
(F)uridylylated Peptides Linked to VPg1 of Foot-and- Mouth Disease Virus (FMDV): Design, Synthesis and X-Ray Crystallography of the Complexes with FMDV RNA-Dependent RNA Polymerase

 ,
, 
Abstract
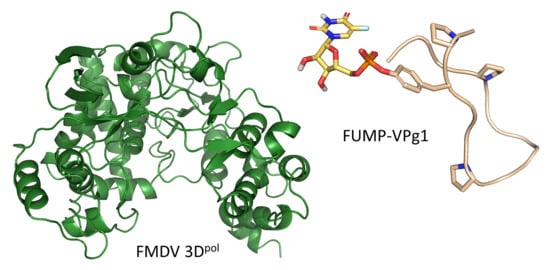
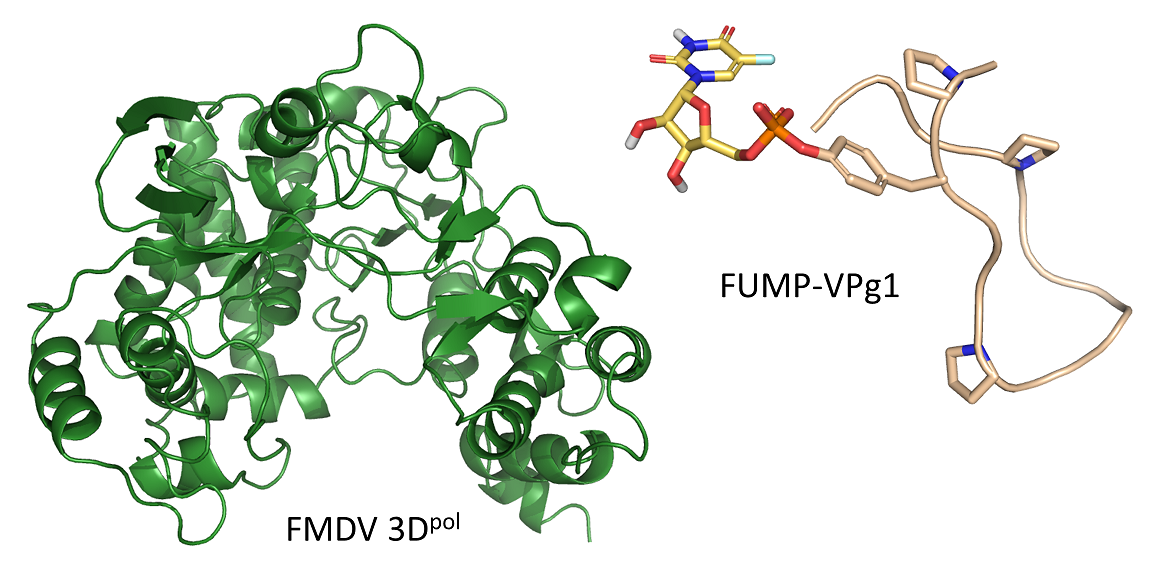
1. Introduction
2. Results and Discussion
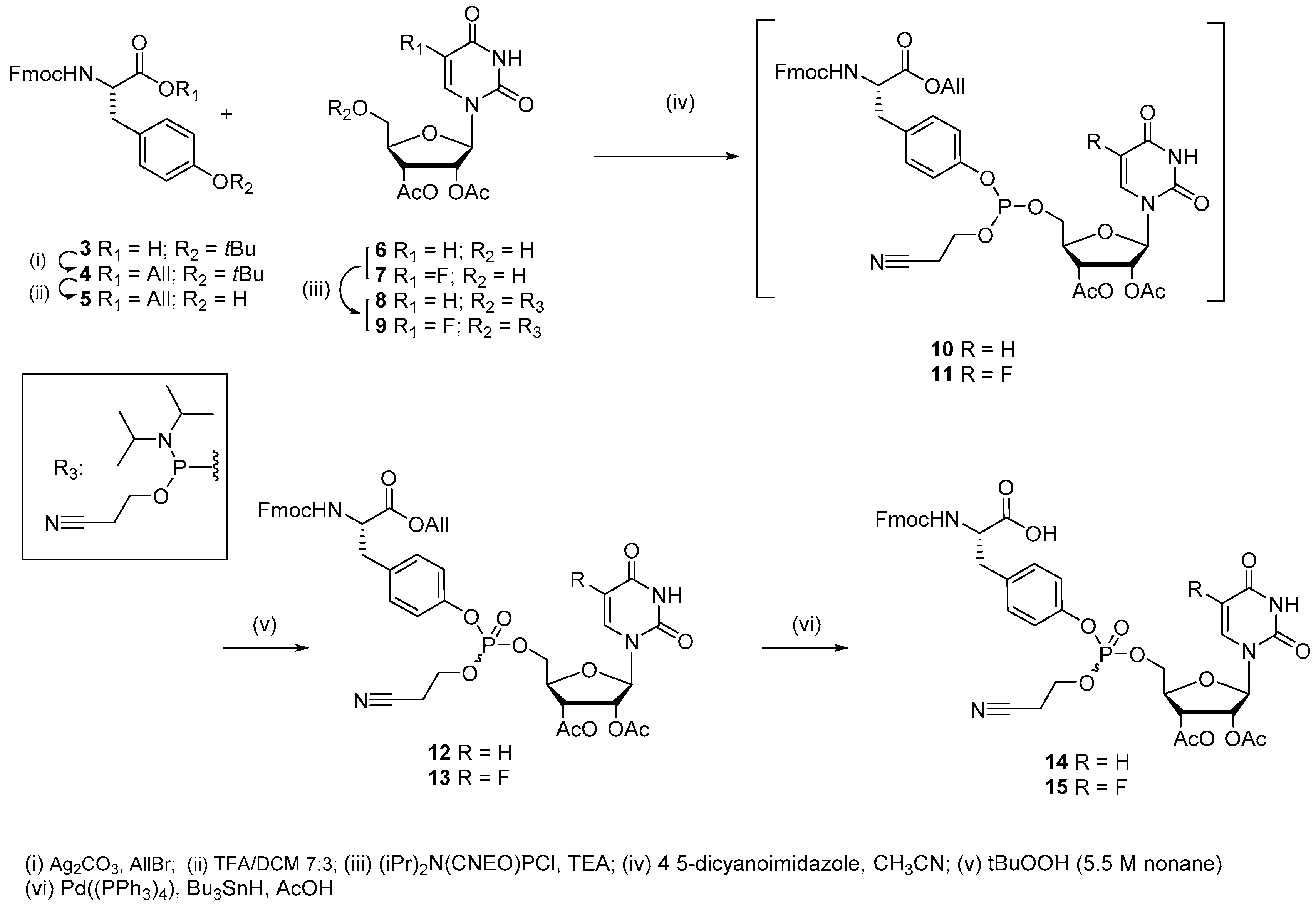
2.1. Chemical Synthesis
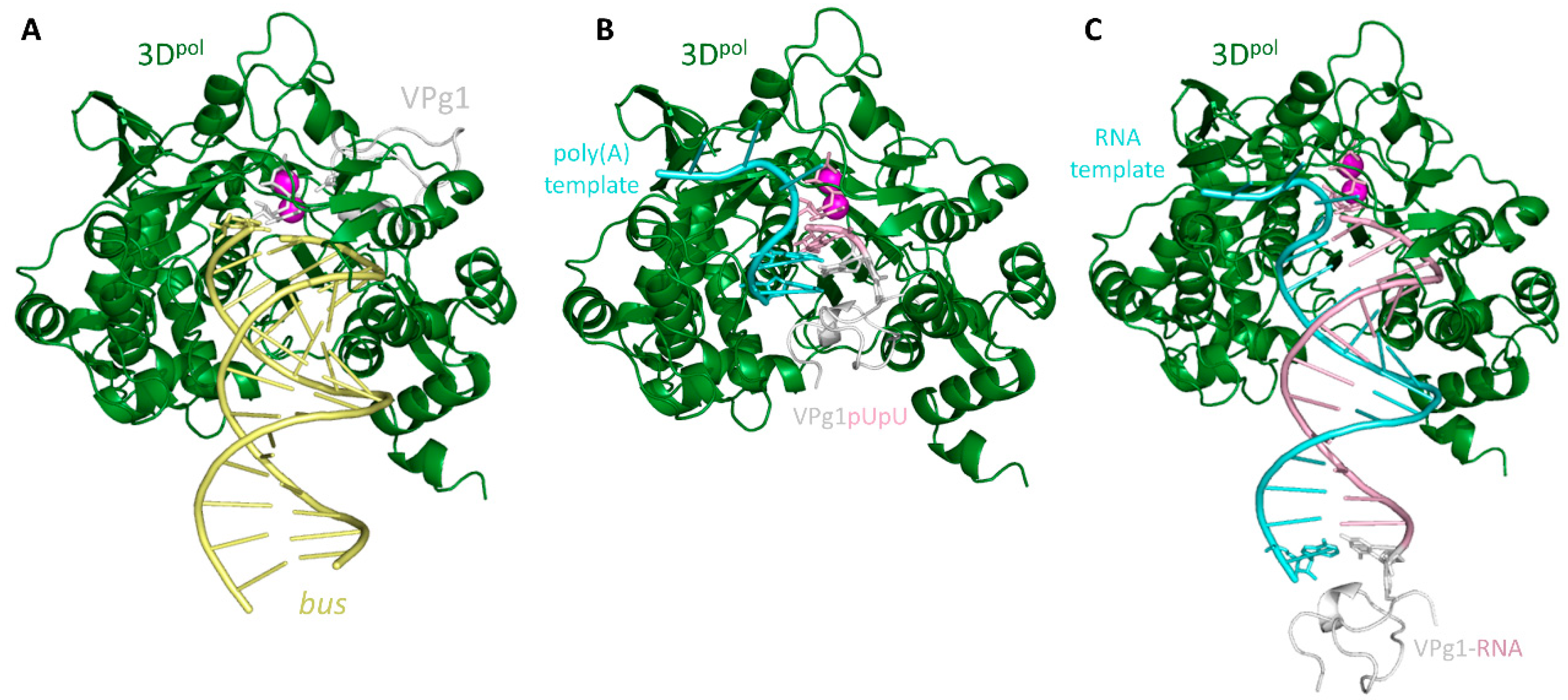
2.2. Crystal Structures of the UMP-VPg1 and FUMP-VPg1 Bound to the 3D Polymerase
2.3. Molecular Modeling Studies
3. Materials and Methods
3.1. Chemistry
3.1.1. General Methods
