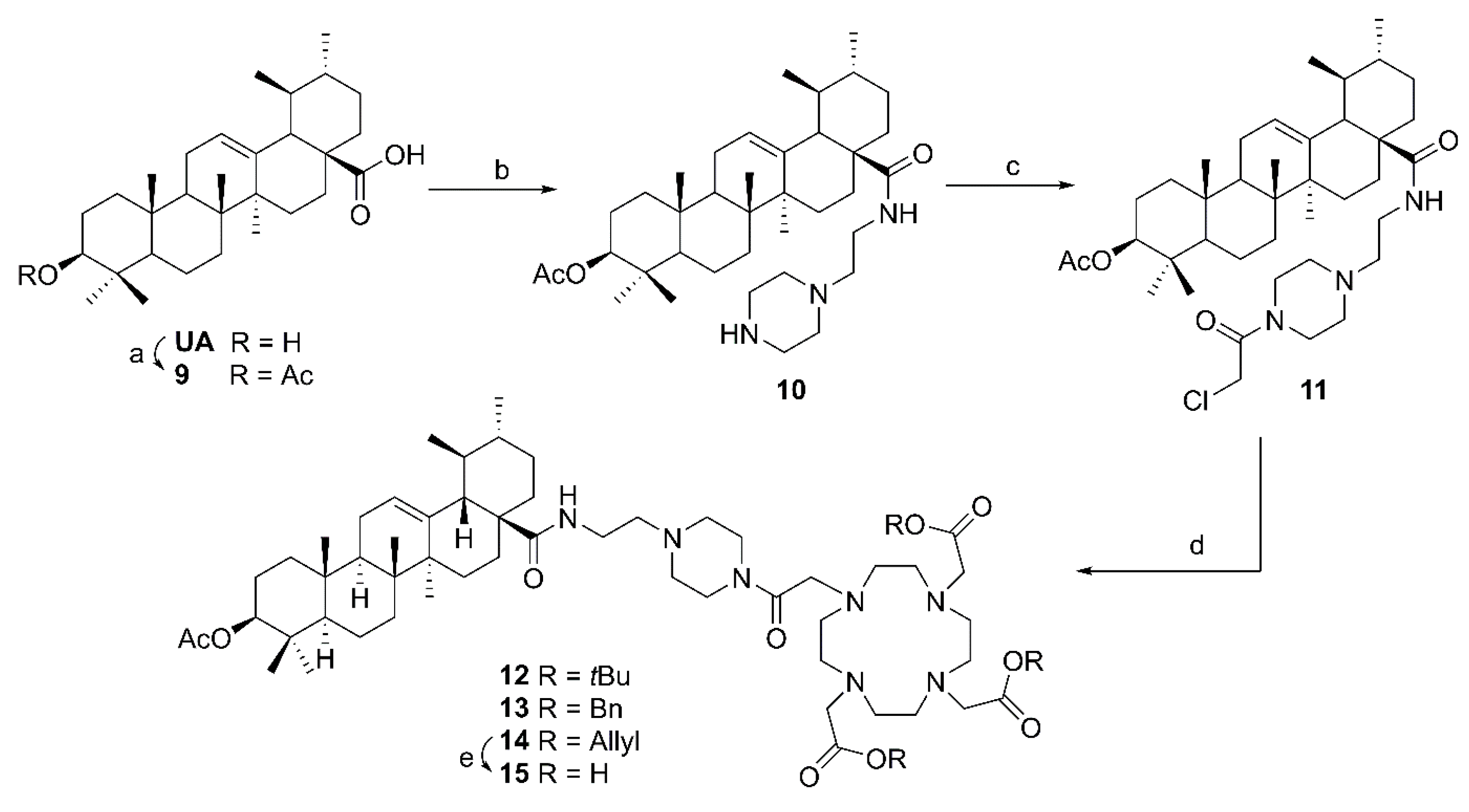
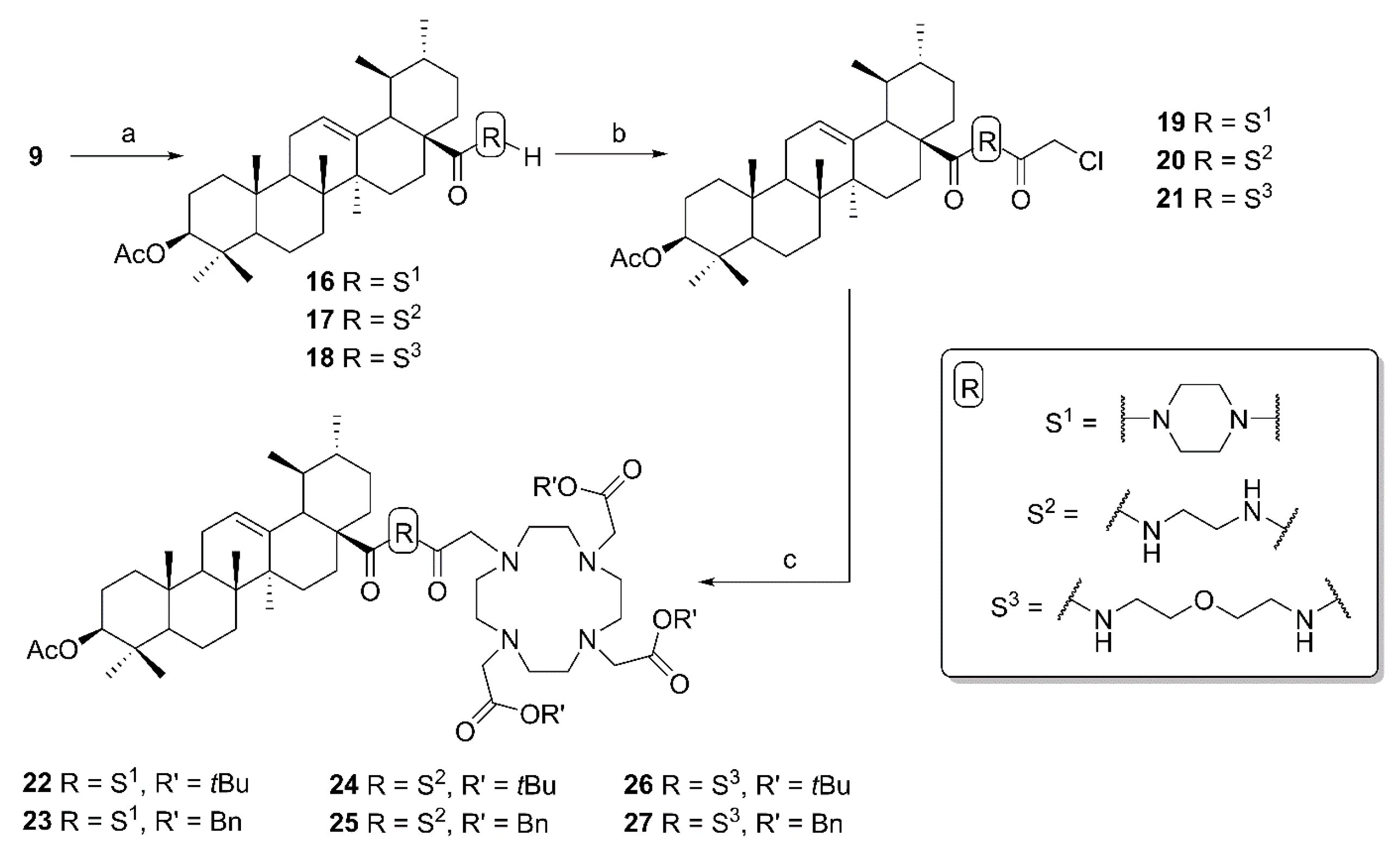
4.3.5. General Procedure D for the Synthesis of Ursolic Acid Chelator Conjugates (12–14, 22–27)
To a solution of the respective chloroacetyl derivative (11, 19–21; 0.44 mmol) and freshly grounded potassium carbonate (0.83 mmol) in dry acetonitrile (15 mL) was added potassium iodide (0.35 mmol) and the respective DOTA precursor (6–8, 0.41 mmol in 5 mL dry acetonitrile). The mixture was stirred for 2–5 days at 25 °C. After completion of the reaction (as indicated by TLC) the mixture was filtered, and the solvent was removed under reduced pressure. The crude products were subjected to column chromatography (silica gel, chloroform/methanol mixtures) to afford compounds 12–14 and 22–27 (yield: 54–88%), respectively.
Allyl bromoacetate (5), To a solution of allyl alcohol (0.62 mol), bromo acetylbromide (0.1 mol) was added dropwise over a period of 30 min at 0 °C under argon atmosphere. The mixture was stirred for 1 h at 0 °C, warmed to 25 °C and stirred for another 3 h. The solvent was removed under reduced pressure and the residue was dissolved in dichloromethane. After usual aqueous work-up, the solvent was removed under reduced pressure, and the crude product was purified by vacuum distillation affording allyl bromoacetate as colorless oil (68%). 1H NMR (400 MHz, CDCl3): δ = 5.88 (ddt, J = 16.5, 11.0, 5.8 Hz, 1H, CH=CH2), 5.38–5.17 (m, 2H, CH=CH2), 4.61 (dt, J = 5.8, 1.4 Hz, 2H, CH2CH=CH2), 3.82 (s, 2H, BrCH2) ppm; 13C NMR (101 MHz, CDCl3): δ = 166.8 (C=O), 131.2 (CH=CH2), 119.0 (CH=CH2), 66.6 (CH2CH=CH2), 25.8 (BrCH2) ppm.
Tri-tert-butyl 2,2′,2’’-(1,4,7,10-tetraazacyclododecane-1,4,7-triyl)triacetate (
6), Compound
6 was prepared from
2 according to general procedure A using
tert-butyl bromoacetate (
3). Column chromatography (SiO
2, CHCl
3/MeOH 9:1) gave
6 (yield: 50%); m.p. 180–182 °C (lit.: 181–183 °C [
25]); R
f = 0.27 (CHCl
3/MeOH 95:5); IR (ATR): ν = 2974
w, 2943
w, 2912
w, 2853
w, 2736
w, 1718
s, 1576
w, 1466
w, 1453
w, 1412
w, 1392
w, 1368
m, 1330
w, 1255
m, 1218
w, 1147
s, 1117
m, 1099
m, 1050
w, 935
m, 873
m, 848
m cm
−1;
1H NMR (400 MHz, CDCl
3): δ = 3.36 (
s, 4H, 2 × CH
2 (acetate)), 3.28 (
s, 2H, CH
2 (acetate)), 3.12–3.06 (
m, 4H, 2 × CH
2 (cyclen)), 2.95–2.84 (
m, 12H, 6 × CH
2 (cyclen)), 1.45 (
s, 18H, 6 × CH
3 (
t-butyl)), 1.45 (
s, 9H, 3 × CH
3 (
t-butyl)) ppm;
13C NMR (101 MHz, CDCl
3): δ = 170.6 (2 × CO, acetate), 169.8 (CO, acetate), 81.9 (C
q,
t-butyl), 81.8 (2 × C
q,
t-butyl), 58.4 (2 × CH
2, acetate), 51.5 (2 × CH
2, cyclen), 51.4 (2 × CH
2, cyclen), 49.4 (2 × CH
2, cyclen), 49.0 (CH
2, acetate), 47.7 (2 × CH
2, cyclen), 28.4 (3 × CH
3,
t-butyl), 28.3 (6 × CH
3,
t-butyl) ppm; MS (ESI, MeOH):
m/
z = 515.3 (100%, [M + H]
+), 537.3 (10%, [M + Na]
+); analysis calcd for C
26H
50N
4O
6 (514.71): C 60.67, H 9.79, N 10.89; found: C 60.51, H 9.98, N 10.67.
