
Populations and Dynamics of Guanine Radicals in DNA strands—Direct versus Indirect Generation



Abstract
1. Introduction
- ➢
- two single strands composed of 30 basesS1: 5′-CGTACTCTTTGGTGGGTCGGTTCTTTCTAT-3′, andS2: 3′-GCATGAGAAACCACCCAGCCAAGAAAGATA-5′,
- ➢
- the duplex D formed by hybridization of S1 with its complementary strand S2, and
- ➢
- the monomolecular G-quadruplex formed by folding of the human telomeric sequence 5′-TAGGG(TTAGGG)3TT-3′ in the presence of Na+ ions, abbreviated as TEL25/Na+.
2. Results and Discussion
2.1. Methodology: Advantages and Limitations
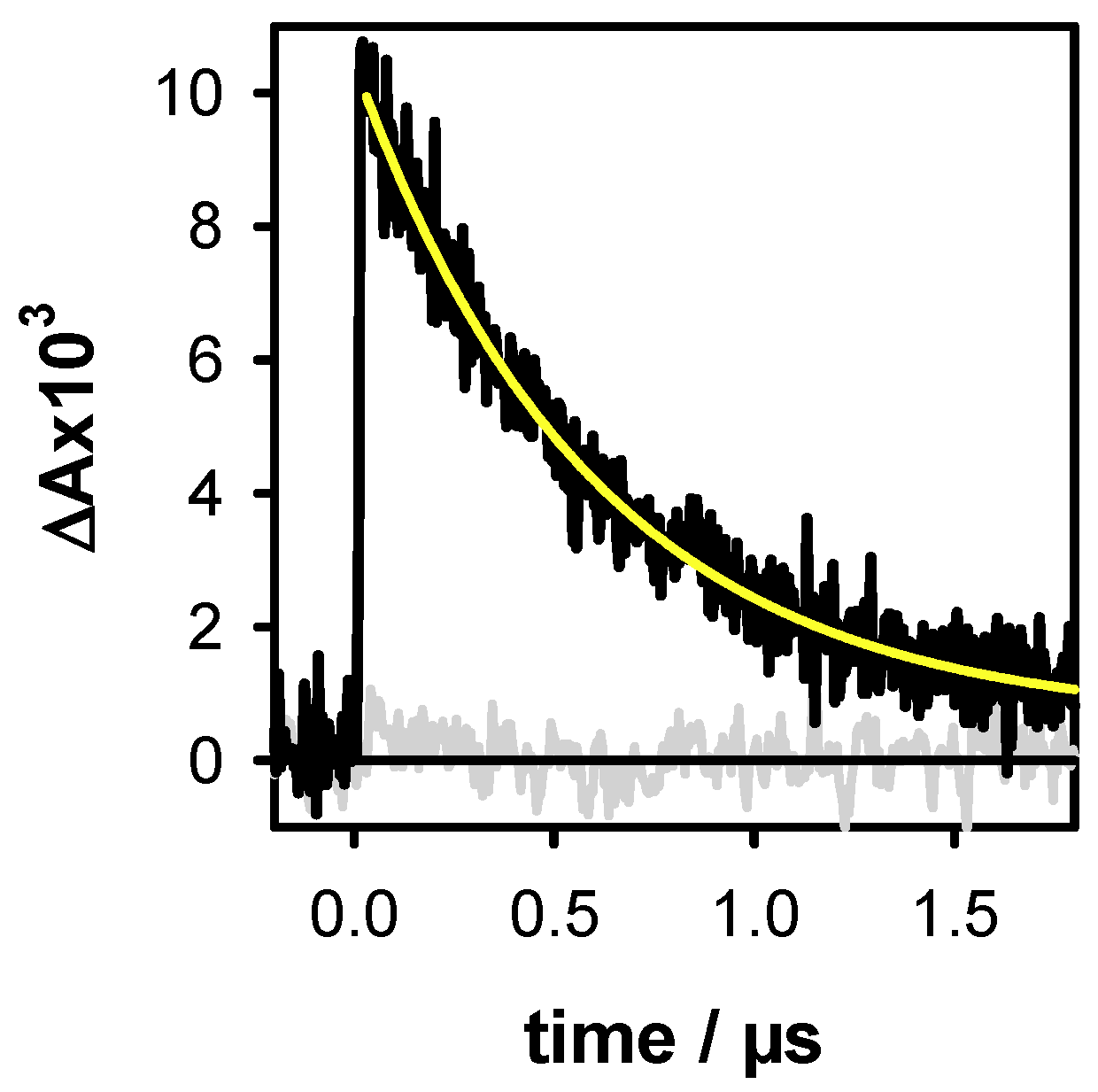
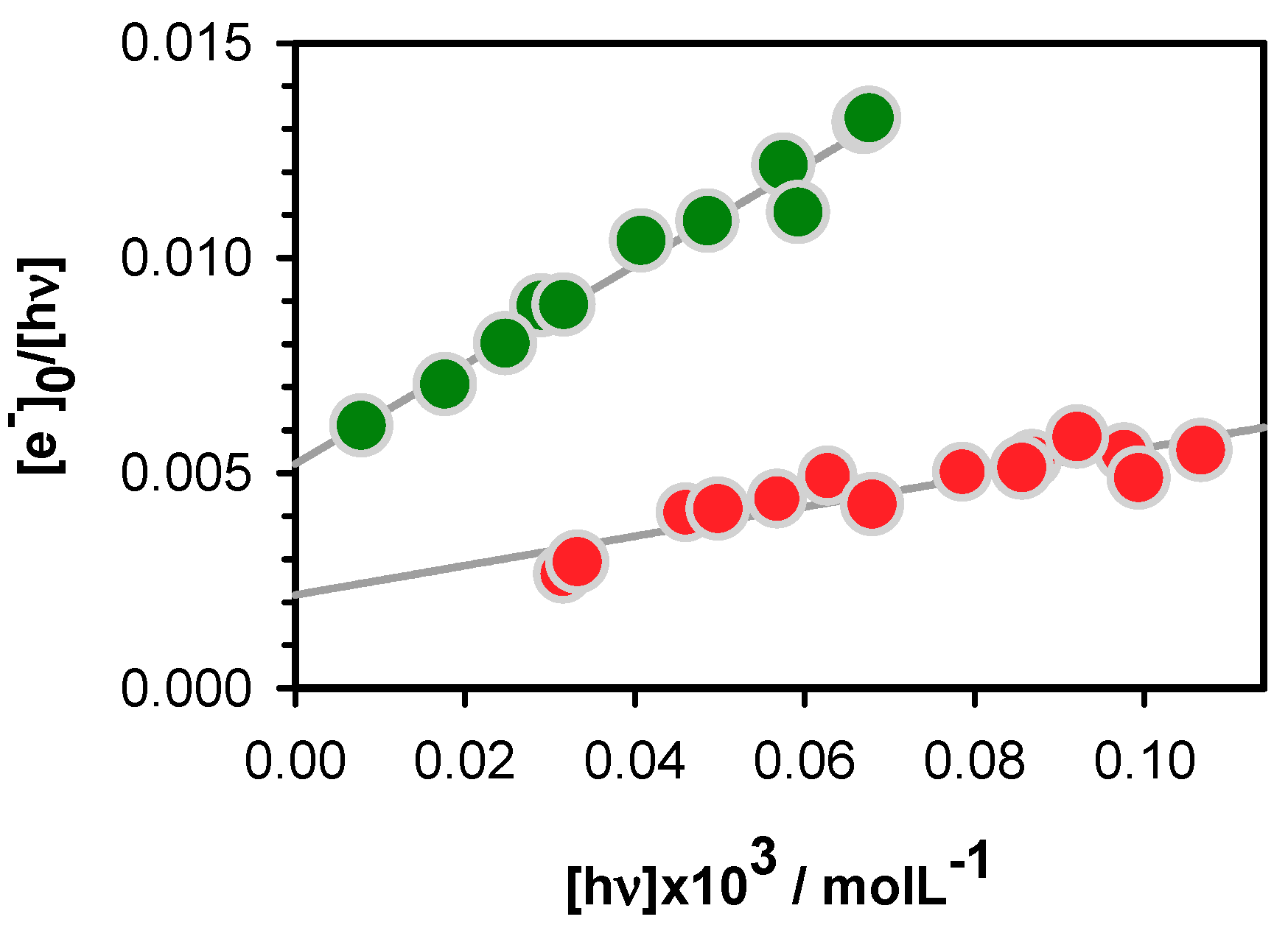
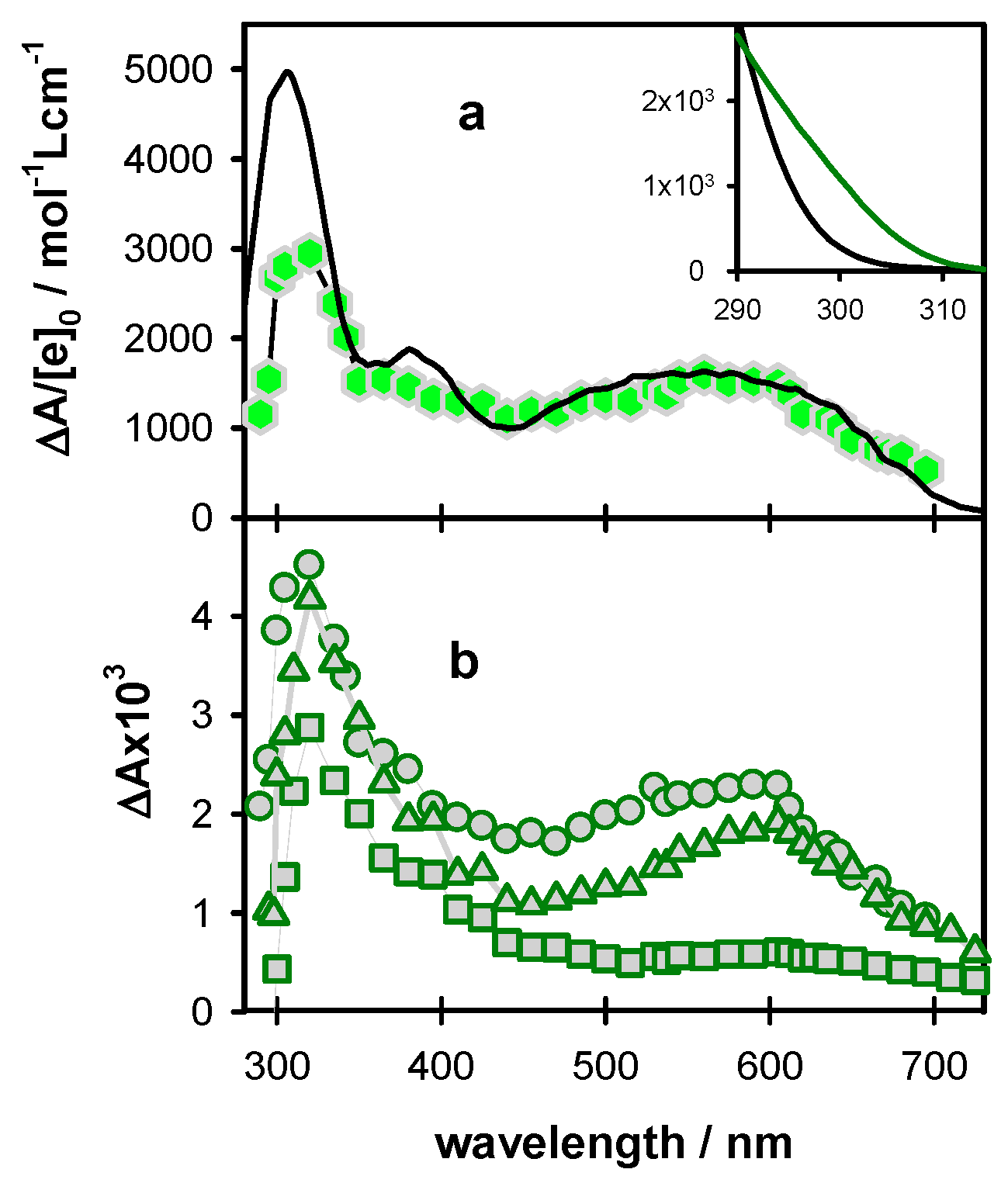
2.2. One- and Two-Photon Ionization
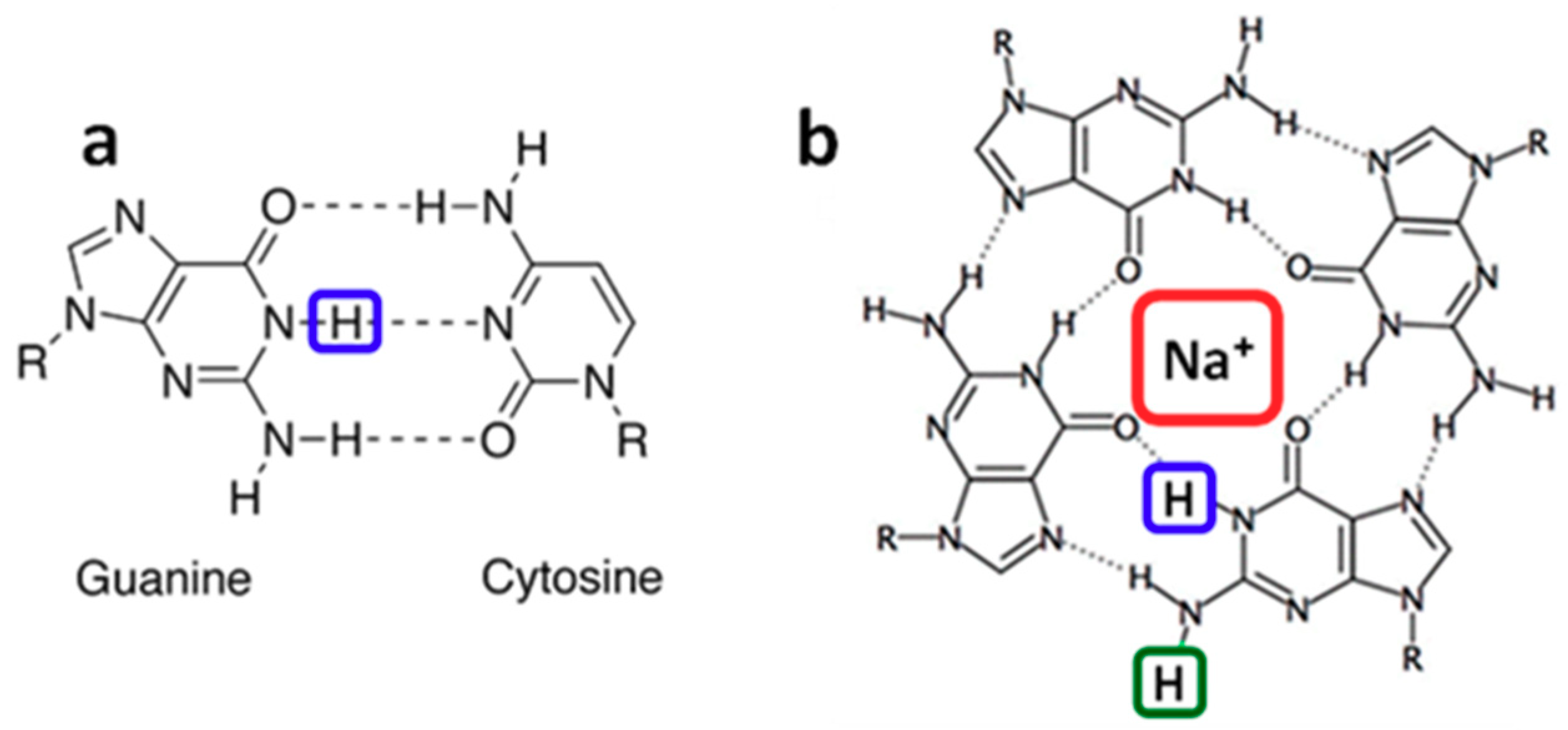
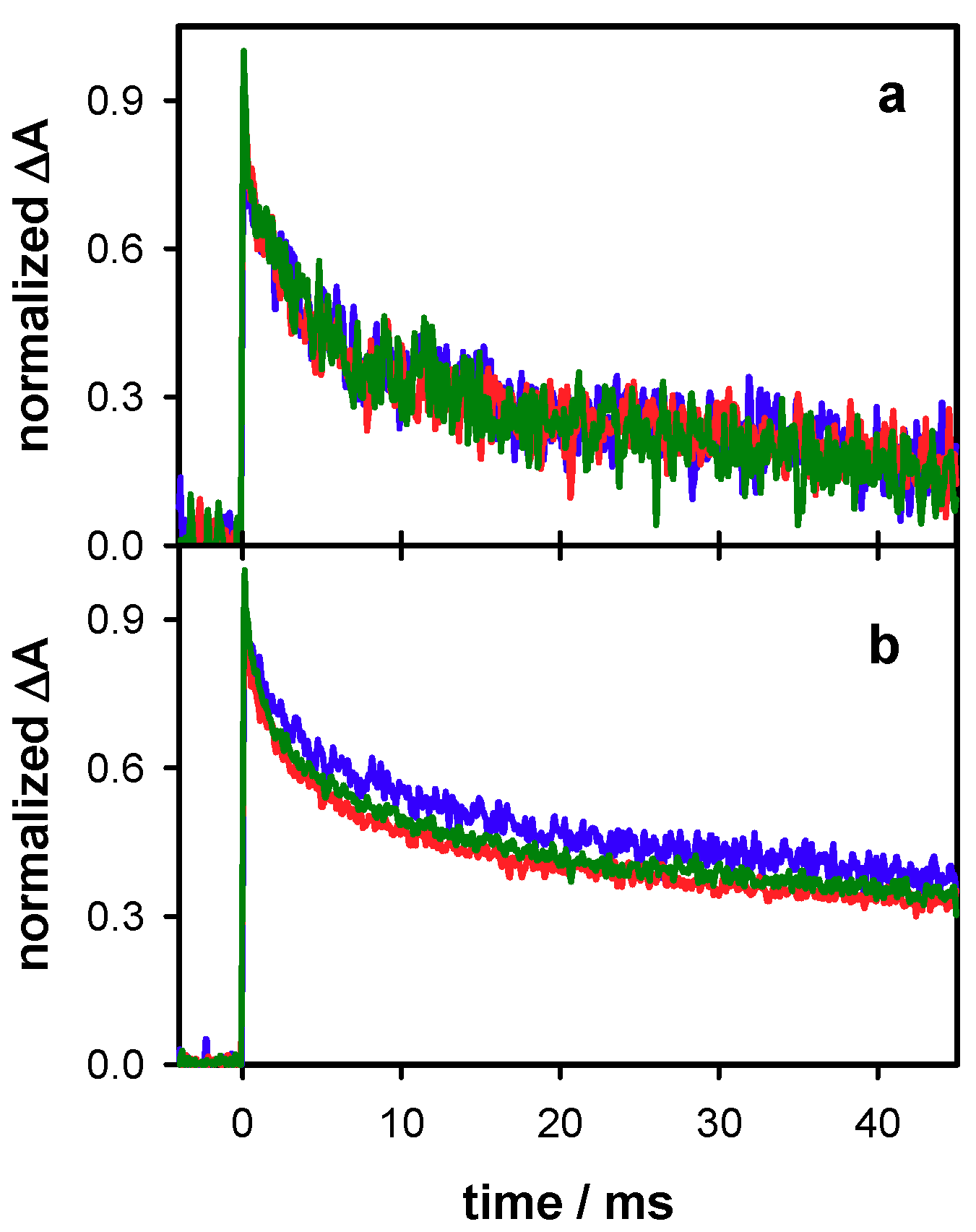
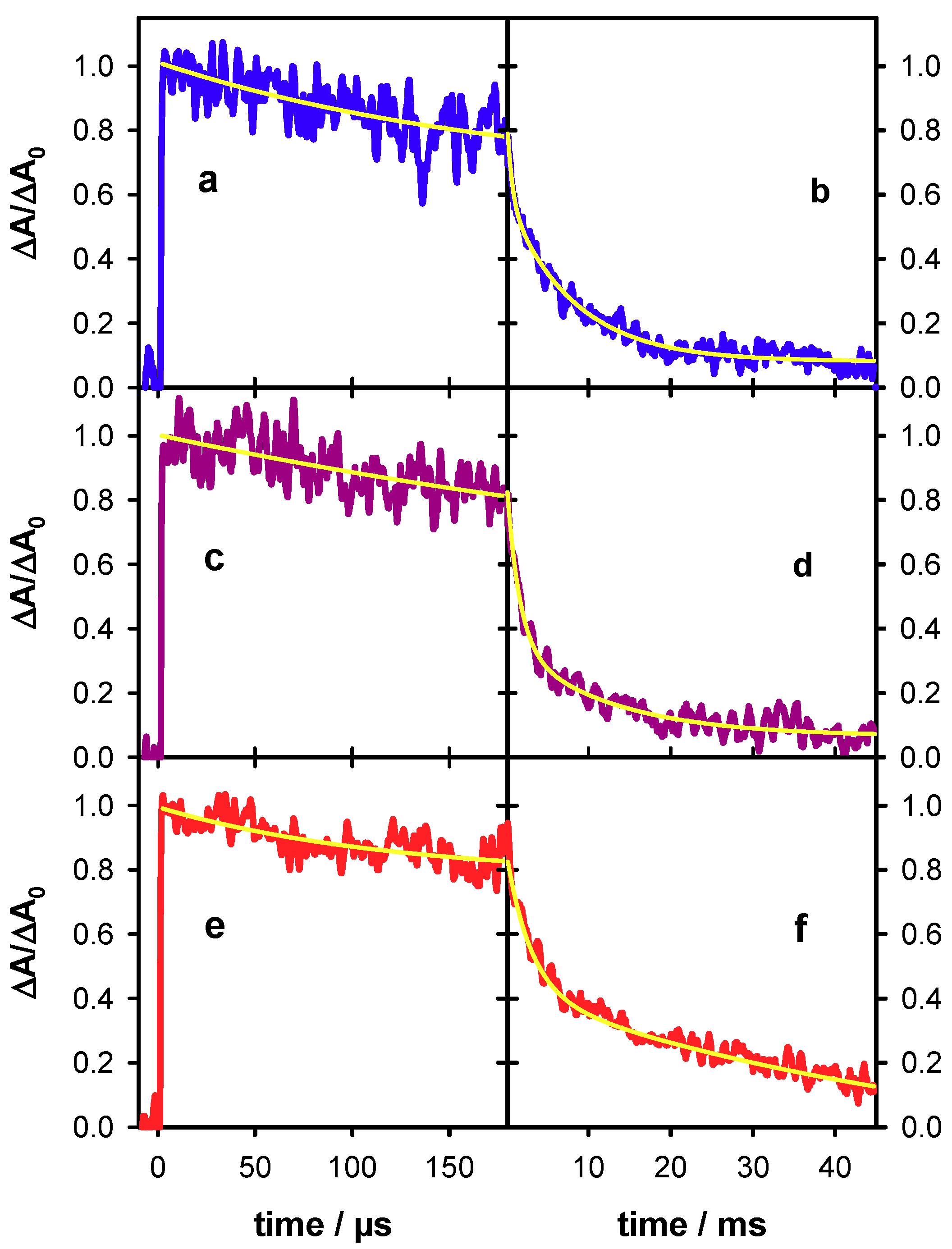
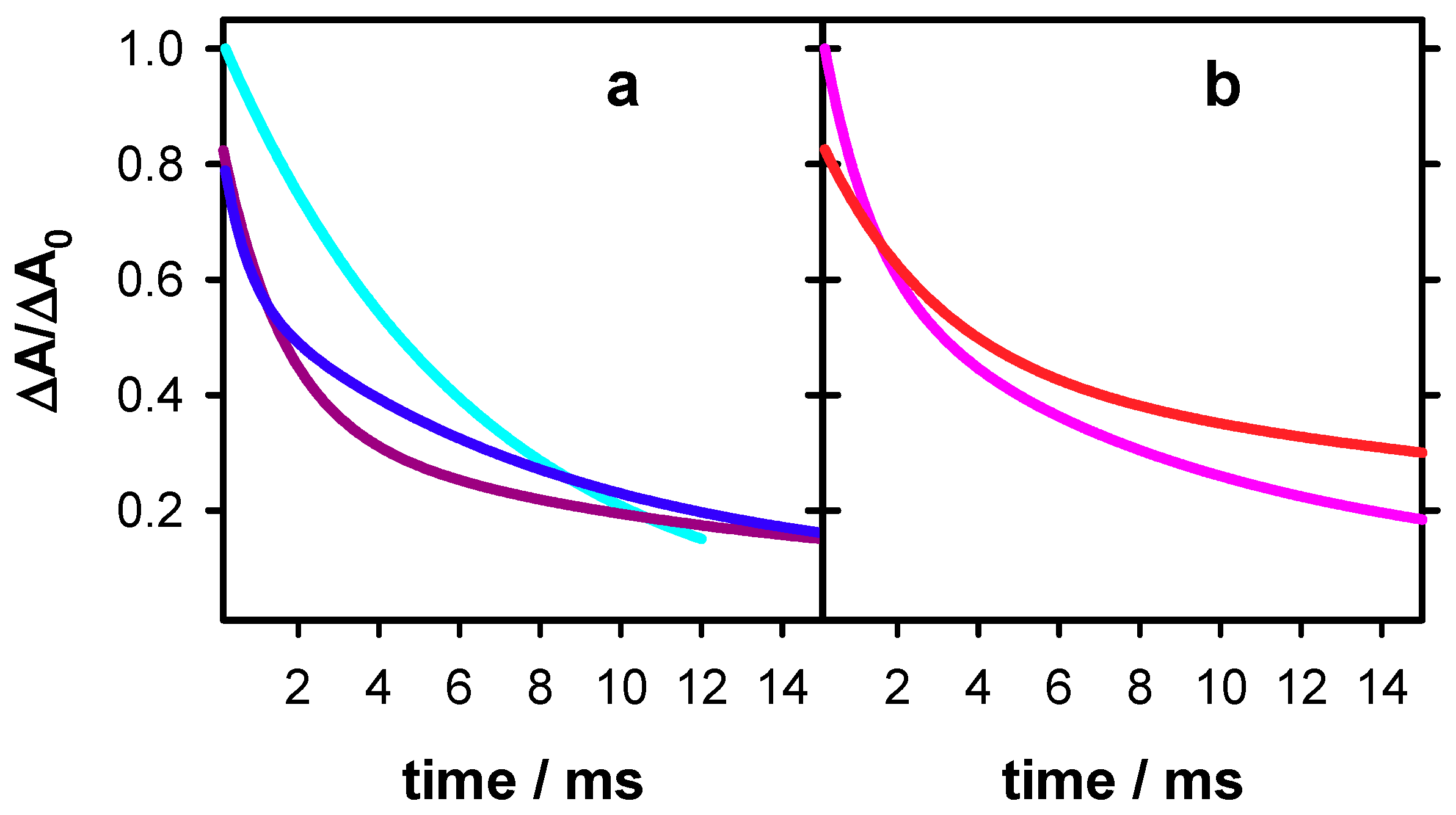
2.3. Radicals in Single and Double Strands
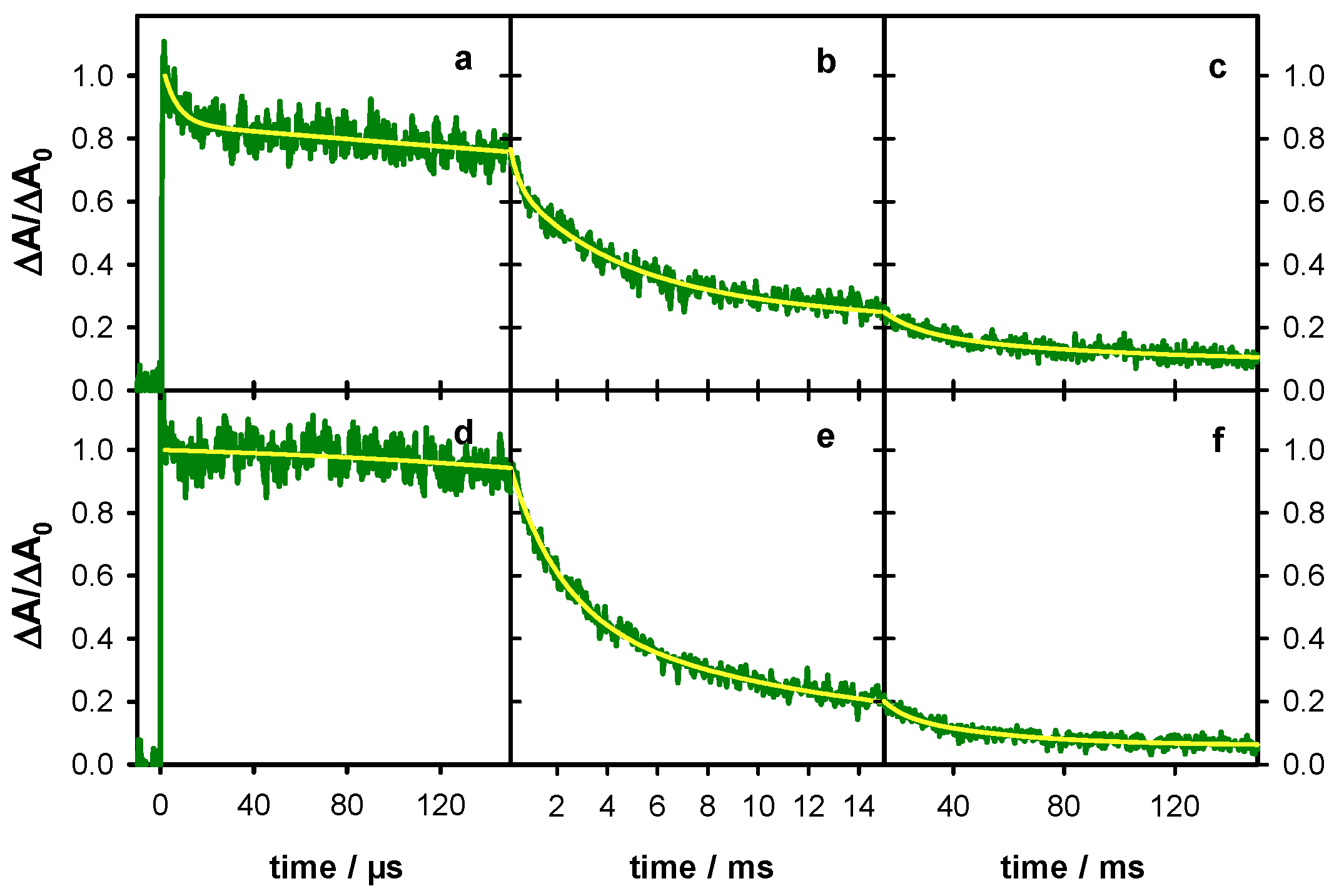
2.4. Radicals in G-Quadruplexes
3. Reaction Schemes of Nucleic Acids
4. Materials and Methods
4.1. Spectroscopic Measurements
4.2. Sample Preparation and Handling
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cadet, J.; Davies, K.J.A. Oxidative DNA damage & repair: An introduction. Free Radic. Biol. Med. 2017, 107, 2–12. [Google Scholar] [PubMed]
- Palecek, E.; Bartosik, M. Electrochemistry of Nucleic Acids. Chem. Rev. 2012, 112, 3427–3481. [Google Scholar] [CrossRef] [PubMed]
- Lewis, F.D.; Letsinger, R.L.; Wasielewski, M.R. Dynamics of photoinduced charge transfer and hole transport in synthetic DNA hairpins. Acc. Chem. Res. 2001, 34, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Kawai, K.; Majima, T. Hole Transfer Kinetics of DNA. Acc. Chem. Res. 2013, 46, 2616–2625. [Google Scholar] [CrossRef] [PubMed]
- Genereux, J.C.; Barton, J.K. Mechanisms for DNA Charge Transport. Chem. Rev. 2010, 110, 1642–1662. [Google Scholar] [CrossRef] [PubMed]
- Kanvah, S.; Joseph, J.; Schuster, G.B.; Barnett, R.N.; Cleveland, C.L.; Landman, U. Oxidation of DNA: Damage to Nucleobases. Acc. Chem. Res. 2010, 43, 280–287. [Google Scholar] [CrossRef]
- Giese, B.; Amaudrut, J.; Köhler, A.-K.; Spormann, M.; Wessely, S. Direct observation of hole transfer through DNA by hopping between adenine bases and by tunneling. Nature 2001, 412, 318–320. [Google Scholar] [CrossRef]