3.1.2. Synthesis of Nα-Fmoc-(O-tert-butyl)-tyrosine allyl ester (4)
3.1.3. Synthesis of Nα-Fmoc-tyrosine-allyl ester (5)
3.1.4. Synthesis of 2-cyanoethyl-(Nα-Fmoc-tyrosin-4-yl-allylester)-(2′,3′-di-O-acetyluridyl-5′-yl)phosphate (12)
3.1.5. Synthesis of 2-cyanoethyl-(Nα-Fmoc-tyrosin-4-yl-allylester)-(2′,3′-di-O-acetyl-5-flurouridyl-5′-yl) phosphate (13)
3.1.6. Synthesis of 2-cyanoethyl-(Nα-Fmoc-tyrosin-4-yl)-(2′,3′-di-O-acetyluridyl-5′-yl)phosphate (14)
3.1.7. Synthesis of 2-cyanoethyl-(Nα-Fmoc-tyrosin-4-yl)-(2′,3′-di-O-acetyl-5-fluorouridyl-5′-yl) phosphate (15)
3.1.8. General Procedure for the SPPS of Nucleopeptides.
- (1)
- Coupling procedure of the first 11 amino acids: After sealing the vial, the reaction was heated in a microwave vial equipped with a magnetic stirrer for 10 min at 40 °C. Then, the vial was opened, the supernatant removed and new coupling mixture added. This process was repeated three times in total (3 × 10 min) until complete coupling. Finally, the resin was transferred to a fritted syringe, drained and washed extensively (DMF/DCM/DMF/DCM, 5 × 0.5 min). This protocol was repeated for the sequential anchoring of each of the first 12 amino acids.