Tribenzyl 2,2’,2’’-(1,4,7,10-tetraazacyclododecane-1,4,7-triyl)triacetate (7). Compound 7 was prepared from 2 according to general procedure A using benzyl bromoacetate (4). Column chromatography (SiO2, CHCl3/MeOH 9:1) gave 7 (yield: 68%); Rf = 0.32 (CHCl3/MeOH 95:5); IR (KBr): ν = 2948w, 2857w, 2738w, 1732s, 1586w, 1498w, 1455m, 1418w, 1381w, 1314w, 1169s, 1096m, 1049m, 994m, 739s, 697s cm−1; UV-Vis (CHCl3): λmax (logε) = 251 nm (2.87), 258 nm (2.86), 263 nm (2.76); 1H NMR (400 MHz, CDCl3): δ = 7.40–7.29 (m, 15H, 15 × CH (Bn)), 5.13 (s, 4H, 2 × CH2 (Bn)), 5.13 (s, 2H, CH2 (Bn)), 3.48 (s, 4H, 2 × CH2 (acetate)), 3.41 (s, 2H, CH2 (acetate)), 3.12–3.05 (m, 4H, 2 × CH2 (cyclen)), 2.93–2.79 (m, 12H, 6 × CH2 (cyclen)) ppm; 13C NMR (101 MHz, CDCl3): δ = 171.1 (2 × CO, acetate), 170.3 (CO, acetate), 135.5 (Ci, Bn), 128.8 (CH, Bn), 128.8 (CH, Bn), 128.7 (CH, Bn), 128.7 (CH, Bn), 128.6 (CH, Bn), 66.8 (CH2, Bn), 57.4 (2 × CH2, acetate), 51.9 (2 × CH2, cyclen), 51.7 (2 × CH2, cyclen), 49.6 (2 × CH2, cyclen), 48.8 (CH2, acetate), 47.5 (2 × CH2, cyclen) ppm; MS (ESI, MeOH): m/z = 309.0 (10%, [M + 2H]2+), 617.4 (100%, [M + H]+), 639.3 (10%, [M + Na]+); analysis calcd for C35H44N4O6 (616.76): C 68.16, H 7.19, N 9.08; found: C 67.84, H 7.39, N 8.81.
Triallyl 2,2’,2’’-(1,4,7,10-tetraazacyclododecane-1,4,7-triyl)triacetate (8), Compound 8 was prepared from 2 according to general procedure A using allyl bromoacetate (5). Column chromatography (SiO2, CHCl3/MeOH 95:5) gave 8 (yield: 63%); Rf = 0.27 (CHCl3/MeOH 95:5); IR (KBr): ν = 2945w, 2858w, 2743w, 1731s, 1673w, 1648w, 1455w, 1420w, 1364w, 1314w, 1179s, 1095m, 985s, 928s cm−1; 1H NMR (400 MHz, CDCl3): δ = 5.93–5.81 (m, 3H, 3 × CH (allyl)), 5.32–5.20 (m, 6H, 3 × CH2 (allyl)), 4.60–4.53 (m, 6H, 3 × CH2 (allyl)), 3.49 (s, 4H, 2 × CH2 (acetate)), 3.41 (s, 2H, CH2 (acetate)), 3.12–3.06 (m, 4H, 2 × CH2 (cyclen)), 2.96–2.81 (m, 12H, 6 × CH2 (cyclen)) ppm; 13C NMR (101 MHz, CDCl3): δ = 170.8 (2 × CO, acetate), 170.0 (CO, acetate), 131.7 (2 × CH, allyl), 131.7 (CH, allyl), 119.2 (CH2, allyl), 119.0 (2 × CH2, allyl), 65.5 (2 × CH2, allyl), 65.4 (CH2, allyl), 57.3 (2 × CH2, acetate), 51.7 (2 × CH2, cyclen), 51.6 (2 × CH2, cyclen), 49.4 (2 × CH2, cyclen), 48.5 (CH2, acetate), 47.4 (CH2, cyclen) ppm; MS (ESI, MeOH): m/z = 234.1 (18%, [M + 2H]2+), 467.3 (100%, [M + H]+), 489.3 (10%, [M + Na]+); analysis calcd for C23H38N4O6 (466.6): C 59.21, H 8.21, N 12.01; found: C 59.03, H 8.44, N 11.78.
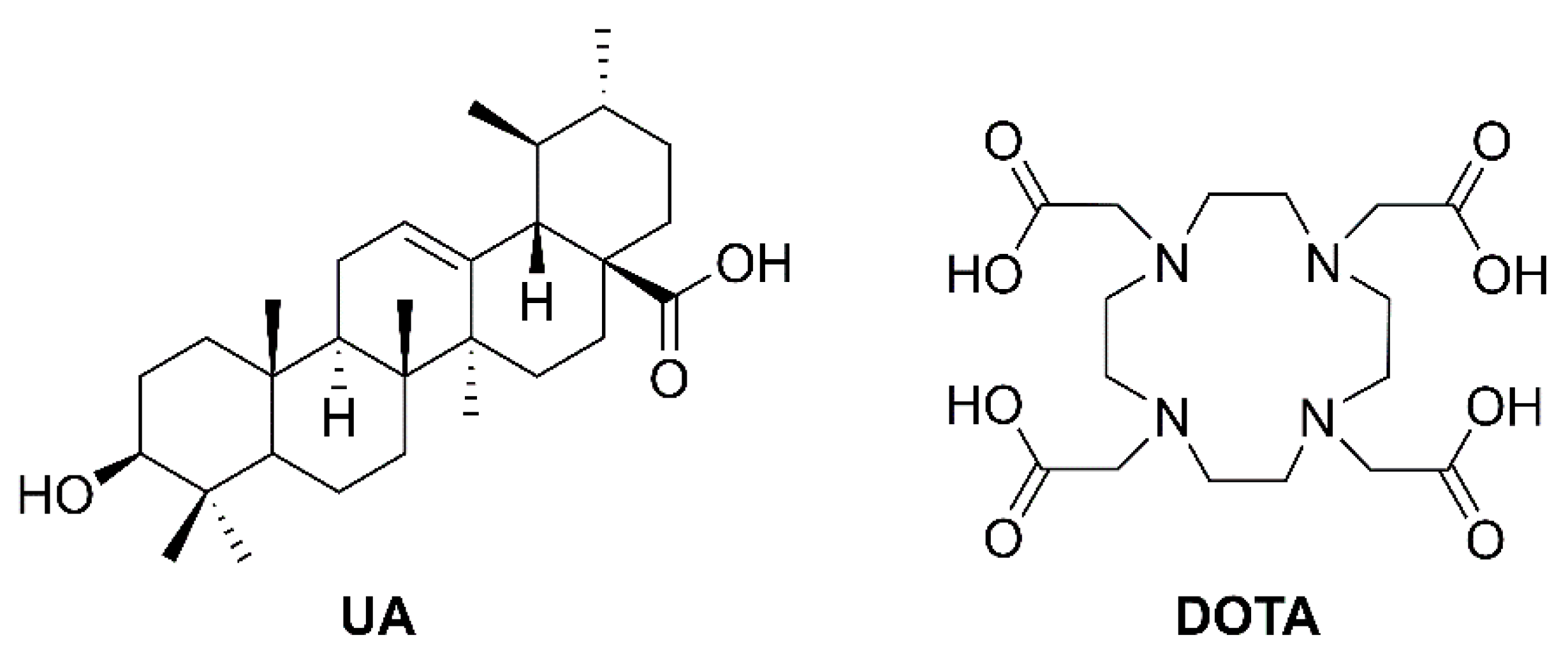
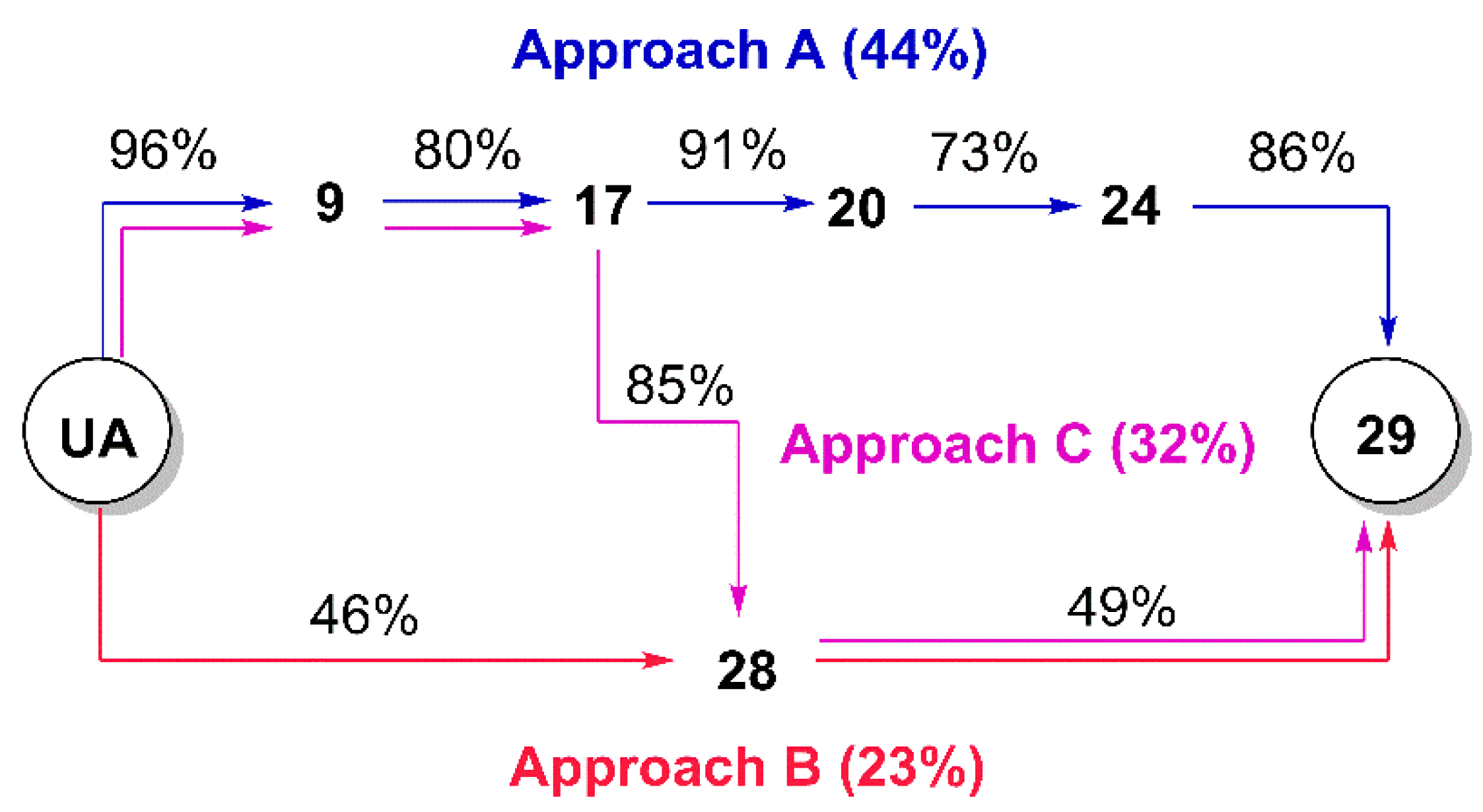
(3β) 3-Acetyloxy-urs-12-en-28-oic acid (
9), Compound
1 was prepared from ursolic acid according to the procedure given in the literature [
26]. Yield: 96%; m.p. 287–290 °C (lit.: 289–290 °C [
27]).