- Cadet, J.; Douki, T.; Ravanat, J.L. Oxidatively generated damage to the guanine moiety of DNA: Mechanistic aspects and formation in cells. Acc. Chem. Res. 2008, 41, 1075–1083. [Google Scholar] [CrossRef]
- Di Mascio, P.; Martinez, G.R.; Miyamoto, S.; Ronsein, G.E.; Medeiros, M.H.G.; Cadet, J. Singlet Molecular Oxygen Reactions with Nucleic Acids, Lipids, and Proteins. Chem. Rev. 2019, 119, 2043–2086. [Google Scholar] [CrossRef]
- Candeias, L.P.; Steenken, S. Stucture and acid-base properties of one-electron-oxidized deoxyguanosine, guanosine, and 1-methylguanosine. J. Am. Chem. Soc. 1989, 111, 1094–1099. [Google Scholar] [CrossRef]
- Kobayashi, K.; Tagawa, S. Direct observation of guanine radical cation deprotonation in duplex DNA using pulse radiolysis. J. Am. Chem. Soc. 2003, 125, 10213–10218. [Google Scholar] [CrossRef] [PubMed]
- Adhikary, A.; Kumar, A.; Becker, D.; Sevilla, M.D. The guanine cation radical: Investigation of deprotonation states by ESR and DFT. J. Phys. Chem. B 2006, 110, 24171–24180. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.; Yamagami, R.; Tagawa, S. Effect of base sequence and deprotonation of guanine cation radical in DNA. J. Phys. Chem. B 2008, 112, 10752–10757. [Google Scholar] [CrossRef] [PubMed]
- Adhikary, A.; Khanduri, D.; Sevilla, M.D. Direct Observation of the Hole Protonation State and Hole Localization Site in DNA-Oligomers. J. Am. Chem. Soc. 2009, 131, 8614–8619. [Google Scholar] [CrossRef] [PubMed]
- Chatgilialoglu, C.; Caminal, C.; Guerra, M.; Mulazzani, Q.G. Tautomers of one-electron-oxidized guanosine. Angew. Chem. Int. Ed. 2005, 44, 6030–6032. [Google Scholar] [CrossRef] [PubMed]
- Chatgilialoglu, C.; Caminal, C.; Altieri, A.; Vougioukalakis, G.C.; Mulazzani, Q.G.; Gimisis, T.; Guerra, M. Tautomerism in the guanyl radical. J. Am. Chem. Soc. 2006, 128, 13796–13805. [Google Scholar] [CrossRef]
- Wu, L.D.; Liu, K.H.; Jie, J.L.; Song, D.; Su, H.M. Direct Observation of Guanine Radical Cation Deprotonation in G-Quadruplex DNA. J. Am. Chem. Soc. 2015, 137, 259–266. [Google Scholar] [CrossRef]
- Banyasz, A.; Martinez-Fernandez, L.; Balty, C.; Perron, M.; Douki, T.; Improta, R.; Markovitsi, D. Absorption of Low-Energy UV Radiation by Human Telomere G-Quadruplexes Generates Long-Lived Guanine Radical Cations. J. Am. Chem. Soc. 2017, 139, 10561–10568. [Google Scholar] [CrossRef]