- (2)
- Coupling of the last three amino acids: The mixture was stirred 1 h at room temperature. Then, the vial was opened, the supernatant removed and new coupling mixture added. Finally, the resin was drained and washed extensively (DMF/DCM/DMF/DCM, 5 × 0.5 min). This protocol was repeated for the sequential anchoring of each of the last three amino acids of the sequence.
3.2. Crystallization, Data Collection and Processing
3.3. Molecular Modeling Methods
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- The Global Foot and Mouth Disease Control Strategy. 2018. Available online: http://www.fao.org/3/I9857EN/i9857en.PDF (accessed on 25 June 2019).
- Ferrer-Orta, C.; Verdaguer, N. The RNA-dependent RNA Polymerase 3D: Structure and Fidelity. In Foot-and-Mouth-Disease Virus Current Research and Emerging Trends; Sobrino, F., Domingo, E., Eds.; Caister Academic Press: Norfolk, UK, 2017; pp. 137–146. [Google Scholar]
- World Organisation for Animal Health (OIE). Available online: http://www.oie.int/en/animal-health-in-the-world/fmd-portal/ (accessed on 25 June 2019).
- Maclachlan, N.; Dubovi, E.J. Fenner’s Veterinary Virology; Academic Press: London, UK, 2017; Chapter 2; pp. 45–47. [Google Scholar]
- Nayak, A.; Goodfellow, I.G.; Belsham, G.J. Factors required for the uridylylation of the Foot-and-Mouth Disease Virus 3B1, 3B2, and 3B3 peptides by the RNA-dependent RNA polymerase (3Dpol) in vitro. J. Virol. 2005, 79, 7698–7706. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Paul, A.V.; Yin, J.; Mugavero, J.; Rierder, E.; Liu, Y.; Wimmer, E. A “slide-back” mechanism for the initiation of protein-primed RNA synthesis by the RNA polymerase of poliovirus. J. Biol. Chem. 2003, 278, 43951–43960. [Google Scholar] [CrossRef] [PubMed]
- Wimmer, E.; Paul, A.V. Initiation of protein-primed picornavirus RNA synthesis. Virus Res. 2015, 206, 12–26. [Google Scholar]
- Forss, S.; Schaller, H. A tandem repeat gene in a picornavirus. Nucleic Acids Res. 1982, 10, 6441–6450. [Google Scholar] [CrossRef] [PubMed]
- King, A.M.; Sangar, D.V.; Harris, T.J.; Brown, F. Heterogeneity of the genome-linked protein of foot-and-mouth disease virus. J. Virol. 1980, 34, 627–634. [Google Scholar] [PubMed]
- Nayak, A.; Goodfellow, I.G.; Woolaway, K.E.; Birtley, J.; Curry, S.; Belsham, G.J. Role of RNA structure and RNA binding activity of Foot-and-Mouth Disease Virus 3C protein in VPguridylylation and virus replication. J. Virol. 2006, 80, 9865–9875. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Falk, M.M.; Sobrino, F.; Beck, E. VPg gene amplification correlates with infective particle formation in foot-and-mouth disease virus. J. Virol. 1992, 66, 2251–2260. [Google Scholar] [PubMed]
- Ferrer-Orta, C.; Arias, A.; Agudo, R.; Pérez-Luque, R.; Escarmís, C.; Domingo, E.; Verdaguer, N. The structure of a protein primer-polymerase complex in the initiation of genome replication. EMBO J. 2006, 25, 880–888. [Google Scholar] [CrossRef] [PubMed]
- Pariente, N.; Sierra, S.; Airaksinen, A. Action of mutagenic agents and antiviral inhibitors on foot-and-mouth disease virus. Virus Res. 2005, 107, 183–193. [Google Scholar] [CrossRef]
- Goris, N.; De Palma, A.; Toussaint, J.-F.; Musch, I.; Neyts, J.; De Clercq, K. 2′-C-methylcytidine as a potent and selective inhibitor of the replication of foot-and mouth disease virus. Antiviral Res. 2007, 73, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Paul, A.V. Possible unifying mechanism of picornavirus genome replication. In Molecular Biology of Picornaviruses; Semler, B.L., Wimmer, E., Eds.; ASM Press: Washington, DC, USA, 2002; pp. 227–246. [Google Scholar]
