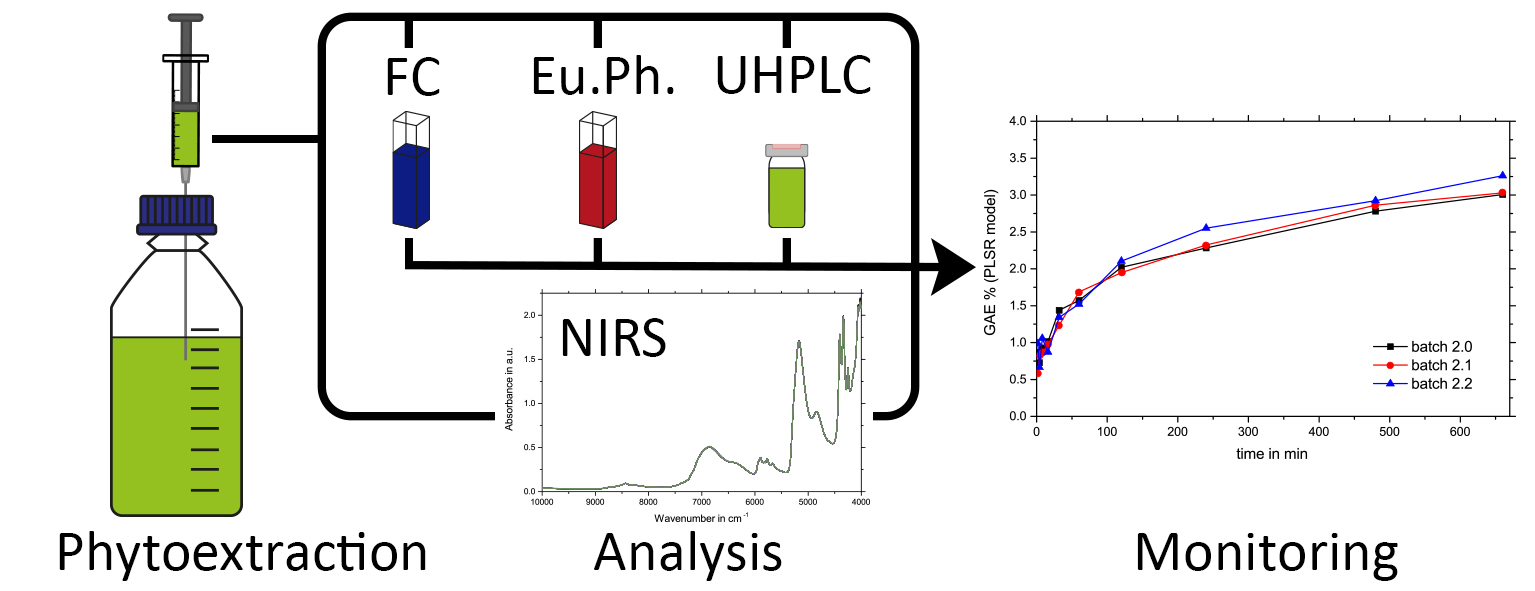
2. Results and Discussion
2.1. Wet Chemical Assays (European Pharmacopoeia and Folin–Ciocalteu)
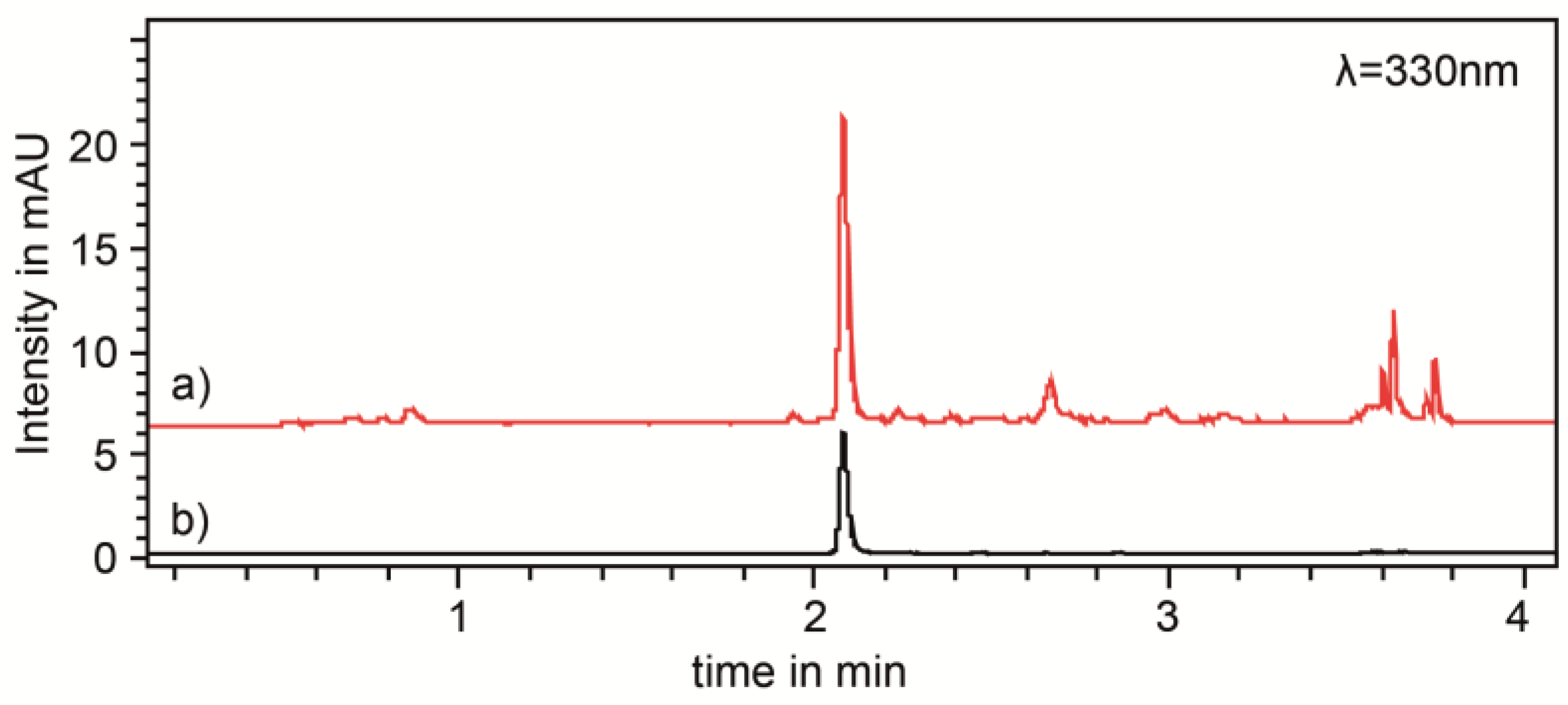
2.2. Ultra-High Performance Liquid Chromatography
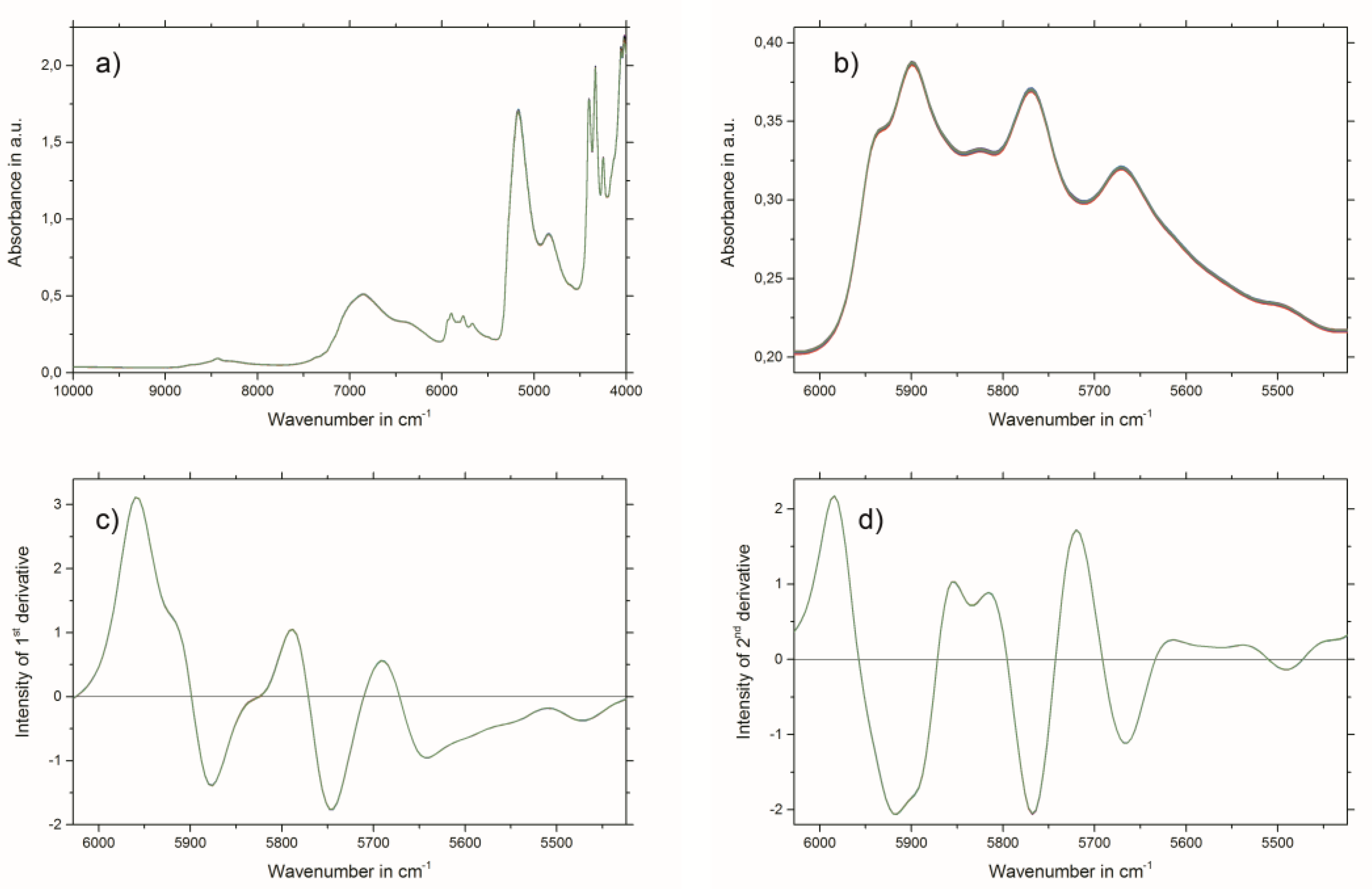
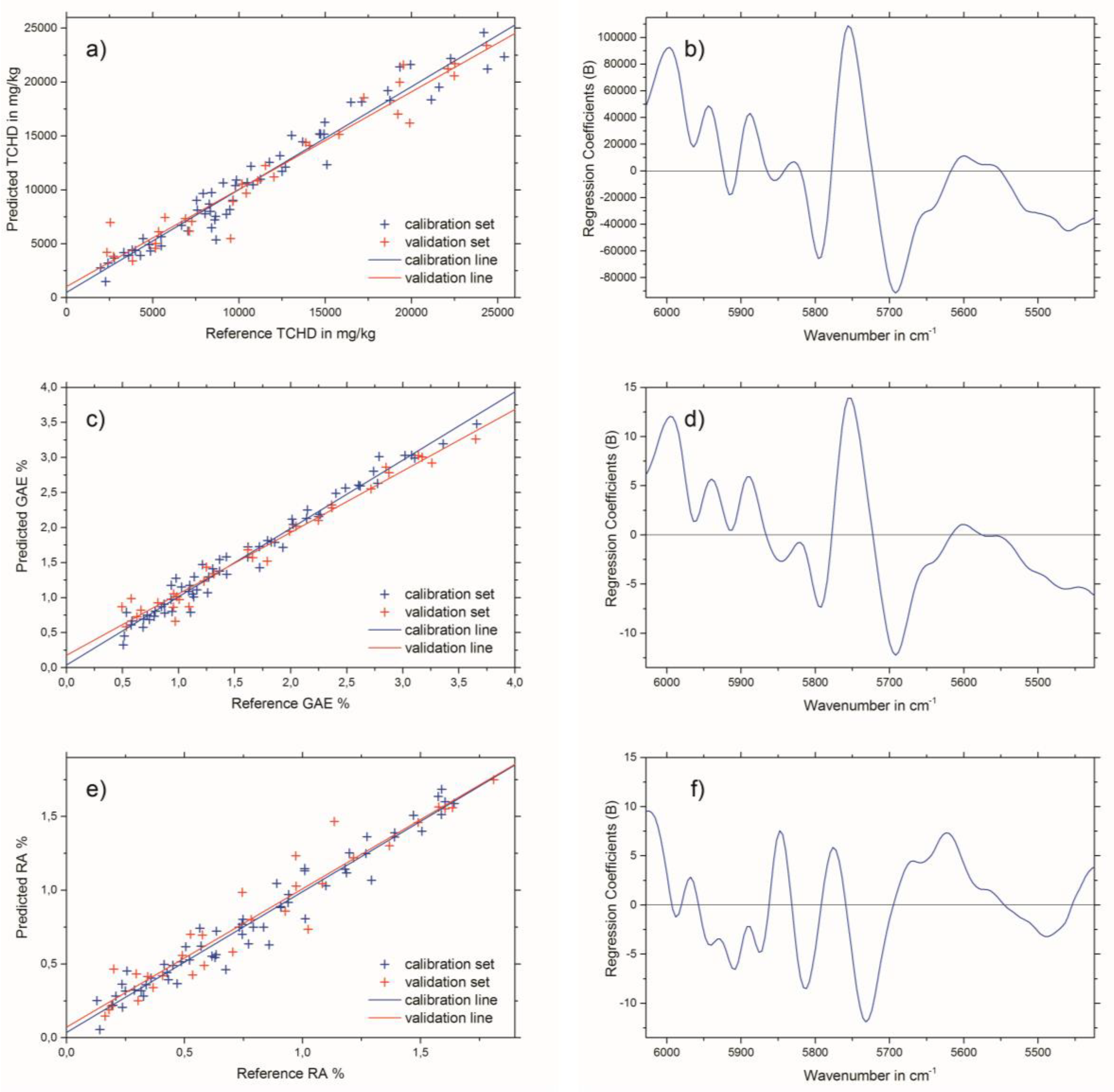
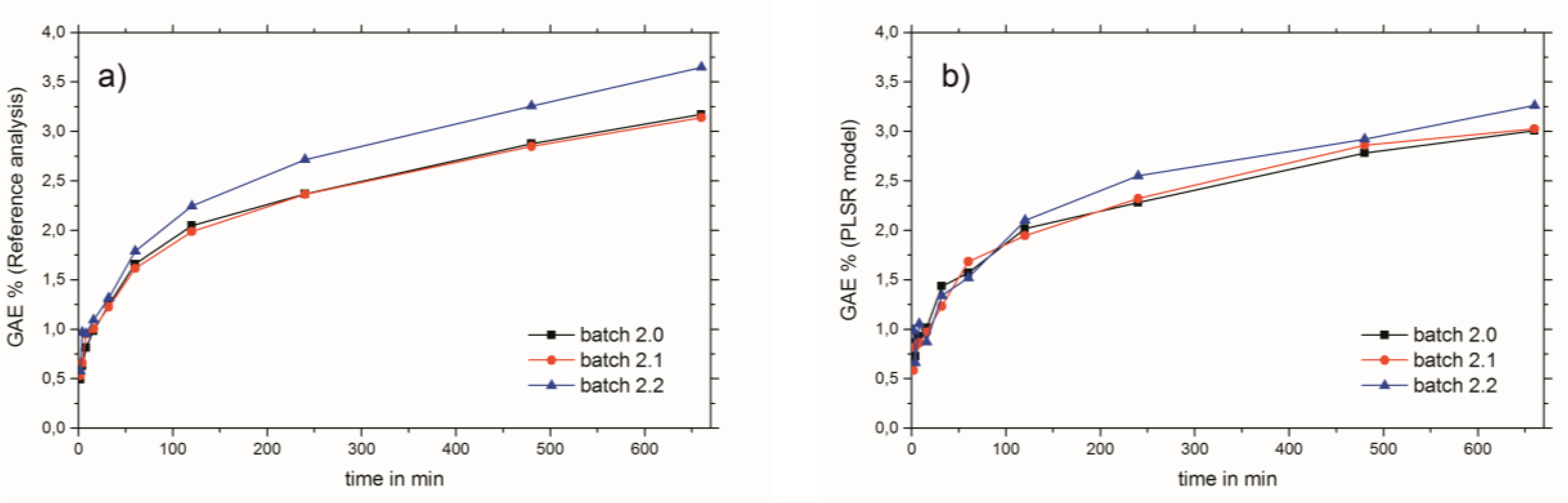
2.3. Near-Infrared Spectroscopy
3. Materials and Methods
3.1. Chemicals
3.2. Extraction and Sampling
3.3. Wet Chemical Assays
3.3.1. European Pharmacopoeia
3.3.2. Folin–Ciocalteu
3.4. Ultra-High Performance Liquid Chromatography
3.5. Near-Infrared Spectroscopy
3.6. Multivariate Data Analysis
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cragg, G.M.; Newman, D.J. Natural products: A continuing source of novel drug leads. Biochim. Et Biophys. Acta (BBA) - Gen. Subj. 2013, 1830, 3670–3695. [Google Scholar] [CrossRef]
- Veeresham, C. Natural products derived from plants as a source of drugs. J. Adv. Pharm. Technol. Res. 2012, 3, 200–201. [Google Scholar] [CrossRef] [PubMed]