- Banyasz, A.; Balanikas, E.; Martinez-Fernadez, L.; Baldacchino, G.; Douki, T.; Improrta, R.; Markovitsi, D. Radicals generated in guanine nanostructures by photo-ionization: Spectral and dynamical features. J. Phys. Chem. B 2019, 123, 4950–4957. [Google Scholar] [CrossRef]
- Wala, M.; Bothe, E.; Görner, H.; Schulte-Frohlinde, D. Quantum yields for the generation of hydrated electrons and single strand breaks in poly(C), poly(A) and single-stranded DNA in aqueous solution on 20 ns laser excitation at 248 nm. J. Photochem. Photobiol. A Chem. 1990, 53, 87–108. [Google Scholar] [CrossRef]
- Candeias, L.P.; Steenken, S. Ionization of purine nucleosides and nucleotides and their components by 193-nm laser photolysis in aqueous solution: Model studies for oxidative damage of DNA. J. Am. Chem. Soc. 1992, 114, 699–704. [Google Scholar] [CrossRef]
- Candeias, L.P.; O’Neill, P.; Jones, G.D.D.; Steenken, S. Ionization of polynucleotides and DNA in aqueous solution by 193 nm pulsed laser light: Identification of base derived radicals. Int. J. Radiat. Biol. 1992, 61, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Kuimova, M.K.; Cowan, A.J.; Matousek, P.; Parker, A.W.; Sun, X.Z.; Towrie, M.; George, M.W. Monitoring the direct and indirect damage of DNA bases and polynucleotides by using time-resolved infrared spectroscopy. Proc. Natl. Acad. Sci. USA 2006, 103, 2150–2153. [Google Scholar] [CrossRef] [PubMed]
- Banyasz, A.; Ketola, T.; Muñoz-Losa, A.; Rishi, S.; Adhikary, A.; Sevilla, M.D.; Martinez-Fernandez, L.; Improrta, R.; Markovitsi, D. UV-induced Adenine Radicals Induced in DNA A-tracts: Spectral and Dynamical Characterization. J. Phys. Chem. Lett. 2016, 7, 3949–3953. [Google Scholar] [CrossRef] [PubMed]
- Banyasz, A.; Ketola, T.; Martinez-Fernandez, L.; Improta, R.; Markovitsi, D. Adenine radicals generated in alternating AT duplexes by direct absorption of low-energy UV radiation. Faraday Discuss. 2018, 207, 181–197. [Google Scholar] [CrossRef]
- Banyasz, A.; Martinez-Fernandez, L.; Improta, R.; Ketola, T.M.; Balty, C.; Markovitsi, D. Radicals generated in alternating guanine-cytosine duplexes by direct absorption of low-energy UV radiation. Phys. Chem. Chem. Phys. 2018, 20, 21381–21389. [Google Scholar] [CrossRef] [PubMed]