(3β) N-(2-(4-(2-Chloroacetyl)piperazin-1-yl)ethyl)-3-acetyloxy-urs-12-en-28-amide (11). Compound 11 was synthesized from 10 according to general procedure C. Column chromatography (SiO2, CHCl3/acetone 4:1) furnished compound 11 (91%); m.p. 124–129 °C; [α]D = +32.3° (c 0.320, CHCl3); Rf = 0.38 (CHCl3/acetone 4:1); IR (KBr): ν = 3423s, 2947s, 1734m, 1654s, 1522m, 1458m, 1370m, 1247s, 1150w, 1027m cm−1; 1H NMR (400 MHz, CDCl3): δ = 6.32 (t, J = 5.0 Hz, 1H, NH), 5.28 (t, J = 3.6 Hz, 1H, 12-H), 4.49 (dd, J = 10.4, 5.4 Hz, 1H, 3-H), 4.06 (s, 2H, 36-H), 3.72–3.47 (m, 4H, 34-H, 34′-H), 3.47–3.36 (m, 1H, 31-Ha), 3.25–3.15 (m, 1H, 31-Hb), 2.58–2.39 (m, 6H, 33-H, 32-H, 33′-H), 2.04 (s, 3H, Ac), 2.02–1.80 (m, 5H, 11-Ha, 11-Hb, 16-Ha, 22-Ha, 18-H), 1.79–1.70 (m, 1H, 16-Hb), 1.69–1.21 (m, 13H, 15-Ha, 1-Ha, 2-Ha, 2-Hb, 9-H, 6-Ha, 21-Ha, 7-Ha, 22-Hb, 19-H, 6-Hb, 21-Hb, 7-Hb), 1.09 (s, 3H, 27-H), 1.08–1.01 (m, 2H, 1-Hb, 15-Hb), 0.96–0.94 (m, 4H, 20-H, 30-H), 0.93 (s, 3H, 25-H), 0.89–0.86 (m, 3H, 29-H), 0.86 (s, 3H, 23-H), 0.85 (s, 3H, 24-H), 0.84–0.79 (m, 1H, 5-H), 0.78 (s, 3H, 26-H) ppm; 13C NMR (101 MHz, CDCl3): δ = 178.1 (C-28), 171.1 (Ac), 165.2 (C-35), 140.0 (C-13), 125.3 (C-12), 80.9 (C-3), 56.7 (C-32), 55.4 (C-5), 54.2 (C-18), 53.0 (C-33), 52.5 (C-33′), 48.0 (C-17), 47.6 (C-9), 46.5 (C-34), 42.6 (C-14), 42.4 (C-34′), 40.9 (C-36), 39.9 (C-19), 39.7 (C-8), 39.3 (C-20) 38.4 (C-1), 37.8 (C-4), 37.5 (C-22), 37.0 (C-10), 35.9 (C-31), 32.8 (C-7), 31.0 (C-21), 28.2 (C-23), 28.0 (C-15), 25.0 (C-16), 23.6 (C-2), 23.6 (C-11), 23.4 (C-27), 21.4 (Ac), 21.3 (C-30), 18.3 (C-6), 17.5 (C-29), 17.1 (C-26), 16.9 (C-24), 15.8 (C-25) ppm; MS (ESI, MeOH): m/z = 686.5 (100%, [M + H]+); analysis calcd for C40H64ClN3O4 (686.42): C 69.99, H 9.40, N 6.12; found: C 69.70, H 9.63, N 6.02.
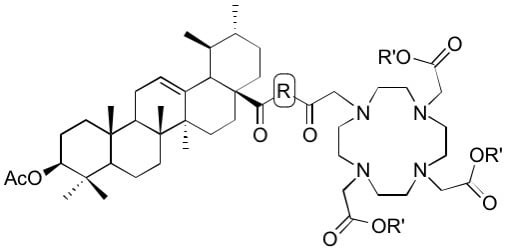
Tris-t-butyl 2’,2’’-[10-[2-[4-[2-(3β-acetyloxy-urs-12-en-28-oylamino)ethyl]piperazin-1-yl]-2-oxoethyl]-1,4,7,10-tetraazacyclododecane-1,4,7-triyl]triacetate (12), Compound 12 was synthesized from 6 and 11 according to general procedure D. Column chromatography (SiO2, CHCl3/MeOH 9:1) furnished compound 12 (54%). m.p. 247–250 °C (decomp.); [α]D = +18.9° (c 0.345, CHCl3); Rf = 0.30 (CHCl3/MeOH 9:1); IR (KBr): ν = 2931m, 1727s, 1644s, 1529w, 1455m, 1425w, 1368s, 1306m, 1228s, 1159s, 1105s, 1005m, 755m cm−1; 1H NMR (400 MHz, CDCl3): δ = 6.38 (s, 1H, NH), 5.26 (t, J = 3.4 Hz, 1H, 12-H), 4.46 (dd, J = 10.5, 5.2 Hz, 1H, 3-H), 3.92–2.04 (m, 36H, 34-H, 34′-H, 36-H, 3 × CH2 (acetate), 31-Ha, 31-Hb, 8 × CH2 (cyclen), 32-H, 33-H, 33′-H), 2.01 (s, 3H, Ac), 2.00–1.68 (m, 4H, 16-Ha, 11-Ha, 11-Hb, 18-H), 1.69–1.14 (m, 14H, 16-Hb, 22-Hb, 15-Ha, 1-Ha, 2-Ha, 2-Hb, 9-H, 6-Ha, 21-Ha, 7-Ha, 19-H, 6-Hb, 21-Hb, 7-Hb), 1.42 (s, 27H, 9 × CH3 (t-Butyl)), 1.05 (s, 3H, 27-H), 1.11–0.95 (m, 3H, 1-Hb, 15-Hb, 20-H), 0.91 (d, J = 6.1 Hz, 3H, 30-H), 0.90 (s, 3H, 25-H), 0.85 (d, J = 6.5 Hz, 3H, 29-H), 0.83 (s, 3H, 23-H), 0.82 (s, 3H, 24-H), 0.80–0.75 (m, 1H, 5-H), 0.74 (s, 3H, 26-H) ppm; 13C NMR (100 MHz, CDCl3): δ = 178.0 (C-28), 172.8 (CO, acetate), 171.0 (Ac), 169.8 (C-35), 139.8 (C-13), 125.3 (C-12), 81.9 (Cq, t-Butyl), 81.7 (2 × Cq, t-Butyl), 80.9 (C-3), 56.8 (C-32), 55.8 (3 × CH2, acetate), 55.3 (C-5), 55.2 (C-36), 53.9 (C-18), 53.4 (8 × CH2, cyclen), 52.2 (C-33, C-33′), 47.8 (C-17), 47.5 (C-9), 44.3 (C-34, C-34′), 42.5 (C-14), 39.8 (C-19), 39.7 (C-8), 39.1 (C-20), 38.4 (C-1), 37.8 (C-4), 37.4 (C-22), 36.9 (C-10), 35.7 (C-31), 32.8 (C-7), 31.0 (C-21), 28.2 (C-23), 28.0 (9 × CH3, t-Butyl), 27.9 (C-15), 24.8 (C-16), 23.6 (C-2), 23.5 (C-11), 23.4 (C-27), 21.4 (Ac), 21.3 (C-30), 18.3 (C-6), 17.4 (C-29), 17.1 (C-26), 16.8 (C-24), 15.7 (C-25) ppm; MS (ESI, MeOH): m/z = 593.9 (100%, [M + Na + H]+), 1186.7 (95%, [M + Na]+); analysis calcd for C66H113N7O10 (1164.67): C 68.06, H 9.78, N 8.42; found: C 67.75, H 9.97, N 8.51.