- Uhlenbrock, L.; Sixt, M.; Tegtmeier, M.; Schulz, H.; Hagels, H.; Ditz, R.; Strube, J. Natural Products Extraction of the Future—Sustainable Manufacturing Solutions for Societal Needs. Processes 2018, 6, 177. [Google Scholar] [CrossRef]
- Kirchler, C.G.; Pezzei, C.K.; Beć, K.B.; Henn, R.; Ishigaki, M.; Ozaki, Y.; Huck, C.W. Critical Evaluation of NIR and ATR-IR Spectroscopic Quantifications of Rosmarinic Acid in Rosmarini folium Supported by Quantum Chemical Calculations. Planta Med. 2017, 83, 1076–1084. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency Committee on Herbal Medicinal Products (HMPC). Available online: https://www.ema.europa.eu/en/committees/committee-herbal-medicinal-products-hmpc#.Accessed (accessed on 11 January 2019).
- EDQM Council of Europe. European Pharmacopoeia, 9th ed.; Deutscher Apotheker Verlag: Stuttgart, Germany, 2016. [Google Scholar]
- Ditz, R.; Gerard, D.; Hagels, H.; Igl, N.; Schäffler, M.; Schulz, H.; Stürtz, M.; Tegtmeier, M.; Treutwein, J.; Chemat, F.; et al. Phytoextracts: Proposal towards a new comprehensive Research Focus; DECHEMA Gesellschaft für Chemische Technik und Biotechnologie e.V.: Frankfurt am Main, Germany, 2017. [Google Scholar]
- Sixt, M.; Gudi, G.; Schulz, H.; Strube, J. In-line Raman spectroscopy and advanced process control for the extraction of anethole and fenchone from fennel (Foeniculum vulgare L. MILL.). Comptes Rendus Chim. 2018, 21, 97–103. [Google Scholar] [CrossRef]
- Gavan, A.; Colobatiu, L.; Mocan, A.; Toiu, A.; Tomuta, I. Development of a NIR Method for the In-Line Quantification of the Total Polyphenolic Content: A Study Applied on Ajuga genevensis L. Dry Extract Obtained in a Fluid Bed Process. Molecules 2018, 23, 2152. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Zhang, H.; Yang, H.; Nie, L.; Zang, H. Rapid determination of major bioactive isoflavonoid compounds during the extraction process of kudzu (Pueraria lobata) by near-infrared transmission spectroscopy. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2015, 137, 1403–1408. [Google Scholar] [CrossRef]
