
Do We Need Anti-Prion Compounds to Treat Alzheimer’s Disease?


Abstract
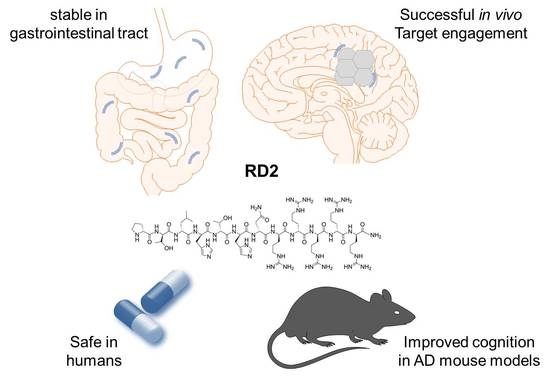
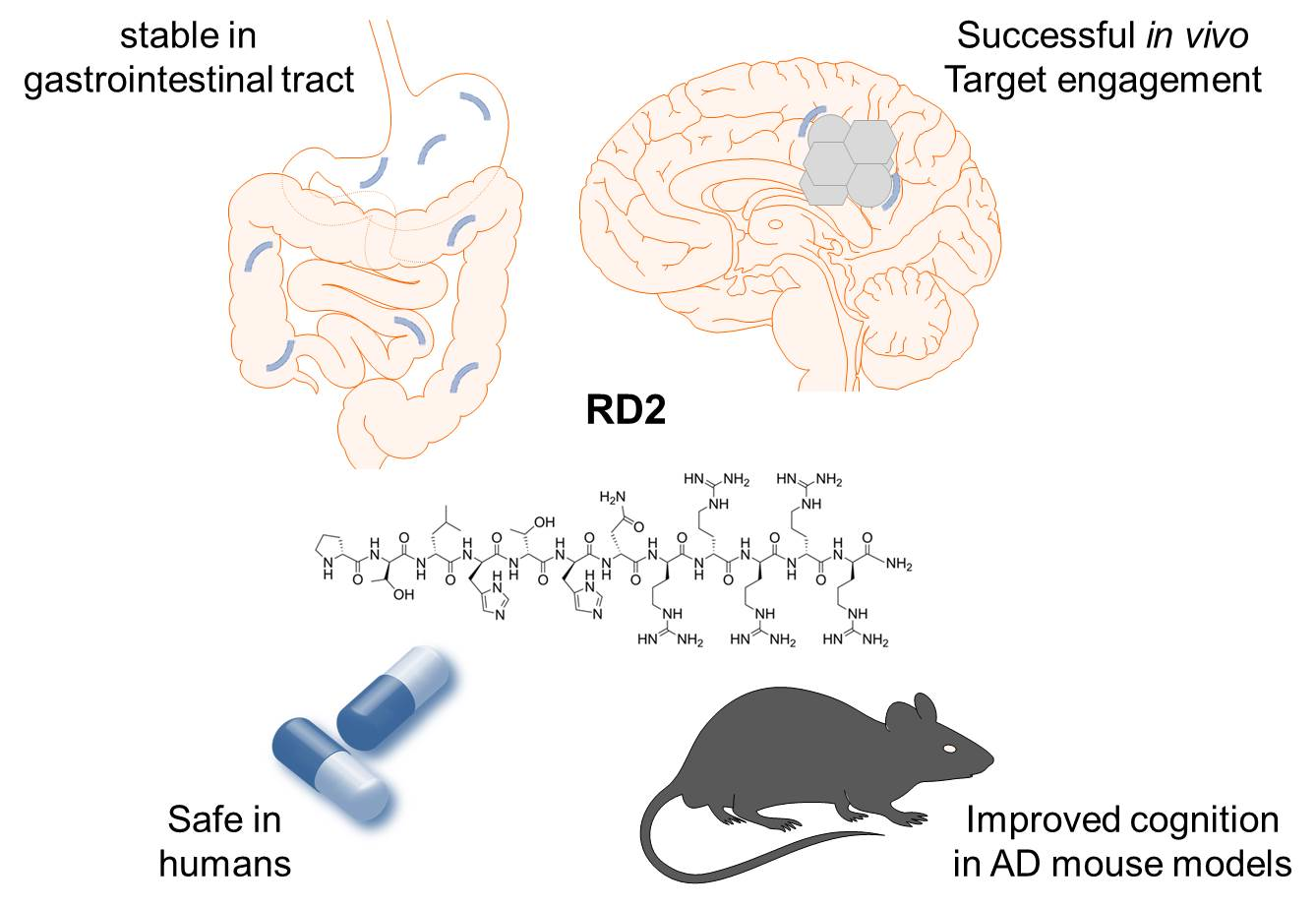{kind=link}
{kind=link}
{kind=link}
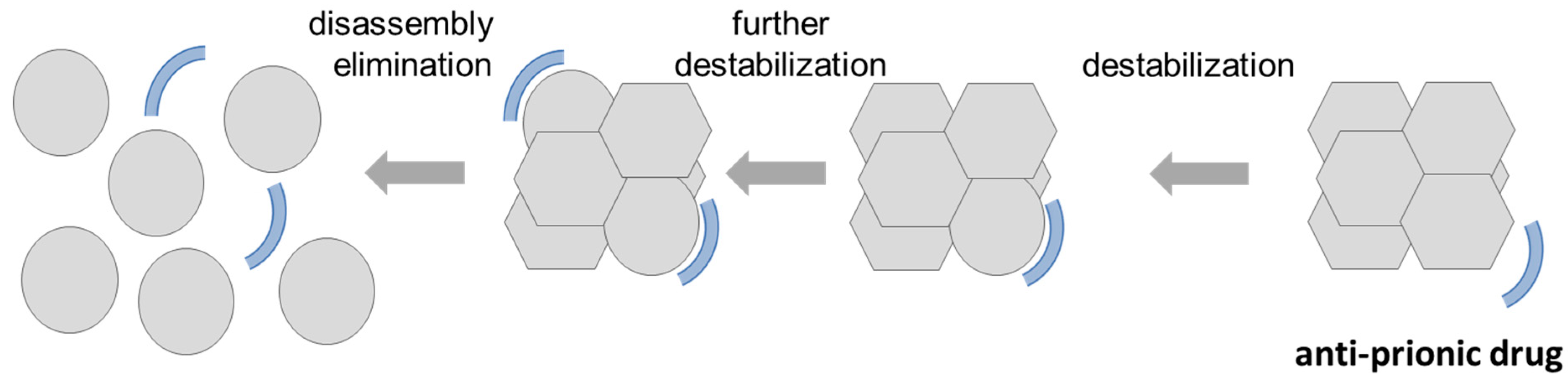{kind=link}
1. Alzheimer’s Disease
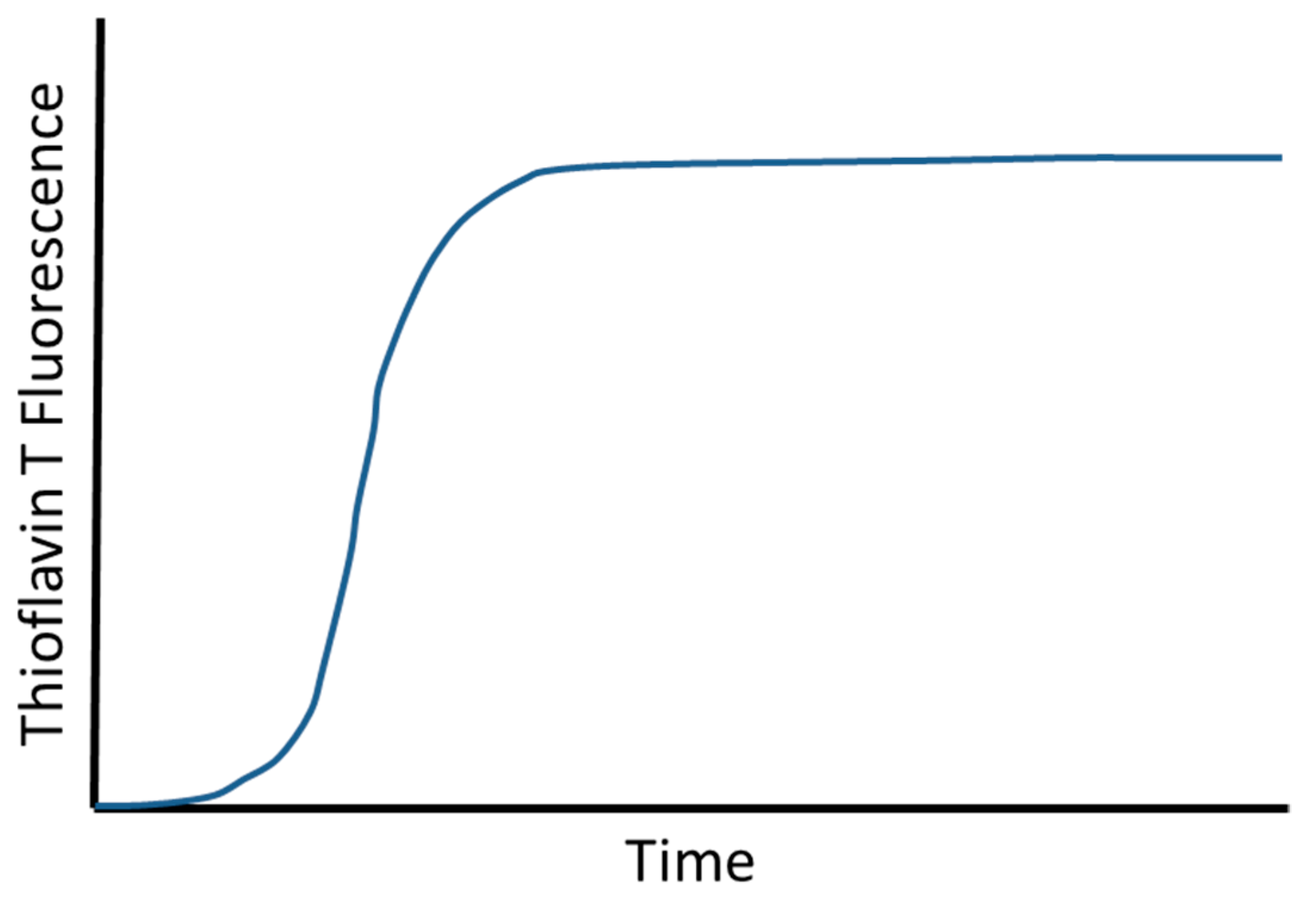
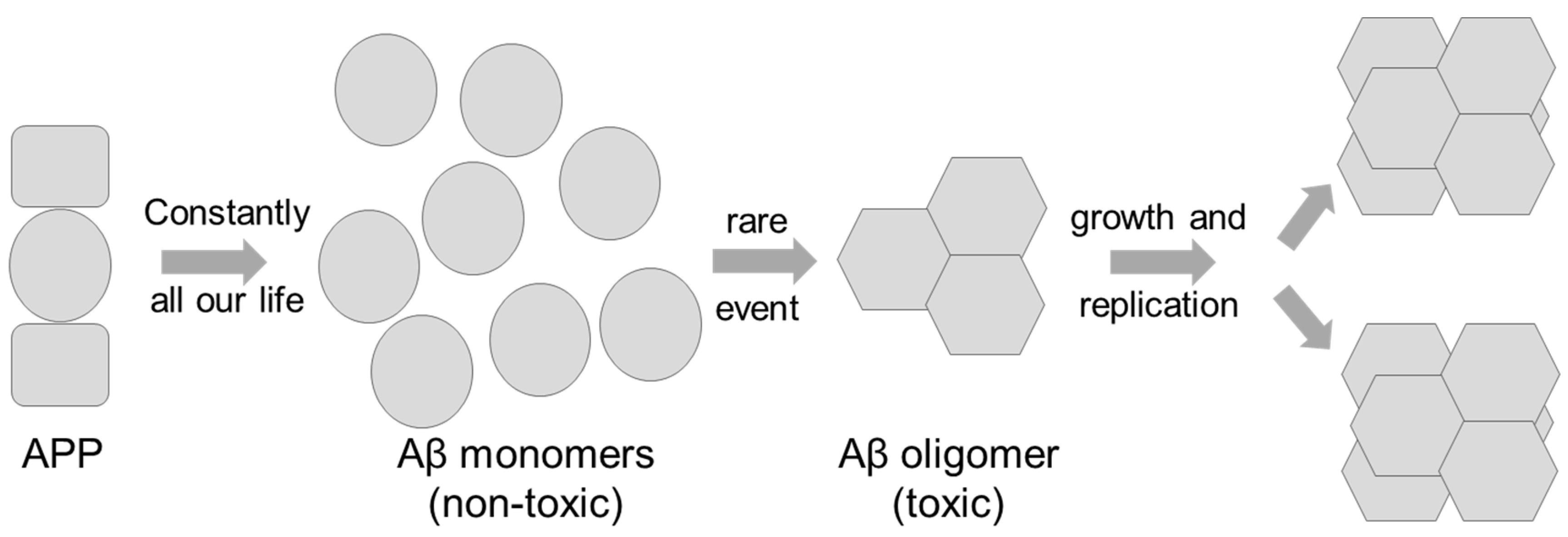
2. Aβ Aggregation
3. Did the Concept of a Replicating Toxic Aβ Assembly Predict the Outcomes of the Failed Phase III Clinical Trials?
4. Consequences of the Concept of a Replicating Toxic Aβ Assembly as the Etiologic Agent of AD for the Potential Treatment of AD
Author Contributions
Funding
Conflicts of Interest
References