Tribenzyl 2,2’,2’’-[10-[2-[4-[2-(3β-acetyloxy-urs-12-en-28-oylamino)ethyl]piperazin-1-yl]-2-oxoethyl]-1,4,7,10-tetraazacyclododecane-1,4,7-triyl]triacetate (13). Compound 13 was synthesized from 7 and 11 according to general procedure D. Column chromatography (SiO2, CHCl3/MeOH 95:5) furnished compound 13 (82%). m.p. 142–146 °C; [α]D = +14.5° (c 0.300, CHCl3); Rf = 0.50 (CHCl3/MeOH 9:1); IR (KBr): ν = 3440s, 2947m, 1734s, 1641s, 1456m, 1371m, 1310w, 1247m, 1197s, 1105m, 1006w, 750m cm−1; UV-Vis (CHCl3): λmax (logε) = 257 nm (3.99); 1H NMR (400 MHz, CDCl3): δ = 7.36–7.24 (m, 15H, CHAr), 6.36–6.28 (m, 1H, NH), 5.27 (t, J = 3.7 Hz, 1H, 12-H), 5.21–5.13 (m, 4H, 2 × CH2Bn), 5.12–5.05 (m, 2H, CH2Bn), 4.47 (dd, J = 10.7, 5.1 Hz, 1H, 3-H), 3.72–2.81 (m, 12H, 34-H, 34′-H, 36-H, 3 × CH2 (acetate)), 3.40–3.30 (m, 1H, 31-Ha), 3.22–3.14 (m, 1H, 31-Hb), 2.46–2.33 (m, 6H, 32-H, 33-H, 33′-H), 2.81–2.06 (m, 16H, 8 × CH2 (cyclen)), 2.03 (s, 3H, Ac), 2.00–1.68 (m, 6H, 16-Ha, 11-Ha, 11-Hb, 18-H, 22-Ha, 16-Hb), 1.68–1.20 (m, 13H, 15-Ha, 1-Ha, 2-Ha, 2-Hb, 9-H, 6-Ha, 21-Ha, 7-Ha, 22-Hb, 19-H, 6-Hb, 21-Hb, 7-Hb), 1.06 (s, 3H, 27-H), 1.05–0.94 (m, 3H, 1-Hb, 15-Hb, 20-H), 0.93 (brs, 3H, 30-H), 0.91 (s, 3H, 25-H), 0.85 (d, J = 5.7 Hz, 3H, 29-H), 0.85 (s, 3H, 23-H), 0.83 (s, 3H, 24-H), 0.82–0.77 (m, 1H, 5-H), 0.76 (s, 3H, 26-H) ppm; 13C NMR (101 MHz, CDCl3): δ = 177.9 (C-28), 173.5 (CO, acetate), 170.9 (Ac), 170.0 (C-35), 139.7 (C-13), 135.4 (CAr), 135.3 (CAr), 135.2 (CAr), 128.7 (CHAr), 128.6 (CHAr), 128.5 (CHAr), 128.3 (CHAr), 128.3 (CHAr), 128.2 (CHAr), 125.3 (CHAr), 80.8 (C-3), 67.0 (CH2, Bn), 66.8 (CH2, Bn), 56.7 (C-32), 55.4 (C-36), 55.3 (CH2, acetate), 55.2 (C-5), 53.9 (C-18), 53.4 (CH2, cyclen), 52.7 (C-33, C-33′), 47.7 (C-17), 47.4 (C-9), 42.4 (C-14), 39.7 (C-19), 39.5 (C-8), 39.0 (C-20), 38.3 (C-1), 37.6 (C-4), 37.3 (C-22), 36.8 (C-10), 35.8 (C-31), 32.7 (C-7), 30.9 (C-21), 28.0 (C-23), 27.8 (C-15), 24.8 (C-16), 23.5 (C-2), 23.4 (C-11), 23.2 (C-27), 21.3 (Ac), 21.2 (C-30), 18.1 (C-6), 17.3 (C-29), 17.0 (C-26), 16.7 (C-24), 15.6 (C-25) ppm; MS (ESI, MeOH): m/z = 634 (20%, [M + 2H]2+), 645 (100%, [M + H + Na]2+), 1289 (62%, [M + Na]+); analysis calcd for C75H107N7O10 (1266.72): C 71.11, H 8.51, N 7.74; found: C 70.73, H 8.70, N 7.49.
Triallyl 2,2’,2’’-[10-[2-[4-[2-(3β-acetyloxy-urs-12-en-28-oylamino)ethyl]piperazin-1-yl]-2-oxoethyl]-1,4,7,10-tetraazacyclododecane-1,4,7-triyl]triacetate (14). Compound 14 was synthesized from 8 and 11 according to general procedure D. Column chromatography (SiO2, CHCl3/MeOH 95:5) furnished compound 14 (80%); m.p. 159–163 °C (decomp.); [α]D = +14.8° (c 0.310, CHCl3); Rf = 0.35 (SiO2, CHCl3/MeOH 9:1); IR (KBr): ν = 3342s, 2946m, 2852w, 1734s, 1642s, 1522w, 1456m, 1386w, 1310w, 1246m, 1202m, 1106m, 1026w cm−1; 1H NMR (400 MHz, CDCl3): δ = 6.35 (s, 1H, NH), 5.97 – 5.83 (m, 3H, 3 × CH (allyl)), 5.34–5.19 (m, 7H, 12-H, 3 × CH2 (allyl)), 4.67–4.55 (m, 6H, 3 × CH2 (allyl)), 4.47 (dd, J = 10.6, 5.3 Hz, 1H, 3-H), 3.72–2.97 (m, 14H, 34-H, 34′-H, 36-H, 3 × CH2 (acetate), 31-Ha, 31-Hb), 2.97–2.14 (m, 22H, 8 × CH2 (cyclen), 32-H, 33-H, 33′-H), 2.03 (s, 3H, Ac), 2.01–1.68 (m, 6H, 16-Ha, 11-Ha, 11-Hb, 18-H, 22-Ha, 16-Hb), 1.68–1.19 (m, 13H, 15-Ha, 1-Ha, 2-Ha, 2-Hb, 9-H, 6-Ha, 21-Ha, 7-Ha, 22-Hb, 19-H, 6-Hb, 21-Hb, 7-Hb), 1.07 (s, 3H, 27-H), 1.06–0.94 (m, 3H, 1-Hb, 15-Hb, 20-H), 0.94 (d, J = 6.5 Hz, 3H, 30-H), 0.92 (s, 3H, 25-H), 0.87 (d, J = 6.5 Hz, 3H, 29-H), 0.85 (s, 3H, 23-H), 0.83 (s, 3H, 24-H), 0.82–0.78 (m, 1H, 5-H), 0.76 (s, 3H, 26-H) ppm; 13C NMR (101 MHz, CDCl3): δ = 178.0 (C-28), 173.4 (2 x CO, acetate), 173.3 (CO, acetate), 171.1 (Ac), 170.0 (C-35), 139.8 (C-13), 131.9 (2 × CH, allyl), 131.7 (CH, allyl), 125.4 (C-12), 119.0 (CH2, allyl), 118.8 (2 × CH2, allyl), 80.9 (C-3), 66.0 (CH2, allyl), 65.9 (2 × CH2, allyl), 56.8 (C-32), 55.4 (C-36), 55.3 (C-5), 55.2 (3 × CH2, acetate), 53.9 (C-18), 53.6 (8 × CH2, cyclen), 52.7 (C-33, C-33′), 47.9 (C-17), 47.5 (C-9), 45.0 (C-34, C-34′), 42.6 (C-24), 39.8 (C-19), 39.7 (C-8), 39.1 (C-20), 38.4 (C-1), 37.8 (C-4), 37.4 (C-22), 37.0 (C-10), 35.9 (C-31), 32.8 (C-7), 31.0 (C-21), 28.2 (C-23), 27.9 (C-15), 24.9 (C-16), 23.6 (C-2), 23.6 (C-11), 23.3 (C-27), 21.4 (Ac), 21.3 (C-30), 18.3 (C-6), 17.4 (C-29), 17.1 (C-26), 16.8 (C-24), 15.7 (C-25) ppm; MS (ESI, MeOH): m/z = 569.8 (100%, [M + Na + H]+), 1138.8 (52%, [M + Na]+); analysis calcd for C63H101N7O10 (1116.54): C 67.77, H 9.12, N 8.78; found: C 67.50, H 9.37, N 8.43.
2,2’,2’’-[10-[2-[4-[2-(3β-Acetyloxy-urs-12-en-28-oylamino)ethyl]piperazin-1-yl]-2-oxoethyl]-1,4,7,10-tetraazacyclododecane-1,4,7-triyl]triacetic acid (15). Triphenylphosphane (0.038 mmol), [(PPh3)4Pd] (0.013 mmol) and pyrrolidine (0.290 mmol) were added to a solution of compound 14 (0.128 mmol) in acetonitrile (4 mL), and the mixture was stirred for 6 days at 25 °C. After filtration, the solvent was removed under reduced pressure, and the crude product was subjected to column chromatography (RP18, MeCN/MeOH/TFA 60:40:0.1) affording compound 15 as colorless solid (96%); m.p. 206–210 °C (decomp.); [α]D = +17.1° (c 0.315, MeOH); Rf = 0.35 (RP18, ACN/TFA 100:1); IR (ATR): ν = 2925w, 1634s, 1371m, 1245s, 1199s, 1127s, 1026m, 829m, 800m, 719m cm−1; 1H NMR (400 MHz, CD3OD): δ = 5.35 (t, J = 3.6 Hz, 1H, 12-H), 4.47 (dd, J = 11.0, 5.3 Hz, 1H, 3-H), 3.72–2.93 (m, 14H, 34-H, 34‘-H, 36-H, 31-Ha, 31-Hb, 3 × CH2 (acetate)), 2.91–2.20 (m, 22H, 32-H, 33-H, 33‘-H, 8 × CH2 (cyclen)), 2.09–2.06 (m, 1H, 18-H), 2.03 (s, 3H, Ac), 2.02–1.93 (m, 3H, 11-Ha, 11-Hb, 16-Ha), 1.84–1.23 (m, 15H, 15-Ha, 22-Ha, 1-Ha, 16-Hb, 2-Ha, 2-Hb, 9-H, 7-Ha, 6-Ha, 21-Ha, 22-Hb, 19-H, 6-Hb, 21-Hb, 7-Hb), 1.15 (s, 3H, 27-H), 1.12–0.96 (m, 3H, 15-Hb, 1-Hb, 20-H), 0.99 (s, 3H, 25-H), 0.97 (brs, 3H, 30-H), 0.92 (d, J = 6.4 Hz, 3H, 29-H), 0.89 (s, 3H, 24-H), 0.88 (s, 3H, 23-H), 0.87–0.84 (m, 1H, 5-H), 0.83 (s, 3H,26-H) ppm; 13C NMR (101 MHz, CD3OD): δ = 180.1 (C-28), 172.8 (Ac), 172.5 (CO, acetate), 172.1 (2 × CO, acetate), 171.2 (C-35), 140.2 (C-13), 127.0 (C-12), 82.4 (C-3), 59.8 (CH2, acetate), 59.7 (CH2, acetate), 59.1 (CH2, acetate), 57.7 (C-32), 56.7 (C-5), 55.3 (C-36), 54.4 (C-18), 54.2 (8 × CH2, cyclen), 53.8 (C-33, C-33‘), 49.0 (C-17), 48.8 (C-9), 46.2 (C-34. C-34‘), 43.4 (C-14), 40.9 (C-8), 40.9 (C-19), 40.3 (C-20), 39.4 (C-1), 38.7 (C-4), 38.7 (C-22), 38.1 (C-10), 37.3 (C-31), 34.0 (C-7), 31.9 (C-21), 29.0 (C-15), 28.6 (C-23), 25.4 (C-16), 24.6 (C-2), 24.5 (C-11), 24.0 (C-27), 21.6 (C-30), 21.1 (Ac), 19.3 (C-6), 18.0 (C-26), 17.8 (C-29), 17.2 (C-24), 16.1 (C-25) ppm; MS (ESI, MeOH, positive ion mode): m/z = 1018.6 (27%, [M + Na]+), 1034.7 (100%, [M + K]+); MS (ESI, MeOH, negative ion mode): m/z = 1017.7 (13%, [M − 2H + Na]–), 1032.6 (100%, [M – 2H + K]–); analysis calcd for C54H89N7O10 (996.35): C 65.10, H 9.00, N 9.84; found: C 64.82, H 9.21, N 9.61.