- Rokhlenko, Y.; Geacintov, N.E.; Shafirovich, V. Lifetimes and Reaction Pathways of Guanine Radical Cations and Neutral Guanine Radicals in an Oligonucleotide in Aqueous Solutions. J. Am. Chem. Soc. 2012, 134, 4955–4962. [Google Scholar] [CrossRef]
- Rokhlenko, Y.; Cadet, J.; Geacintov, N.E.; Shafirovich, V. Mechanistic Aspects of Hydration of Guanine Radical Cations in DNA. J. Am. Chem. Soc. 2014, 136, 5956–5962. [Google Scholar] [CrossRef]
- Merta, T.J.; Geacintov, N.E.; Shafirovich, V. Generation of 8-oxo-7,8-dihydroguanine in G-Quadruplexes Models of Human Telomere Sequences by One-electron Oxidation. Photochem. Photobiol. 2019, 95, 244–251. [Google Scholar] [CrossRef]
- Latus, A.; Alam, M.S.; Mostafavi, M.; Marignier, J.L.; Maisonhaute, E. Guanosine radical reactivity explored by pulse radiolysis coupled with transient electrochemistry. Chem. Commun. 2015, 51, 9089–9092. [Google Scholar] [CrossRef]
- Crespo-Hernandez, C.E.; Arce, R. Near threshhold photo-oxidation of dinucleotides containing purines upon 266 nm nanosecond laser excitation. The role of base stacking, conformation and sequence. J. Phys. Chem. B 2003, 107, 1062–1070. [Google Scholar] [CrossRef]
- Gabelica, V.; Rosu, F.; Tabarin, T.; Kinet, C.; Antoine, R.; Broyer, M.; De Pauw, E.; Dugourd, P. Base-dependent electron photodetachment from negatively charged DNA strands upon 260-nm laser irradiation. J. Am. Chem. Soc. 2007, 129, 4706–4713. [Google Scholar] [CrossRef] [PubMed]
- Marguet, S.; Markovitsi, D.; Talbot, F. One and two photon ionization of DNA single and double helices studied by laser flash photolysis at 266 nm. J. Phys. Chem. B 2006, 110, 11037–11039. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gomez-Mendoza, M.; Banyasz, A.; Douki, T.; Markovitsi, D.; Ravanat, J.L. Direct Oxidative Damage of Naked DNA Generated upon Absorption of UV Radiation by Nucleobases. J. Phys. Chem. Lett. 2016, 7, 3945–3948. [Google Scholar] [CrossRef] [PubMed]
- Gauduel, Y.; Migus, A.; Chambaret, J.P.; Antonetti, A. Femtosecond Reactivity of Electron in Aqueous Solutions. Rev. Phys. Appl. 1987, 22, 1755–1759. [Google Scholar] [CrossRef]
- Torche, F.; Marignier, J.L. Direct Evaluation of the Molar Absorption Coefficient of Hydrated Electron by the Isosbestic Point Method. J. Phys. Chem. B 2016, 120, 7201–7206. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Wang, F.; Denisov, S.A.; Adhikary, A.; Mostafavi, M. Reactivity of prehydrated electrons toward nucleobases and nucleotides in aqueous solution. Sci. Adv. 2017, 3, e1701669. [Google Scholar] [CrossRef]