- Furuta, Y.; Takahashi, K.; Shiraki, K.; Sakamoto, K.; Smee, D.F.; Barnard, D.L.; Gowen, B.B.; Julander, J.G.; Morrey, J.D. T-705 (favipiravir) and related compounds: Novel broad-spectrum inhibitors of RNA viral infections. Antiviral Res. 2009, 82, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Durk, R.C.; Singh, K.; Cornelison, C.A.; Rai, D.K.; Matzek, K.B.; Leslie, M.D.; Schafer, E.; Marchand, B.; Adedeji, A.; Michailidis, E.; et al. Inhibitors of foot and mouth disease virus targeting a novel pocket of the RNA-dependent RNA polymerase. PLoS ONE 2010, 5, e15049. [Google Scholar] [CrossRef]
- Agudo, R.; Arias, A.; Pariente, N.; Perales, C.; Escarmís, C.; Jorge, A.; Marina, A.; Domingo, E. Molecular characterization of a dual inhibitory and mutagenic activity of 5-fluorouridine triphosphate on viral RNA synthesis. Implications for lethal mutagenesis. J. Mol. Biol. 2008, 382, 652–666. [Google Scholar] [CrossRef]
- Gordon, M.P.; Staehelin, M. Studies on the incorporation of 5-fluorouracil into a virus nucleic acid. Biochim. Biophys. Acta 1959, 36, 351–361. [Google Scholar] [CrossRef]
- Munyon, W.; Salzman, N.P. The incorporation of 5-fluoro-uracil into poliovirus. Virology 1962, 18, 95–101. [Google Scholar] [CrossRef]
- Ghosh, A.; Nayak, R.; Shaila, M.S. Inhibition of replication of rinderpest virus by 5-fluorouracil. Antiviral Res. 1996, 31, 35–44. [Google Scholar] [CrossRef]
- Sierra, S.; Dávila, M.; Lowenstein, P.R.; Domingo, E. Response of foot-and-mouth disease virus to increased mutagenesis. Influence of viral load and fitness in loss of infectivity. J. Virol. 2000, 74, 8316–8323. [Google Scholar] [CrossRef]
- Kriek, N.M.A.J.; Filipov, D.V.; Van den Elst, H.; Meeuwenoord, N.J.; Tesser, G.; Van Bomm, H.; van der Marel, G. Stepwise solid phase synthesis of uridylylated viral genome-linked peptides using uridylylated amino acid building blocks. Tetrahedron 2003, 59, 1589–1597. [Google Scholar] [CrossRef]
- Van der Heden van Noort, G.J.; Schein, C.H.; Overkleeft, H.S.; Van der Marel, G.A.; Filippov, D.V. A general synthetic method toward uridylylated picornavirus VPg proteins. J. Pept. Sci. 2013, 19, 333–336. [Google Scholar] [CrossRef] [PubMed]
- Terada, T.; KatsuhikoFujimoto, J.Y.; Yasumoto, M.; Takeda, S.; Uchida, J.; Wierzba, K.; Ymada, Y. 5-Substituted Uridine Derivatives. US5420117A, 30 May 1995. [Google Scholar]
- Kenten, J.H.; Casadei, J.M.; Kamireddy, B.; Mark, M.; Massey, R. Prodrugs Activated by Targeted Catalytic Proteins. WO9302703A1, 18 February 1993. [Google Scholar]
- Van der Heden van Noort, G.J.; Overkleeft, H.S.; Van der Marel, G.A.; Filippov, D.V. Synthesis of nucleotidylated poliovirus VPg proteins. J. Org. Chem. 2010, 75, 5733–5736. [Google Scholar] [CrossRef] [PubMed]
- Filippov, D.; Kuyl-Yeheskiely, E.; Van der Marel, G.A.; Tesser, G.I.; Van Boom, J.H. Synthesis of a nucleopeptide fragment from poliovirus genome. Tetrahedron Lett. 1998, 39, 3597–3600. [Google Scholar] [CrossRef]
- Ferrer-Orta, C.; Arias, A.; Pérez-Luque, R.; Escarmís, C.; Domingo, E.; Verdaguer, N. Sequential structures provide insights into the fidelity of RNA replication. Proc. Natl. Acad. Sci. USA 2007, 104, 9463–9468. [Google Scholar] [CrossRef] [PubMed]
- Gong, P.; Peersen, O.B. Structural basis for active site closure by the poliovirus RNA-dependent RNA polymerase. Proc. Natl. Acad. Sci. USA 2010, 107, 22505–22510. [Google Scholar] [CrossRef] [PubMed]