1-(3β-Acetyloxy-urs-12-en-28-oyl)-4-(2-chloroacetyl) piperazine (19). Compound 19 has been synthesized from 16 according to general procedure C. Column chromatography (SiO2, CHCl3/acetone/hexanes 95:5:20) furnished compound 19 (94%); m.p. 155–158 °C; [α]D = +34.3° (c 0.370, CHCl3); Rf = 0.66 (CHCl3/acetone 9:1); IR (ATR): ν = 2924m, 2871w, 1731m, 1658s, 1455m, 1392m, 1370m, 1243s, 1200m, 1145m, 1025m, 985m, 752m cm−1; 1H NMR (400 MHz, CDCl3): δ = 5.21 (t, J = 3.6 Hz, 1H, 12-H), 4.52–4.45 (m, 1H, 3-H), 4.06 (s, 2H, 34-H), 3.73–3.46 (m, 8H, 31-H, 31′-H, 32-H, 32′-H), 2.41 (d, J = 11.6 Hz, 1H, 18-H), 2.24–2.11 (m, 1H, 16-Ha), 2.03 (s, 3H, Ac), 1.91 (dd, J = 8.9, 3.6 Hz, 2H, 11-Ha, 11-Hb), 1.80–1.24 (m, 15H, 15-Ha, 16-Hb, 22-Ha, 1-Ha, 2-Ha, 2-Hb, 22-Hb, 9-H, 6-Ha, 21-Ha, 7-Ha, 19-H, 6-Hb, 21-Hb, 7-Hb), 1.07 (s, 3H, 27-H), 1.12–0.98 (m, 3H, 1-Hb, 15-Hb, 20-H), 0.95 (d, J = 6.2 Hz, 3H, 30-H), 0.93 (s, 3H, 25-H), 0.88 (d, J = 6.4 Hz, 3H, 29-H), 0.85 (s, 3H, 23-H), 0.84 (s, 3H, 24-H), 0.84–0.77 (m, 1H, 5-H), 0.73 (s, 3H, 26-H) ppm; 13C NMR (101 MHz, CDCl3): δ = 175.8 (C-28), 171.1 (Ac), 165.5 (C-33), 138.6 (C-13), 125.5 (C-12), 81.0 (C-3), 55.5 (C-5), 55.1 (C-18), 48.8 (C-17), 47.7 (C-9), 46.3 (C-31), 45.5 (C-31′), 45.1 (C-32), 42.3 (C-32′, C-14), 40.9 (C-34), 39.6 (C-19), 39.6 (C-8), 38.9 (C-20), 38.4 (C-1), 37.8 (C-4), 37.1 (C-10), 34.6 (C-22), 33.1 (C-7), 30.6 (C-21), 28.3 (C-15), 28.2 (C-23), 23.9 (C-27), 23.7 (C-2, C-16), 23.4 (C-11), 21.4 (Ac), 21.4 (C-30), 18.3 (C-6), 17.6 (C-29), 17.0 (C-26), 16.9 (C-24), 15.6 (C-25) ppm; MS (ESI, MeOH): m/z = 643.5 (100%, [M + H]+), 665.4 (56%, [M + Na]+), 1307.3 78%, [2M + Na]+); analysis calcd for C38H59ClN2O4 (643.4): C 70.94, H 9.24, N 4.35; found: C 70.72, H 9.51, N 4.09.
(3β) N-(2-(2-Chloroacetyl)aminoethyl)-3-acetyloxy-urs-12-en-28-amide (20). Compound 20 was synthesized from 17 according to general procedure C. Column chromatography (SiO2, CHCl3/acetone/hexanes 95:5:20) furnished compound 20 (91%); m.p. 103–107 °C; [α]D = +25.2° (c 0.300, CHCl3); Rf = 0.40 (CHCl3/acetone 9:1); IR (KBr): ν = 3422br s, 2948s, 2872m, 1734s, 1640s, 1532s, 1456m, 1370m, 1246s, 1148w, 1092w, 1028m, 756m cm−1; 1H NMR (400 MHz, CDCl3): δ = 7.43 (t, J = 5.1 Hz, 1H, NH), 6.28 (t, J = 5.7 Hz, 1H, NH), 5.32 (t, J = 3.6 Hz, 1H, 12-H), 4.48 (dd, J = 9.7, 6.1 Hz, 1H, 3-H), 4.00 (s, 2H, 34-H), 3.56–3.46 (m, 1H, 31-Ha), 3.41–3.35 (m, 2H, 32-H), 3.26–3.18 (m, 1H, 31-Hb), 2.04 (s, 3H, Ac), 2.03–1.80 (m, 5H, 16-Ha, 11-Ha, 11-Hb, 18-H, 22-Ha), 1.80–1.22 (m, 14H, 16-Hb, 2-Ha, 2-Hb, 1-Ha, 15-Ha, 9-H, 6-Ha, 21-Ha, 7-Ha, 22-Hb, 19-H, 6-Hb, 21-Hb, 7-Hb), 1.08 (s, 3H, 27-H), 1.08–0.95 (m, 3H, 1-Hb, 15-Hb, 20-H), 0.94 (s, 3H, 30-H), 0.93 (s, 3H, 25-H), 0.87 (d, J = 6.5 Hz, 3H, 29-H), 0.86 (s, 3H, 23-H), 0.84 (s, 3H, 24-H), 0.84–0.79 (m, 1H, 5-H), 0.75 (s, 3H, 26-H) ppm; 13C NMR (101 MHz, CDCl3): δ = 180.0 (C-28), 171.1 (Ac), 167.0 (C-33), 139.7 (C-13), 125.9 (C-12), 81.0 (C-3), 55.4 (C-5), 53.8 (C-18), 48.0 (C-17), 47.6 (C-9), 42.6 (C-14), 42.6 (C-34), 41.3 (C-32), 39.9 (C-19), 39.7 (C-8), 39.2 (C-31), 39.2 (C-20), 38.4 (C-1), 37.8 (C-4), 37.4 (C-22), 37.0 (C-10), 32.8 (C-7), 31.0 (C-21), 28.2 (C-23), 27.9 (C-15), 24.9 (C-16), 23.7 (C-2), 23.5 (C-11), 23.4 (C-27), 21.4 (Ac), 21.3 (C-30), 18.3 (C-6), 17.4 (C-29), 17.0 (C-26), 16.8 (C-24), 15.7 (C-25) ppm; MS (ESI, MeOH): m/z = 617.3 (48%, [M + H]+), 639.5 (52%, [M + Na]+), 1255.4 (100%, [2M + Na]+); analysis calcd for C36H57ClN2O4 (617.31): C 70.04, H 9.31, N 4.54; found: C 69.83, H 9.52, N 4.11.
(3β) N-(2-(2-(2-Chloroacetyl)aminoethoxy)ethyl)-3-acetyloxy-urs-12-en-28-amide (21). Compound 21 has been synthesized from 18 according to general procedure C. Column chromatography (SiO2, CHCl3/acetone/hexanes 95:5:20) furnished compound 21 (91%); m.p. 94–97 °C; [α]D = +33.7° (c 0.335, CHCl3); Rf = 0.35 (CHCl3/acetone 9:1); IR (KBr): ν = 3426br s, 2928m, 2872m, 1734m, 1638s, 1528m, 1458w, 1386w, 1248s, 1124w, 1028m cm−1; 1H NMR (400 MHz, CDCl3): δ = 7.00–6.90 (m, 1H, NH), 6.22 (t, J = 5.0 Hz, 1H, NH), 5.30 (t, J = 3.6 Hz, 1H, 12-H), 4.49 (dd, J = 10.0, 5.7 Hz, 1H, 3-H), 4.06 (s, 2H, 36-H), 3.57–3.47 (m, 7H, 32-H, 31-Ha, 33-H, 34-H), 3.31–3.21 (m, 1H, 31-Hb), 2.04 (s, 3H, Ac), 2.02–1.76 (m, 5H, 16-Ha, 11-Ha, 11-Hb, 18-H, 22-Ha), 1.77–1.21 (m, 14H, 16-Hb, 15-Ha, 1-Ha, 2-Ha, 2-Hb, 9-H, 6-Ha, 21-Ha, 7-Ha, 22-Hb, 19-H, 6-Hb, 21-Hb, 7-Hb), 1.09 (s, 3H, 27-H), 1.08–0.95 (m, 3H, 1-Hb, 15-Hb, 20-H), 0.94 (d, J = 6.2 Hz, 3H, 30-H), 0.93 (s, 3H, 25-H), 0.88 (d, J = 6.2 Hz, 3H, 29-H), 0.86 (s, 3H, 23-H), 0.85 (s, 3H, 24-H), 0.84–0.80 (m, 1H, 5-H), 0.78 (s, 3H, 26-H) ppm; 13C NMR (101 MHz, CDCl3): δ = 178.4 (C-28), 171.1 (Ac), 166.1 (C-35), 139.9 (C-13), 125.6 (C-12), 80.9 (C-3), 70.0 (C-33), 69.4 (C-32), 55.4 (C-5), 54.0 (C-18), 48.0 (C-17), 47.6 (C-9), 42.8 (C-36), 42.6 (C-14), 39.9 (C-29), 39.8 (C-34), 39.7 (C-8), 39.2 (C-31), 39.2 (C-20), 38.5 (C-1), 37.8 (C-4), 37.4 (C-22), 37.0 (C-10), 32.8 (C-7), 31.0 (C-21), 28.2 (C-23), 28.0 (C-15), 25.0 (C-16), 23.7 (C-2), 23.6 (C-11), 23.4 (C-27), 21.4 (Ac), 21.4 (C-30), 18.3 (C-6), 17.4 (C-29), 17.1 (C-26), 16.9 (C-24), 15.7 (C-25) ppm; MS (ESI, MeOH): m/z = 661.4 (60%, [M + H]+), 685.5 (86%, [M + Na]+), 1343.3 (100%, [2M + Na]+); analysis calcd for C38H61ClN2O5 (661.37): C 69.01, H 9.30, N 4.24, Cl 5.36; found: C 68.80, H 9.61, N 4.01.