- Buxton, G.V.; Greenstock, C.L.; Helman, W.P.; Ross, A.B. Critical review of rate constants for reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals (.OH/O−) in aqueous solution. J. Phys. Chem. Ref. Data 1988, 17, 513–886. [Google Scholar] [CrossRef]
- Cadet, J.; Grand, A.; Douki, T. Solar UV radiation-induced DNA Bipyrimidine photoproducts: Formation and mechanistic insights. Top. Curr. Chem. 2015, 356, 249–275. [Google Scholar]
- Douki, T.; Voituriez, L.; Cadet, J. Measurement of pyrimidine (6-4) photoproducts in DNA by a mild acidic hydrolysis-HPLC fluorescence detection assay. Chem. Res. Toxicol. 1995, 8, 244–253. [Google Scholar] [CrossRef]
- Marguet, S.; Markovitsi, D. Time-resolved study of thymine dimer formation. J. Am. Chem. Soc. 2005, 127, 5780–5781. [Google Scholar] [CrossRef] [PubMed]
- Porschke, D. Analysis of a Specific Photoreaction in Oligodoxyadenylic and Polydeoxyadenylic acids. J. Am. Chem. Soc. 1973, 95, 8440–8446. [Google Scholar] [CrossRef] [PubMed]
- Bose, S.N.; Davies, R.J.H.; Sethi, S.K.; McCloskey, J.A. Formation of an adenine- thymine photoadduct in the deoxydinucleosides monophosphate d(TpA) and in DNA. Science 1983, 220, 723–725. [Google Scholar] [CrossRef] [PubMed]
- Bose, S.N.; Kumar, S.; Davies, R.J.H.; Sethi, S.K.; McCloskey, J.A. The Photochemistry of d(T-A) in Aqueous Solution and Ice. Nucleic Acids Res. 1984, 12, 7929–7947. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Sharma, N.D.; Davies, R.J.H.; Phillipson, D.W.; McCloskey, J.A. The isolation and and characterization of a new type of dimeric adenine photoproduct in UV-irradiated deoxyadenylates. Nucleic Acids Res. 1987, 15, 1199–1216. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Joshi, P.C.; Sharma, N.D.; Bose, S.N.; Davies, R.J.H.; Takeda, N.; McCloskey, J.A. Adenine Photodimerization in Deoxyadenylate Sequences—Elucidation of the Mechanism through Structural Studies of a Major d(ApA) Photoproduct. Nucleic Acids Res. 1991, 19, 2841–2847. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.D.; Nadji, S.; Kao, J.L.F.; Taylor, J.S. The structure of d(TpA)*, the major photoproduct of thymidylyl-(3′-5′)-deoxyadenosine. Nucleic Acids Res. 1996, 24, 1554–1560. [Google Scholar] [CrossRef] [PubMed]