- Garriga, D.; Ferrer-Orta, C.; Querol-Audí, J.; Oliva, B.; Verdaguer, N. Role of motif B loop in allosteric regulation of RNA-dependent RNA polymerization activity. J. Mol. Biol. 2013, 425, 2279–2287. [Google Scholar] [CrossRef] [PubMed]
- Sholders, A.J.; Peersen, O.B. Distinct conformations of a putative translocation element in poliovirus polymerase. J. Mol. Biol. 2014, 426, 1407–1419. [Google Scholar] [CrossRef] [PubMed]
- Garriga, D.; Navarro, A.; Querol-Audí, J.; Abaitua, F.; Rodríguez, J.F.; Verdaguer, N. Activation mechanism of a noncanonical RNA-dependent RNA polymerase. Proc. Natl. Acad. Sci. USA 2007, 104, 20540–20545. [Google Scholar] [CrossRef] [PubMed]
- Sosunov, V.; Zorov, S.; Sosunova, E.; Nikolaev, A.; Zakeyeva, I.; Bass, I.; Goldfarb, A.; Nikiforov, V.; Severinov, K.; Mustaev, A. The involvement of the aspartate triad of the active center in all catalytic activities of multisubunit RNA polymerase. Nucleic. Acids. Res. 2005, 33, 4202–4211. [Google Scholar] [CrossRef] [PubMed]
- Mason, P.W.; Bezborodova, S.V.; Henry, T.M. Identification and characterization of a cis-acting replication element (cre) adjacent to the internal ribosome entry site of foot-and-mouth disease virus. J. Virol. 2002, 76, 9686–9694. [Google Scholar] [CrossRef]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef]
- Popenda, M.; Szachniuk, M.; Antczak, M.; Purzycka, K.J.; Lukasiak, P.; Bartol, N.; Blazewicz, J.; Adamiak, R.W. Automated 3D structure composition for large RNAs. Nucleic Acids Res. 2012, 40, e112. [Google Scholar] [CrossRef]
- Salas, M. Protein-priming of DNA replication. Annu. Rev. Biochem. 1991, 60, 39–71. [Google Scholar] [CrossRef] [PubMed]
- Gmeiner, W.H. Novel chemical strategies for thymidylate synthase inhibition. Curr. Med. Chem. 2005, 12, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W. XDS. Acta Cryst. 2010, D66, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Potterton, E.; Briggs, P.; Turkenburg, M.; Dodson, E. A graphical user interface to the CCP4 program suite. Acta. Crystallogr. D. Biol. Crystallogr 2003, 59, 1131–1137. [Google Scholar] [CrossRef] [PubMed]
- Murshudov, G.N.; Vagin, A.A.; Dodson, E.J. Refinement of macromolecular structures by the maximum-likelihood method. Acta. Crystallogr. D. Biol. Crystallogr 1997, 53, 240–255. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta. Crystallogr. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [PubMed]
- Adams, P.D.; Afonine, P.V.; Bunkoczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr 2010, 66, 213–221. [Google Scholar] [CrossRef]
- Das, R.; Karanicolas, J.; Baker, D. Atomic accuracy in predicting and designing noncanonical RNA structure. Nat. Methods 2010, 7, 291–294. [Google Scholar] [CrossRef]
- Sripakdeevong, P.; Kladwang, W.; Das, R. An enumerative stepwise ansatz enables atomic-accuracy RNA loop modeling. Proc. Natl. Acad. Sci. USA 2011, 108, 20573–20578. [Google Scholar] [CrossRef]
- Raman, S.; Vernon, R.; Thompson, J.; Tyka, M.; Sadreyev, R.; Pei, J.; Kim, D.; Kellogg, E.; DiMaio, F.; Lange, O.; et al. Structure prediction for CASP8 with all-atom refinement using Rosetta. Proteins 2009, 77, 89–99. [Google Scholar] [CrossRef]
- Kim, D.E.; Chivian, D.; Baker, D. Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res. 2004, 32, W526–W531. [Google Scholar] [CrossRef] [PubMed]
- The PyMOL Molecular Graphics System, Version 2.0. Schrödinger, LLC. Available online: https://pymol.org/2/ (accessed on 25 June 2019).