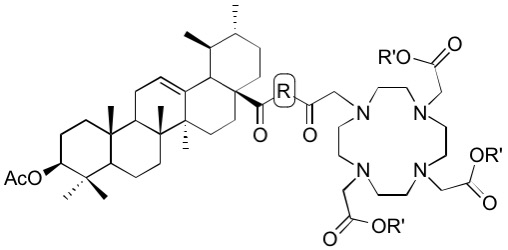
Tri-tert-butyl 2,2’,2’’-[10-[2-[4-(3β-acetyloxy-urs-12-en-28-oyl)piperazin-1-yl]-2-oxoethyl]-1,4,7,10-tetraazacyclododecane-1,4,7-triyl]triacetate (22). Compound 22 was synthesized from 6 and 19 according to general procedure D. Column chromatography (SiO2, CHCl3/MeOH 95:5) furnished compound 22 (88%); m.p. 237–240 °C (decomp.); [α]D = +15.0° (c 0.3, CHCl3); Rf = 0.42 (CHCl3/MeOH 9:1); IR (ATR): ν = 2927w, 2928w, 2871w, 2829w, 1726s, 1646m, 1453m, 1424w, 1368s, 1305m, 1227s, 1160s, 1105s, 1004m, 975m, 754m cm−1; 1H NMR (400 MHz, CDCl3): δ = 5.19 (t, J = 3.5 Hz, 1H, 12-H), 4.47 (dd, J = 9.8, 6.0 Hz, 1H, 3-H), 3.93–2.05 (m, 32H, 31-H, 31′-H, 32-H, 32′-H, 8 × CH2 (cyclen), 3 × CH2 (acetate), 34-H), 2.40 (d, J = 11.0 Hz, 1H, 18-H), 2.19–2.09 (m, 1H, 16-Ha), 2.02 (s, 3H, Ac), 1.93–1.85 (m, 2H, 11-Ha, 11-Hb), 1.82–1.67 (m, 3H, 15-Ha, 16-Hb, 22-Ha), 1.65–1.21 (m, 12H, 2-Ha, 2-Hb, 22-Hb, 1-Ha, 9-H, 21-Ha, 6-Ha, 7-Ha, 19-H, 21-Hb, 6-Hb, 7-Hb), 1.43 (s, 9H, CH3 (tButyl)), 1.42 (s, 9H; CH3 (tButyl)), 1.42 (s, 9H, CH3 (tButyl)), 1.10–0.96 (m, 3H, 1-Hb, 15-Hb, 20-H), 1.05 (s, 3H, 27-H), 0.92 (d, J = 6.3 Hz, 3H, 30-H), 0.91 (s, 3H, 25-H), 0.86 (d, J = 6.3 Hz, 3H, 29-H), 0.84 (s, 3H, 23-H), 0.82 (s, 3H, 24-H), 0.82–0.76 (m, 1H, 5-H), 0.71 (s, 3H, 26-H) ppm; 13C NMR (101 MHz, CDCl3): δ = 175.9 (C-28), 172.8 (CO, acetate), 172.7 (CO, acetate), 171.0 (Ac), 170.7 (C-33), 138.7 (C-13), 125.1 (C-12), 81.9 (Cq, tButyl), 81.7 (Cq, tButyl), 81.7 (Cq, tButyl), 81.0 (C-3), 55.8 (CH2, acetate), 55.8 (CH2, acetate), 55.7 (CH2, cyclen), 55.4 (C-5), 55.1 (C-18), 48.8 (C-17), 47.6 (C-9), 44.6 (C-31, C-31′, C-32, C-32′), 42.3 (C-14), 41.7 (C-34), 39.5 (C-8), 39.4 (C-19), 38.8 (C-20), 38.3 (C-1), 37.8 (C-4), 37.0 (C-10), 34.4 (C-22), 33.1 (C-7), 30.5 (C-21), 28.3 (C-15), 28.2 (C-23), 28.1 (CH3, tButyl), 28.0 (CH3, tButyl), 23.8 (C-27), 23.6 (C-2, C-16), 23.4 (C-11), 21.4 (Ac), 21.3 (C-30), 18.2 (C-6), 17.5 (C-29), 17.0 (C-26), 16.8 (C-24), 15.6 (C-25) ppm; MS (ESI, MeOH): m/z = 1143.7 (100%, [M + Na]+); analysis calcd for C64H108N6O10 (1121.60): C 68.54, H 9.71, N 7.49; found: C 68.31, H 10.03, N 7.27.
Tribenzyl 2,2’,2’’-[10-[2-[4-(3β-acetyloxy-urs-12-en-28-oyl)piperazin-1-yl]-2-oxoethyl]-1,4,7,10-tetraazacyclododecane-1,4,7-triyl]triacetate (23). Compound 23 was synthesized from 19 and 7 according to general procedure D. Column chromatography (SiO2, CHCl3/MeOH 95:5) furnished compound 23 (72%); m.p. 157–161 °C; [α]D = +4.2° (c 0.300, CHCl3); Rf = 0.37 (CHCl3/MeOH 9:1); IR (ATR): ν = 2945w, 2836w, 1730s, 1638m, 1454m, 1424w, 1392m, 1370m, 1302m, 1243s, 1194s, 1105s, 1005m, 966m, 743m, 697s cm−1; UV-Vis (CHCl3): λmax (logε) = 246 nm (3.96), 294 nm (3.47), 364 nm (3.23); 1H NMR (400 MHz, CDCl3): δ = 7.39–7.26 (m, 15H, 15 × CH (Bn)), 5.33–4.97 (m, 7H, 12-H, 3 × CH2 (Bn)), 4.45 (dd, J = 10.1, 5.9 Hz, 1H, 3-H), 4.02–2.05 (m, 34H, 31-H, 31‘-H, 32-H, 32‘-H, 8 × CH2 (cyclen), 3 × CH2 (acetate), 34-H, 16-Ha, 18-H), 2.02 (s, 3H, Ac), 1.95–1.65 (m, 5H, 11-Ha, 11-Hb, 15-Ha, 16-Hb, 22-Ha), 1.65–1.09 (m, 12H, 2-Ha, 2-Hb, 22-Hb, 1-Ha, 9-H, 21-Ha, 6-Ha, 7-Ha, 19-H, 21-Hb, 6-Hb, 7-Hb), 1.03 (s, 3H, 27-H), 1.08–0.95 (m, 3H, 1-Hb, 15-Hb, 20-H), 0.93 (d, J = 6.0 Hz, 3H, 30-H), 0.85 (d, J = 6.3 Hz, 3H, 29-H), 0.85 (s, 3H, 25-H), 0.82 (s, 3H, 23-H), 0.79 (s, 3H, 24-H), 0.79–0.71 (m, 1H, 5-H), 0.66 (s, 3H, 26-H) ppm; 13C NMR (101 MHz, CDCl3): δ = 175.9 (C-28), 173.6 (CO, acetate), 171.0 (Ac), 170.8 (C-33), 138.6 (C-13), 135.5 (Ci, Bn), 135.3 (Ci, Bn), 128.7 (CH, Bn), 128.7 (CH, Bn), 128.6 (CH, Bn), 128.6 (CH, Bn), 128.4 (CH, Bn), 128.4 (CH, Bn), 125.1 (C-12), 81.0 (C-3), 67.1 (CH2, Bn), 66.9 (CH2, Bn), 55.7 (CH2, acetate), 55.4 (CH2, cyclen), 55.2 (C-5), 55.1 (C-18), 48.7 (C-17), 47.6 (C-9), 44.8 (C-31, C-31‘, C-32, C-32‘), 42.2 (C-14), 42.0 (C-34), 39.5 (C-19), 39.5 (C-8), 38.8 (C-20), 38.3 (C-1), 37.7 (C-4), 36.9 (C-10), 34.5 (C-22), 33.1 (C-7), 30.6 (C-21), 28.2 (C-15), 28.1 (C-23), 23.6 (C-2, C-16), 23.5 (C-27), 23.4 (C-11), 21.4 (Ac), 21.3 (C-30), 18.2 (C-6), 17.5 (C-29), 17.0 (C-26), 16.8 (C-24), 15.5 (C-25) ppm; MS (ESI, MeOH): m/z = 1245.8 (100%, [M + Na]+); analysis calcd for C73H102N6O10 (1223.65): C 71.65, H 8.40, N 6.87; found: C 71.42, H 8.69, N 6.56.
Tri-tert-butyl 2,2’,2’’-[10-[2-[2-(3β-acetyloxy-urs-12-en-28-oylamino)ethyl]amino-2-oxoethyl]-1,4,7,10-tetraazacyclododecane-1,4,7-triyl]triacetate (24). Compound 24 was synthesized from 20 and 6 according to general procedure D. Column chromatography (SiO2, CHCl3/MeOH 95:5) furnished compound 24 (73%); m.p. 254–257 °C (decomp.); [α]D = +33.8° (c 0.300, CHCl3); Rf = 0.40 (CHCl3/MeOH 9:1); IR (KBr): ν = 2971m, 2929m, 2829w, 1727s, 1668m, 1520m, 1454m, 1426w, 1368s, 1307m, 1228s, 1159s, 1106s, 1027m, 1006m, 975m, 755m cm−1; 1H NMR (400 MHz, CDCl3): δ = 8.03 (s, 1H, NH), 7.08 (s, 1H, NH), 5.46 (t, J = 3.4 Hz, 1H, 12-H), 4.47 (dd, J = 9.8, 6.1 Hz, 1H, 3-H), 3.80–2.05 (m, 28H, 31-H, 32-H, 34-H, 3 × CH2 (acetate), 8 × CH2 (cyclen)), 2.44–2.38 (m, 1H, 18-H), 2.02 (s, 3H, Ac), 2.00–1.67 (m, 6H, 15-Ha, 11-Ha, 11-Hb, 16-Ha, 16-Hb, 22-Ha), 1.66–1.19 (m, 12H, 1-Ha, 2-Ha, 2-Hb, 22-Hb, 9-H, 6-Ha, 7-Ha, 21-Ha, 19-H, 6-Hb, 21-Hb, 7-Hb), 1.44 (s, 9H, CH3 (tButyl)), 1.43 (s, 9H, CH3 (tButyl)), 1.43 (s, 9H, CH3 (tButyl)), 1.15–0.98 (m, 3H, 20-H, 1-Hb, 15-Hb), 1.04 (s, 3H, 27-H), 0.91 (s, 3H, 25-H), 0.90 (d, J = 6.1 Hz, 3H, 30-H), 0.88 (d, J = 6.1 Hz, 3H, 29-H), 0.84 (s, 3H, 23-H), 0.83 (s, 3H, 24-H), 0.82–0.77 (m, 1H, 5-H), 0.74 (s, 3H, 26-H) ppm; 13C NMR (101 MHz, CDCl3): δ = 178.7 (C-28), 172.4 (CO, acetate), 171.7 (C-33), 171.1 (Ac), 139.0 (C-13), 125.5 (C-12), 82.1 (Cq, tButyl), 82.1 (Cq, tButyl), 82.1 (Cq, tButyl), 81.1 (C-3), 56.4 (3 × CH2, acetate), 55.8 (C-34), 55.8 (CH2, cyclen), 55.4 (C-5), 52.3 (C-18), 47.7 (C-9), 47.6 (C-17), 42.2 (C-14), 40.0 (C-32), 39.7 (C-19), 39.7 (C-8), 38.5 (C-20), 38.4 (C-31, C-1), 37.8 (C-4), 37.4 (C-22), 37.0 (C-10), 32.9 (C-7), 31.2 (C-21), 28.2 (C-23), 28.2 (CH3, tButyl), 28.1 (CH3, tButyl), 28.1 (CH3, tButyl), 27.9 (C-15), 24.4 (C-16), 23.7 (C-2), 23.5 (C-27), 23.4 (C-11), 21.5 (C-30), 21.4 (Ac), 18.4 (C-6), 17.2 (C-29), 17.0 (C-26), 16.9 (C-24), 15.7 (C-25) ppm; MS (ESI, MeOH): m/z = 1117.7 (100%, [M + Na]+); analysis calcd for C62H106N6O10 (1095.56): C 67.97, H 9.75, N 7.67; found: C 67.68, H 10.02, N 7.41.