- Banyasz, A.; Martinez-Fernandez, L.; Ketola, T.; Muñoz-Losa, A.; Esposito, L.; Markovitsi, D.; Improta, R. Excited State Pathways Leading to Formation of Adenine Dimers. J. Phys. Chem. Lett. 2016, 7, 2020–2023. [Google Scholar] [CrossRef]
- Clingen, P.H.; Davies, R.J.H. Quantum yields of adenine photodimerization in poly(deoxyadenylic acid) and DNA. J. Photochem. Photobiol. B Biol. 1997, 38, 81–87. [Google Scholar] [CrossRef]
- Douki, T. Effect of denaturation on the photochemistry of pyrimidine bases in isolated DNA. J. Photochem. Photobiol. B 2006, 82, 45–52. [Google Scholar] [CrossRef]
- McCullagh, M.; Lewis, F.; Markovitsi, D.; Douki, T.; Schatz, G.C. Conformational control of TT dimerization in DNA conjugates. A molecular dynamics study. J. Phys. Chem. B 2010, 114, 5215–5221. [Google Scholar] [CrossRef] [PubMed]
- Görner, H. Photochemistry of DNA and related biomolecules: Quantum yields and consequences of photoionization. J. Photochem. Photobiol. B Biol. 1994, 26, 117–139. [Google Scholar] [CrossRef]
- Cadet, J.; Wagner, J.R.; Angelov, D. Biphotonic Ionization of DNA: From Model Studies to Cell. Photochem. Photobiol. 2019, 95, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Meggers, E.; Michel-Beyerle, M.E.; Giese, B. Sequence dependent long range hole transport in DNA. J. Am. Chem. Soc. 1998, 120, 12950–12955. [Google Scholar] [CrossRef]
- Yoshioka, Y.; Kitagawa, Y.; Takano, Y.; Yamaguchi, K.; Nakamura, T.; Saito, I. Experimental and theoretical studies on the selectivity of GGG triplets toward one-electron oxidation in B-form DNA. J. Am. Chem. Soc. 1999, 121, 8712–8719. [Google Scholar] [CrossRef]
- Barnett, R.N.; Cleveland, C.L.; Joy, A.; Landman, U.; Schuster, G.B. Charge migration in DNA: Ion-gated transport. Science 2001, 294, 567–571. [Google Scholar] [CrossRef]
- Takada, T.; Kawai, K.; Fujitsuka, M.; Majima, T. Direct observation of hole transfer through double-helical DNA over 100 A. Proc. Natl. Acad. Sci. USA 2004, 101, 14002–14006. [Google Scholar] [CrossRef] [PubMed]
- Renaud, N.; Harris, M.A.; Singh, A.P.N.; Berlin, Y.A.; Ratner, M.A.; Wasielewski, M.R.; Lewis, F.D.; Grozema, F.C. Deep-hole transfer leads to ultrafast charge migration in DNA hairpins. Nat. Chem. 2016, 8, 1015–1021. [Google Scholar] [CrossRef]
- Stemp, E.D.A.; Arkin, M.R.; Barton, J.K. Oxidation of guanine in DNA by Ru(phen)2(dppz)3+ using the flash-quench technique. J. Am. Chem. Soc. 1997, 119, 2921–2925. [Google Scholar] [CrossRef]