- Case, D.A.; Cheatham, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M.J.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The AMBER biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds (1, 2) are available from the authors upon request. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Collection | 3Dpol-FUMP-VPg1 |
| Beamline | ID29 (ESRF) |
| Resolution (Å) | 74.23–2.30 (2.42–2.30) |
| Space Group | P41212 |
| Cell dimensions | |
| a, b, c (Å) | 93.85 93.85 121.31 |
| α, β, γ (°) | 90 90 90 |
| Rmerge | 0.092 (0.77) |
| I/σI | 12.7 (2.5) |
| Completeness (%) | 99.9 (100) |
| Multiplicity | 6.6 (6.9) |
| Refinement | |
| Resolution (Å) | 74.23–2.3 |
| No. reflections (total/unique) | 162,185/24,747 |
| Rwork †/Rfree ‡ | 22.88/26.91 |
| No. Atoms/Residues | |
| 3Dpol | 3918 (476) |
| Waters | 148 |
| B-factors (Å2) | |
| All atoms | 27.41 |
| 3Dpol | 27.54 |
| Waters | 26.94 |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.006 |
| Bond angles (°) | 0.892 |
| Ramachandran plot | |
| Residues in preferred regions | 462 (97.5%) |
| Residues in allowed regions | 10 (2.1%) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Castro, S.; Ferrer-Orta, C.; Mills, A.; Fernández-Cureses, G.; Gago, F.; Verdaguer, N.; Camarasa, M.-J. (F)uridylylated Peptides Linked to VPg1 of Foot-and- Mouth Disease Virus (FMDV): Design, Synthesis and X-Ray Crystallography of the Complexes with FMDV RNA-Dependent RNA Polymerase. Molecules 2019, 24, 2360. https://doi.org/10.3390/molecules24132360
de Castro S, Ferrer-Orta C, Mills A, Fernández-Cureses G, Gago F, Verdaguer N, Camarasa M-J. (F)uridylylated Peptides Linked to VPg1 of Foot-and- Mouth Disease Virus (FMDV): Design, Synthesis and X-Ray Crystallography of the Complexes with FMDV RNA-Dependent RNA Polymerase. Molecules. 2019; 24(13):2360. https://doi.org/10.3390/molecules24132360
Chicago/Turabian Stylede Castro, Sonia, Cristina Ferrer-Orta, Alberto Mills, Gloria Fernández-Cureses, Federico Gago, Nuria Verdaguer, and María-José Camarasa. 2019. "(F)uridylylated Peptides Linked to VPg1 of Foot-and- Mouth Disease Virus (FMDV): Design, Synthesis and X-Ray Crystallography of the Complexes with FMDV RNA-Dependent RNA Polymerase" Molecules 24, no. 13: 2360. https://doi.org/10.3390/molecules24132360
APA Stylede Castro, S., Ferrer-Orta, C., Mills, A., Fernández-Cureses, G., Gago, F., Verdaguer, N., & Camarasa, M.-J. (2019). (F)uridylylated Peptides Linked to VPg1 of Foot-and- Mouth Disease Virus (FMDV): Design, Synthesis and X-Ray Crystallography of the Complexes with FMDV RNA-Dependent RNA Polymerase. Molecules, 24(13), 2360. https://doi.org/10.3390/molecules24132360