- Wu, Z.; Sui, C.; Xu, B.; Ai, L.; Ma, Q.; Shi, X.; Qiao, Y. Multivariate detection limits of on-line NIR model for extraction process of chlorogenic acid from Lonicera japonica. J. Pharm. Biomed. Anal. 2013, 77, 16–20. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Jin, Y.; Ding, H.; Luan, L.; Chen, Y.; Liu, X. In-line monitoring of extraction process of scutellarein from Erigeron breviscapus (vant.) Hand-Mazz based on qualitative and quantitative uses of near-infrared spectroscopy. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2011, 79, 934–939. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, Y.; Chen, Y.; Wang, L.; Sun, C.; Liu, X. Study on fast quality control in extracting process of Paeonia lactiflora using near infrared spectroscopy. China J. Chin. Mater. Med. 2009, 34, 1355–1358. [Google Scholar]
- Hu, T.; Li, T.; Nie, L.; Zang, L.; Zang, H.; Zeng, Y. Rapid monitoring the water extraction process of Radix Paeoniae Alba using near infrared spectroscopy. J. Innov. Opt. Health Sci. 2017, 10, 1750002. [Google Scholar] [CrossRef]
- Siesler, H.W.; Ozaki, Y.; Kawata, S.; Heise, H.M. Near-infrared spectroscopy: Principles, Instruments, Applications; 2nd reprint ed.; Wiley-VCH Verlag: Weinheim, Germany, 2002. [Google Scholar]
- Aeschbach, R.; Löliger, J.; Scott, B.C.; Mucia, A.; Butler, J.; Halliwell, B.; Aruoma, O.I. Antioxidant actions of thymol, carvacrol, 6-gingerol, zingerone and hydroxytyrosol. Food Chem. Toxicol. 1994, 32, 31–36. [Google Scholar] [CrossRef]
- Schönbichler, S.A.; Falser, G.F.J.; Hussain, S.; Bittner, L.K.; Abel, G.; Popp, M.; Bonn, G.K.; Huck, C.W. Comparison of NIR and ATR-IR spectroscopy for determination of the capaxity of Primulae flox cum calycibus. Anal. Methods 2014, 16, 6343–6351. [Google Scholar] [CrossRef]
- Rover, M.R.; Brown, R.C. Quantification of total phenols in bio-oil using the Folin–Ciocalteu method. J. Anal. Appl. Pyrolysis 2013, 104, 366–371. [Google Scholar] [CrossRef]
- Colowick, S.P.; Kaplan, N.O.; Abelson, J.N.; Simon, M.I.; Packer, L. Methods in Enzymology; Academic Press: San Diego, CA, USA, 1999. [Google Scholar]
- Waterhouse, A.L. Determination of Total Phenolics. Curr. Protoc. Food Anal. Chem. 2002, 6, I1.1.1–I1.1.8. [Google Scholar]
- Kirchler, C.G.; Pezzei, C.K.; Bec, K.B.; Mayr, S.; Ishigaki, M.; Ozaki, Y.; Huck, C.W. Critical evaluation of spectral information of benchtop vs. portable near-infrared spectrometers: Quantum chemistry and two-dimensional correlation spectroscopy for a better understanding of PLS regression models of the rosmarinic acid content in Rosmarini folium. Analyst 2017, 142, 455–464. [Google Scholar]
- Guidance for Industry: Q2B Validation of Analytical Procedures: Methodology. Available online: https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM073384.pdf (accessed on 24 May 2018).