- Steenken, S. Purine-Bases, Nucleosides and Nucleotides—Aqueous-Solution Redox Chemistry and transformation Reactions of their Radical Cations and e- and OH Adducts. Chem. Rev. 1989, 89, 503–520. [Google Scholar] [CrossRef]
- Kumar, A.; Sevilla, M.D. Excited States of One-Electron Oxidized Guanine-Cytosine Base Pair Radicals: A Time Dependent Density Functional Theory Study. J. Phys. Chem. A 2019, 123, 3098–3108. [Google Scholar] [CrossRef] [PubMed]
- Onidas, D.; Markovitsi, D.; Marguet, S.; Sharonov, A.; Gustavsson, T. Fluorescence properties of DNA nucleosides and nucleotides: A refined steady-state and femtosecond investigation. J. Phys. Chem. B 2002, 106, 11367–11374. [Google Scholar] [CrossRef]
- Laage, D.; Elsaesser, T.; Hynes, J.T. Water Dynamics in the Hydration Shells of Biomolecules. Chem. Rev. 2017, 117, 10694–10725. [Google Scholar] [CrossRef] [PubMed]
- Borrego-Varillas, R.; Cerullo, G.; Markovitsi, D. Exciton Trapping Dynamics in DNA Multimers. J. Phys. Chem. Lett. 2019, 10, 1639–1643. [Google Scholar] [CrossRef] [PubMed]
- Banyasz, A.; Gustavsson, T.; Onidas, D.; Changenet-Barret, P.; Markovitsi, D.; Importa, R. Multi-Pathway Excited State Relaxation of Adenine Oligomers in Aqueous Solution: A Joint Theoretical and Experimental Study. Chem. Eur. J. 2013, 19, 3762–3774. [Google Scholar] [CrossRef] [PubMed]
- Markovitsi, D.; Talbot, F.; Gustavsson, T.; Onidas, D.; Lazzarotto, E.; Marguet, S. Complexity of excited state dynamics in DNA. Nature 2006, 441, E7. [Google Scholar] [CrossRef] [PubMed]
- Blumen, A.; Klafter, J.; Zumofen, G. Models for reaction dynamics in glasses. In Optical Spectroscopy of Glasses; Zschokke, I., Ed.; Springer: Dordrecht, the Netherlands, 1986; pp. 199–265. [Google Scholar]
- Markovitsi, D.; Germain, A.; Millie, P.; Lécuyer, I.; Gallos, L.; Argyrakis, P.; Bengs, H.; Ringsdorf, H. Triphenylene columnar liquid crystals: Excited states and energy transfer. J. Phys. Chem. 1995, 99, 1005–1017. [Google Scholar] [CrossRef]
- Emelianova, E.V.; Athanasopoulos, S.; Silbey, R.J.; Beljonne, D. 2D Excitons as Primary Energy Carriers in Organic Crystals: The Case of Oligoacenes. Phys. Rev. Lett. 2010, 104, 206405. [Google Scholar] [CrossRef]
- Benichou, O.; Chevalier, C.; Klafter, J.; Meyer, B.; Voituriez, R. Geometry-controlled kinetics. Nat. Chem. 2010, 2, 472–477. [Google Scholar] [CrossRef]