Tribenzyl 2,2’,2’’-[10-[2-[2-(3β-acetyloxy-urs-12-en-28-oylamino)ethyl]amino-2-oxoethyl]-1,4,7,10-tetraazacyclododecane-1,4,7-triyl]triacetate (25). Compound 25 was synthesized from 7 and 20 according to general procedure D. Column chromatography (SiO2, CHCl3/MeOH 95:5) furnished compound 25 (75%); m.p. 142–146 °C; [α]D = +20.5° (c 0.390, CHCl3); Rf = 0.36 (CHCl3/MeOH 9:1); IR (ATR): ν = 2945w, 2832w, 1732s, 1663m, 1519w, 1454m, 1370m, 1305m, 1245s, 1194s, 1176s, 1105s, 1007m, 965m, 747m, 697s cm−1; UV-Vis (CHCl3): λmax (logε) = 241 nm (3.89), 295 nm (3.29), 364 nm (3.08); 1H NMR (400 MHz, CDCl3): δ = 8.20 (t, J = 4.5 Hz, 1H, NH), 7.39–7.27 (m, 15H, CH (Bn)), 7.01 (t, J = 4.9 Hz, 1H, NH), 5.48 (s, 1H, 12-H), 5.26–5.05 (m, 6H, CH2 (Bn)), 4.47 (dd, J = 10.0, 5.7 Hz, 1H, 3-H), 3.77–2.07 (m, 28H, 31-Ha, 31-Hb, 32-H, 34-H, 3 × CH2 (acetate), 8 × CH2 (cyclen)), 2.41 (d, J = 10.7 Hz, 1H, 18-H), 2.02 (s, 3H, Ac), 2.00–1.64 (m, 6H, 16-Ha, 16-Hb, 11-Ha, 11-Hb, 22-Ha, 15-Ha), 1.64–1.19 (m, 12H, 1-Ha, 2-Ha, 2-Hb, 22-Hb, 9-H, 6-Ha, 7-Ha, 21-Ha, 6-Hb, 19-H, 21-Hb, 7-Hb), 1.17–0.97 (m, 3H, 20-H, 1-Hb, 15-Hb), 1.04 (s, 3H, 27-H), 0.89 (d, J = 6.3 Hz, 6H, 29-H, 30-H), 0.87 (s, 3H, 25-H), 0.83 (s, 3H, 23-H), 0.81 (s, 3H, 24-H), 0.80–0.76 (m, 1H, 5-H), 0.72 (s, 3H, 26-H) ppm; 13C NMR (101 MHz, CDCl3): δ = 178.8 (C-28), 173.2 (CO, acetate), 172.0 (C-33), 171.0 (Ac), 139.0 (C-13), 135.4 (Ci, Bn), 135.4 (Ci, Bn), 135.3 (Ci, Bn), 128.8 (CH, Bn), 128.8 (CH, Bn), 128.7 (CH, Bn), 128.6 (CH, Bn), 125.6 (12-H), 81.0 (3-H), 67.3 (CH2, Bn), 67.3 (CH2, Bn), 67.2 (CH2, Bn), 56.8 (CH2, acetate), 55.4 (C-34), 55.3 (C-5), 55.3 (CH2, cyclen), 52.3 (C-18), 47.7 (C-17), 47.6 (C-9), 42.2 (C-14), 40.1 (C-32), 39.8 (C-19), 39.7 (C-8), 38.5 (C-20), 38.5 (C-31, C-1), 37.8 (C-4), 37.4 (C-22), 37.0 (C-10), 32.9 (C-7), 31.2 (C-21), 28.2 (C-23), 28.0 (C-15), 24.5 (C-16), 23.7 (C-2), 23.5 (C-27), 23.4 (C-11), 21.4 (Ac), 21.4 (C-30), 18.4 (C-6), 17.2 (C-29), 17.1 (C-26), 16.8 (C-24), 15.6 (C-25) ppm; MS (ESI, MeOH): m/z = 1219.8 (100%, [M + Na]+); analysis calcd for C71H100N6O10 (1197.61): C 71.21, H 8.42, N 7.02; found: 70.93, H 8.56, N 6.82.
Tri-tert-butyl 2,2’,2’’-[10-[2-[2-[2-(3β-acetyloxy-urs-12-en-28-oylamino)ethoxy]ethyl]amino-2-oxoethyl]-1,4,7,10-tetraazacyclododecane-1,4,7-triyl]triacetate (26). Compound 26 was synthesized from 6 and 21 according to general procedure D. Column chromatography (SiO2, CHCl3/MeOH 95:5) furnished compound 26 (74%); m.p. 263–266 °C (decomp.); [α]D = +21.9° (c 0.315, CHCl3); Rf = 0.38 (CHCl3/MeOH 9:1); IR (ATR): ν = 2972m, 2930m, 1726s, 1166m, 1523w, 1453m, 1368s, 1307m, 1228s, 1160s, 1106s, 1026m, 1006m, 975m, 755m cm−1; 1H NMR (400 MHz, CDCl3): δ = 7.82 (t, J = 5.6 Hz, 1H, NH), 6.46 (t, J = 5.1 Hz, 1H, NH), 5.33 (t, J = 3.7 Hz, 1H, 12-H), 4.47 (dd, J = 10.2, 5.9 Hz, 1H, 3-H), 3.56–3.30 (m, 9H, 32-H, 31-Ha, 33-H, 36-H, 34-H), 3.28–3.16 (m, 1H, 31-Hb), 3.15–2.05 (m, 22H, 3 × CH2 (acetate), 8 × CH2 (cyclen)), 2.02 (s, 3H, Ac), 2.02–1.21 (m, 19H, 18-H, 16-Ha, 11-Ha, 11-Hb, 22-Hb, 16-Hb, 15-Ha, 1-Ha, 2-Ha, 2-Hb, 9-H, 6-Ha, 21-Ha, 7-Ha, 22-Hb, 19-H, 6-Hb, 21-Hb, 7-Hb), 1.44 (s, 9H, CH3 (tButyl)), 1.43 (s, 18H, CH3 (tButyl)), 1.06 (s, 3H, 27-H), 1.05–0.94 (m, 3H, 1-Hb, 15-Hb, 20-H), 0.92 (s, 3H, 25-H), 0.92 (d, J = 6.2 Hz, 3H, 30-H), 0.86 (d, J = 6.5 Hz, 3H, 29-H), 0.84 (s, 3H, 23-H), 0.83 (s, 3H, 24-H), 0.82–0.77 (m, 1H, 5-H), 0.76 (s, 3H, 26-H) ppm; 13C NMR (101 MHz, CDCl3): δ = 178.3 (C-28), 173.0 (CO, acetate), 172.5 (CO, acetate), 172.1 (C-35), 171.1 (Ac), 139.3 (C-13), 125.7 (C-12), 82.1 (Cq, tButyl), 82.0 (Cq, tButyl), 81.9 (Cq, tButyl), 81.0 (C-3), 69.5 (C-33), 69.0 (C-32), 56.5 (C-36), 55.9 (CH2, acetate), 55.8 (CH2, acetate), 55.7 (CH2, cyclen), 55.4 (C-5), 53.4 (C-18), 47.8 (C-17), 47.6 (C-9), 42.4 (C-14), 39.8 (C-19), 39.7 (C-8), 39.2 (C-31), 39.0 (C-34), 39.0 (C-20), 38.4 (C-1), 37.8 (C-4), 37.3 (C-22), 37.0 (C-10), 32.9 (C-7), 31.1 (C-21), 28.2 (CH3, tButyl), 28.0 (CH3, tButyl), 28.0 (C-23), 28.0 (C-15), 24.9 (C-16), 23.7 (C-2), 23.5 (C-11), 23.4 (C-27), 21.4 (Ac), 21.4 (C-30), 18.3 (C-6), 17.3 (C-29), 17.0 (C-26), 16.8 (C-24), 15.7 (C-25) ppm; MS (ESI, MeOH): m/z = 1161.7 (100%, [M + Na]+); analysis calcd for C64H110N6O11 (1139.6): C 67.45, H 9.73, N 7.37; found: C 67.31, H 9.87, N 7.09.