- Shabir, G.A. A practical approach to validation of HPLC methods under current good manufacturing practices. J. Valid. Technol. 2004, 10, 210–218. [Google Scholar]
- Singleton, V.L.; Orthofer, R.; Lamuela-Raventós, R.M. [14] Analysis of total phenols and other oxidation substrates and antioxidants by means of folin-ciocalteu reagent. Methods Enzym. 1999, 299, 152–178. [Google Scholar]
- Barnes, R.J.; Dhanoa, M.S.; Lister, S.J. Standard Normal Variate Transformation and De-trending of Near-Infrared Diffuse Reflectance Spectra. Appl. Spectrosc. 1989, 43, 772–777. [Google Scholar] [CrossRef]
- Geladi, P.; MacDougall, D.; Martens, H. Linearization and Scatter-Correction for Near-Infrared Reflectance Spectra of Meat. Appl. Spectrosc. 1985, 39, 491–500. [Google Scholar] [CrossRef]
- Savitzky, A.; Golay, M.J.E. Smoothing and Differentiation of Data by Simplified Least Squares Procedures. Anal. Chem. 1964, 36, 1627–1639. [Google Scholar] [CrossRef]
- Haaland, D.M.; Thomas, E.V. Partial least-squares methods for spectral analyses. 1. Relation to other quantitative calibration methods and the extraction of qualitative information. Anal. Chem. 1988, 60, 1193–1202. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ph. Eur. | FC | UHPLC | |
|---|---|---|---|
| Ph. Eur. | 1 | - | - |
| FC | 0.966 | 1 | - |
| UHPLC | 0.955 | 0.953 | 1 |
| Ph. Eur. | FC | UHPLC | |
|---|---|---|---|
| Repeatability in % RSD | 0.12 | 1.1 | 0.28 |
| Intermediate precision in % RSD | 4.1 | 1.3 | 0.55 |
| Repeatability (absolute) | 16 * | 0.028 ** | 0.0017 *** |
| Intermediate precision (absolute) | 593 * | 0.033 ** | 0.0028 *** |
| Reference Analysis | Ph. Eur. | FC | UHPLC | |||
|---|---|---|---|---|---|---|
| Samples | 90 | 90 | 90 | |||
| Outliers | 0 | 0 | 0 | |||
| CV | TSV | CV | TSV | CV | TSV | |
| R2calibration | 0.95 | 0.95 | 0.97 | 0.97 | 0.94 | 0.95 |
| R2validation | 0.94 | 0.94 | 0.96 | 0.96 | 0.94 | 0.93 |
| RMSEC (a) | 1425 * | 1308 * | 0.14 ** | 0.13 ** | 0.11 *** | 0.09 *** |
| RMSECV (b) or RMSEP (c) | 1527 * | 1632 * | 0.16 ** | 0.18 ** | 0.12 *** | 0.13 *** |
| Factor | 3 | 4 | 3 | 4 | 4 | 4 |
| Calibration range | 1975–25378 * | 0.494–3.660 ** | −1.810 *** | |||
| Sampling 1 | Sampling 2 | Sampling 3 |
|---|---|---|
| 1.5 min | 2 min | 2.5 min |
| 3 min | 4 min | 5 min |
| 6 min | 8 min | 10 min |
| 12 min | 16 min | 20 min |
| 24 min | 32 min | 40 min |
| 45 min | 60 min | 50 min |
| 90 min | 120 min | 80 min |
| 180 min | 240 min | 150 min |
| 360 min | 480 min | 300 min |
| 720 min | 660 min | 600 min |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Delueg, S.; Kirchler, C.G.; Meischl, F.; Ozaki, Y.; Popp, M.A.; Bonn, G.K.; Huck, C.W. At-Line Monitoring of the Extraction Process of Rosmarini Folium via Wet Chemical Assays, UHPLC Analysis, and Newly Developed Near-Infrared Spectroscopic Analysis Methods. Molecules 2019, 24, 2480. https://doi.org/10.3390/molecules24132480
Delueg S, Kirchler CG, Meischl F, Ozaki Y, Popp MA, Bonn GK, Huck CW. At-Line Monitoring of the Extraction Process of Rosmarini Folium via Wet Chemical Assays, UHPLC Analysis, and Newly Developed Near-Infrared Spectroscopic Analysis Methods. Molecules. 2019; 24(13):2480. https://doi.org/10.3390/molecules24132480
Chicago/Turabian StyleDelueg, Stefanie, Christian G. Kirchler, Florian Meischl, Yukihiro Ozaki, Michael A. Popp, Günther K. Bonn, and Christian W. Huck. 2019. "At-Line Monitoring of the Extraction Process of Rosmarini Folium via Wet Chemical Assays, UHPLC Analysis, and Newly Developed Near-Infrared Spectroscopic Analysis Methods" Molecules 24, no. 13: 2480. https://doi.org/10.3390/molecules24132480
APA StyleDelueg, S., Kirchler, C. G., Meischl, F., Ozaki, Y., Popp, M. A., Bonn, G. K., & Huck, C. W. (2019). At-Line Monitoring of the Extraction Process of Rosmarini Folium via Wet Chemical Assays, UHPLC Analysis, and Newly Developed Near-Infrared Spectroscopic Analysis Methods. Molecules, 24(13), 2480. https://doi.org/10.3390/molecules24132480