- Dolgushev, M.; Guerin, T.; Blumen, A.; Benichou, O.; Voituriez, R. Contact Kinetics in Fractal Macromolecules. Phys. Rev. Lett. 2015, 115, 208301. [Google Scholar] [CrossRef]
- Candeias, L.P.; Steenken, S. Electron transfer in di(deoxy)nucleoside phosphates in aqueous solution: Rapid migration of oxidative damage (via adenine) to guanine. J. Am. Chem. Soc. 1993, 115, 2437–2440. [Google Scholar] [CrossRef]
- Amand, B.; Bensasson, R. Determination of triplet quantum yields by laser flash absorption spectroscopy. Chem. Phys. Lett. 1975, 34, 44–48. [Google Scholar] [CrossRef]
- Bhat, V.; Cogdell, R.; Crespo-Hernández, C.E.; Datta, A.; De, A.; Haacke, S.; Helliwell, J.; Improta, R.; Joseph, J.; Karsili, T.; et al. Photocrosslinking between nucleic acids and proteins: General discussion. Faraday Discuss. 2018, 207, 283–306. [Google Scholar] [CrossRef] [PubMed]
- Gervasio, F.L.; Laio, A.; Iannuzzi, M.; Parrinello, M. Influence of DNA structure on the reactivity of the guanine radical cation. Chem. Eur. J. 2004, 10, 4846–4852. [Google Scholar] [CrossRef] [PubMed]
- Dumont, E.; Monari, A. Understanding DNA under oxidative stress and sensitization: The role of molecular modeling. Front. Chem. 2015, 3, 43. [Google Scholar] [CrossRef]
Sample Availability: Samples not available. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balanikas, E.; Banyasz, A.; Baldacchino, G.; Markovitsi, D. Populations and Dynamics of Guanine Radicals in DNA strands—Direct versus Indirect Generation. Molecules 2019, 24, 2347. https://doi.org/10.3390/molecules24132347
Balanikas E, Banyasz A, Baldacchino G, Markovitsi D. Populations and Dynamics of Guanine Radicals in DNA strands—Direct versus Indirect Generation. Molecules. 2019; 24(13):2347. https://doi.org/10.3390/molecules24132347
Chicago/Turabian StyleBalanikas, Evangelos, Akos Banyasz, Gérard Baldacchino, and Dimitra Markovitsi. 2019. "Populations and Dynamics of Guanine Radicals in DNA strands—Direct versus Indirect Generation" Molecules 24, no. 13: 2347. https://doi.org/10.3390/molecules24132347
APA StyleBalanikas, E., Banyasz, A., Baldacchino, G., & Markovitsi, D. (2019). Populations and Dynamics of Guanine Radicals in DNA strands—Direct versus Indirect Generation. Molecules, 24(13), 2347. https://doi.org/10.3390/molecules24132347