Tribenzyl 2,2’,2’’-[10-[2-[2-[2-(3β-acetyloxy-urs-12-en-28-oylamino)ethoxy]ethyl]amino-2-oxoethyl]-1,4,7,10-tetraazacyclododecane-1,4,7-triyl]triacetate (27). Compound 27 has been synthesized from 7 and 21 according to general procedure D. Column chromatography (SiO2, CHCl3/MeOH 95:5) furnished compound 27 (62%); m.p. 137–141 °C; [α]D = +15.4° (c 0.350, CHCl3); Rf = 0.35 (CHCl3/MeOH 9:1); IR (ATR): ν = 2945w, 2830w, 1731s, 1662m, 1523w, 1454m, 1390m, 1370m, 1305m, 1245s, 1193s, 1105s, 1026m, 1008m, 967m, 747m, 697m cm−1; UV-Vis (CHCl3): λmax (logε) = 244 nm (3.73), 294 nm (3.36), 363 nm (3.12); 1H NMR (400 MHz, CDCl3): δ = 8.02 (t, J = 5.4 Hz, 1H, NH), 7.37–7.27 (m, 15H, CH (Bn)), 6.48 (t, J = 5.1 Hz, 1H, NH), 5.32 (t, J = 3.5 Hz, 1H, 12-H), 5.24–5.14 (m, 4H, CH2 (Bn)), 5.14–5.05 (m, 2H, CH2 (Bn)), 4.46 (dd, J = 10.2, 5.7 Hz, 1H, 3-H), 3.59–3.33 (m, 9H, 32-H, 33-H, 31-Ha, 34-H, 36-H), 3.33–2.01 (m, 23H, 31-Hb, 8 × CH2 (cyclen), 3 × CH2 (acetate)), 2.02 (s, 3H, Ac), 2.00–1.17 (m, 19H, 18-H, 16-Ha, 11-Ha, 11-Hb, 22-Ha, 16-Hb, 15-Ha, 1-Ha, 2-Ha, 2-Hb, 9-H, 6-Ha, 22-Hb, 7-Ha, 21-Ha, 19-H, 6-Hb, 7-Hb, 21-Hb), 1.05 (s, 3H, 27-H), 1.04 – 0.94 (m, 3H, 1-Hb, 15-Hb, 20-H), 0.90 (s, 3H, 25-H), 0.90 (d, J = 5.8 Hz, 3H, 30-H), 0.85 (d, J = 6.6 Hz, 3H, 29-H), 0.84 (s, 3H, 23-H), 0.82 (s, 3H, 24-H), 0.81–0.76 (m, 1H, 5-H), 0.75 (s, 3H, 26-H) ppm; 13C NMR (101 MHz, CDCl3): δ = 178.3 (C-28), 173.3 (CO, acetate), 173.1 (CO, acetate), 172.3 (C-35), 171.1 (Ac), 139.3 (C-13), 135.4 (Ci, Bn), 135.3 (Ci, Bn), 128.7 (CH, Bn), 128.7 (CH, Bn), 128.7 (CH, Bn), 128.6 (CH, Bn), 128.5 (CH, Bn), 125.7 (C-12), 81.0 (C-3), 69.6 (C-33), 68.9 (C-32), 67.2 (CH2, Bn), 67.2 (CH2, Bn), 56.9 (C-36), 55.4 (3 × CH2, acetate), 55.4 (C-5), 55.3 (8 × CH2, cyclen), 53.3 (C-18), 47.8 (C-17), 47.6 (C-9), 42.4 (C-14), 39.8 (C-19), 39.7 (C-8), 39.2 (C-31, C-34), 38.9 (C-20), 38.4 (C-1), 37.8 (C-4), 37.3 (C-22), 36.9 (C-10), 32.9 (C-7), 31.1 (C-21), 28.2 (C-23), 28.0 (C-15), 24.8 (C-16), 23.6 (C-2), 23.5 (C-11), 23.4 (C-27), 21.4 (Ac), 21.4 (C-30), 18.3 (C-6), 17.3 (C-29), 17.1 (C-26), 16.8 (C-24), 15.7 (C-25) ppm; MS (ESI, MeOH): m/z = 1263.9 (100%, [M + Na]+); analysis calcd for C73H104N6O11 (1241.67): C 70.62, H 8.44, N 6.77; found: C 70.41, H 8.69, N 6.41.
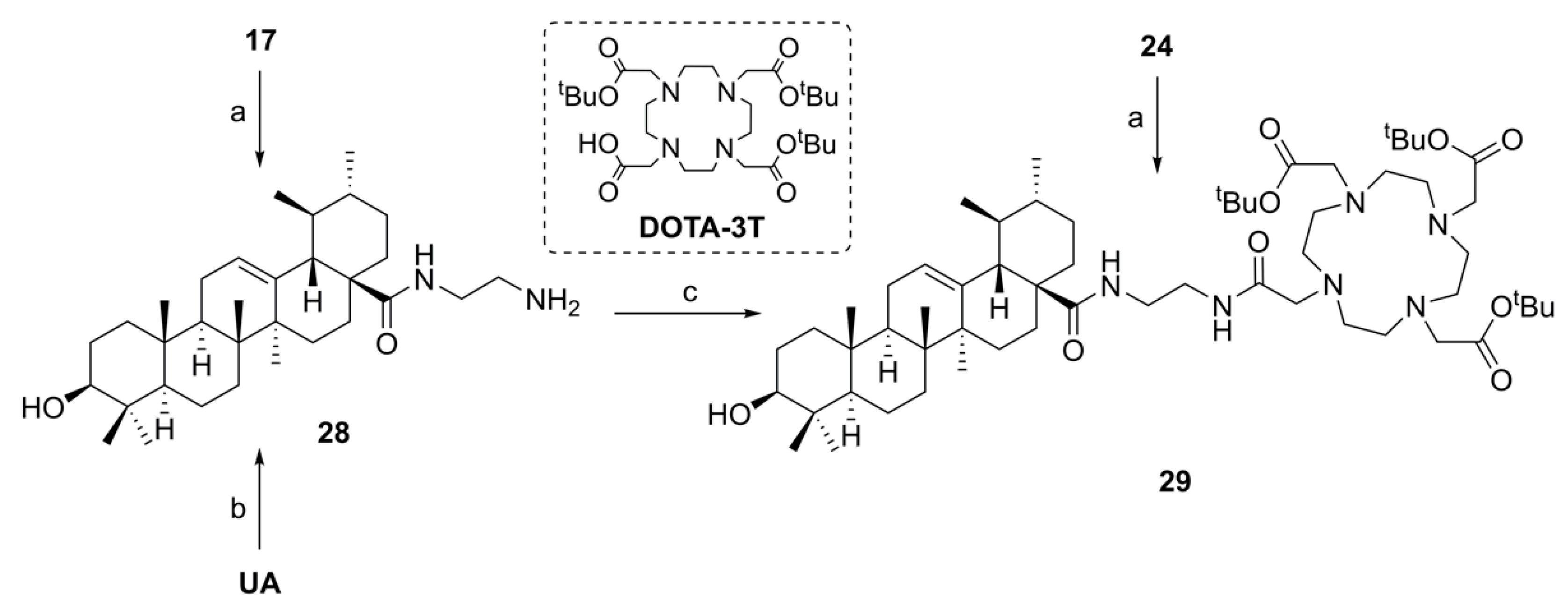
(3β) N-(2-Aminoethyl)-3-hydroxy-urs-12-en-28-amide (
28). Method A: The synthesis was performed according to the procedure given in the
Supplementary material in 82%.
Method B: Ursolic acid (100 mg, 0.20 mmol), HOBt·H
2O (37 mg, 0.24 mmol) and EDC·HCl (46 mg, 0.24 mmol) were dissolved in dry DMF (5 mL), and the mixture was stirred for 30 min at 25 °C. Ethylene diamine (55 µL, 0.82 mmol) was added to the mixture, and stirring was continued for 24 h at 25 °C. Usual aqueous work-up followed by column chromatography (silica gel, CHCl
3/MeOH/NH
4OH 90:10:0.1) gave
28 (46%). Analytical data of this compound can be found in the
supplementary material.
Tri-tert-butyl 2,2’,2’’-[10-[2-[2-(3β-hydroxy-urs-12-en-28-oylamino)ethyl]amino-2-oxoethyl]-1,4,7,10-tetraazacyclododecane-1,4,7-triyl]triacetate (29). Method A: To a solution of DOTA-tris(tert-butyl ester) (58 mg, 0.10 mmol) in dry DMF (8 mL) were added HOBt·H2O (29 mg, 0.19 mmol) and EDC·HCl (29 mg, 0.15 mmol). After stirring for 30 min at 25 °C, a solution of 28 (71 mg, 0.14 mmol) in dry DMF (2 mL) was added and stirring was continued for 5 days. After usual aqueous work-up, the solvent was removed under reduced pressure and the crude product was subjected to column chromatography (silica gel, CHCl3/MeOH 9:1) yielding compound 29 as colorless solid. Yield: 49%. Method B: Compound 24 (50 mg, 0.10 mmol) was dissolved in methanol (7 mL) and a solution of potassium hydroxide (12 mg, 0.21 mmol) in methanol (1 mL) was added. The mixture was stirred at 25 °C for 24 h. After completion of the reaction (as indicated by TLC) and usual work-up, the solvent was removed under reduced pressure, and the residue was subjected to column chromatography (silica gel, CHCl3/MeOH 9:1) affording 29 (yield: 86%); m.p. 136–139 °C; [α]D = −44.4° (c 0.330, MeOH); Rf = 0.29 (CHCl3/MeOH 9:1); IR (KBr): ν = 3300br w, 2973w, 2928m, 2869w, 1728s, 1668m, 1525m, 1455m, 1425w, 1368s, 1307m, 1227s, 1159s, 1121m, 1106s, 1047w, 1006w cm−1; 1H NMR (400 MHz, CDCl3): δ = 8.84 (s, 1H, NH), 7.55 (s, 1H, NH), 5.36 (t, J = 3.5 Hz, 1H, 12-H), 3.59–2.05 (m, 28H, 31-H, 32-H, 33-H, 3 × CH2 (acetate), 8 × CH2 (cyclen)), 3.20 (dd, J = 11.1, 4.8 Hz, 1H, 3-H), 2.34 (d, J = 11.0 Hz, 2H, 18-H), 2.03–1.96 (m, 1H, 16-Ha), 1.95–1.83 (m, 3H, 11-Ha, 11-Hb, 16-Hb), 1.83–1.72 (m, 1H, 22-Ha), 1.64–1.21 (m, 13H, 1-Ha, 15-Ha, 2-Ha, 2-Hb, 22-Hb, 6-Ha, 9-H, 7-Ha, 21-Ha, 19-H, 6-Hb, 7-Hb, 21-Hb), 1.45 (s, 9H, CH3 (tButyl)), 1.43 (s, 18H, CH3 (tButyl)), 1.05 (s, 3H, 27-H), 1.04–0.97 (m, 3H, 1-Hb, 15-Hb, 20-H), 0.97 (s, 3H, 23-H), 0.89 (d, J = 6.3 Hz, 3H, 30-H), 0.89 (s, 3H, 25-H), 0.85 (d, J = 6.4 Hz, 3H, 29-H), 0.76 (s, 3H, 24-H), 0.75 (s, 3H, 26-H), 0.72–0.68 (m, 1H, 5-H) ppm; 13C NMR (101 MHz, CDCl3): δ = 178.4 (C-28), 172.4 (CO, acetate), 171.8 (C-33), 138.9 (C-13), 125.3 (C-12), 82.2 (Cq, tButyl), 82.1 (Cq, tButyl), 82.1 (Cq, tButyl), 79.2 (C-3), 56.0 (CH2, acetate), 55.9 (C-34), 55.8 (CH2, cyclen), 55.3 (C-5), 52.5 (C-18), 47.8 (C-9), 47.5 (C-17), 42.2 (C-14), 40.0 (C-32), 39.7 (C-8), 39.6 (C-19), 38.9 (C-4), 38.7 (C-20), 38.7 (C-31, C-1), 37.4 (C-22), 37.1 (C-10), 33.2 (C-7), 31.3 (C-21), 28.3 (C-23), 28.2 (CH3, tButyl), 28.2 (CH3, tButyl), 28.1 (CH3, tButyl), 28.0 (C-15), 27.4 (C-2), 24.2 (C-16), 23.6 (C-27), 23.5 (C-11), 21.5 (C-30), 18.6 (C-6), 17.2 (C-29), 17.1 (C-26), 15.8 (C-24), 15.7 (C-25) ppm; MS (ESI, MeOH): m/z = 1075.7 (100%, [M + Na]+); analysis calcd for C60H104N6O9 (1053.53): C 68.40, H 9.95, N 7.98; found: C 60.09, H 10.11, N 7.83.














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}