- Andreadis, A.; Brown, W.M.; Kosik, K.S. Structure and novel exons of the human. tau. gene. Biochemistry 1992, 31, 10626–10633. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Crowther, R.A.; Garner, C.C. Molecular characterization of microtubule-associated proteins tau and MAP2. Trends Neurosci. 1991, 14, 193–199. [Google Scholar] [CrossRef]
- Goedert, M. Tau gene mutations and their effects. Mov. Disord. 2005, 20 (Suppl. 12), S45–S52. [Google Scholar] [CrossRef]
- Selkoe, D.J. The cell biology of beta-amyloid precursor protein and presenilin in Alzheimer’s disease. Trends Cell Biol. 1998, 8, 447–453. [Google Scholar] [CrossRef]
- Campion, D.; Dumanchin, C.; Hannequin, D.; Dubois, B.; Belliard, S.; Puel, M.; Thomas-Anterion, C.; Michon, A.; Martin, C.; Charbonnier, F.; et al. Early-onset autosomal dominant Alzheimer disease: Prevalence, genetic heterogeneity, and mutation spectrum. Am. J. Hum. Genet. 1999, 65, 664–670. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Selkoe, D.J. The Amyloid Hypothesis of Alzheimer’s Disease: Progress and Problems on the Road to Therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef]
- Matsunaga, S.; Kishi, T.; Nomura, I.; Sakuma, K.; Okuya, M.; Ikuta, T.; Iwata, N. The efficacy and safety of memantine for the treatment of Alzheimer’s disease. Expert Opin. Drug Saf. 2018, 17, 1053–1061. [Google Scholar] [CrossRef]
- Snowdon, D.A. Healthy aging and dementia: Findings from the Nun Study. Ann. Intern. Med. 2003, 139, 450–454. [Google Scholar] [CrossRef]
- Gandy, S.; Simon, A.J.; Steele, J.W.; Lublin, A.L.; Lah, J.J.; Walker, L.C.; Levey, A.I.; Krafft, G.A.; Levy, E.; Checler, F.; et al. Days to criterion as an indicator of toxicity associated with human Alzheimer amyloid-beta oligomers. Ann. Neurol. 2010, 68, 220–230. [Google Scholar] [CrossRef]
- Sengupta, U.; Nilson, A.N.; Kayed, R. The Role of Amyloid-β Oligomers in Toxicity, Propagation, and Immunotherapy. EBioMedicine 2016, 6, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Forny-Germano, L.; Lyra e Silva, N.M.; Batista, A.F.; Brito-Moreira, J.; Gralle, M.; Boehnke, S.E.; Coe, B.C.; Lablans, A.; Marques, S.A.; Martinez, A.M.; et al. Alzheimer’s disease-like pathology induced by amyloid-beta oligomers in nonhuman primates. J. Neurosci. 2014, 34, 13629–13643. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.M.; Klyubin, I.; Fadeeva, J.V.; Cullen, W.K.; Anwyl, R.; Wolfe, M.S.; Rowan, M.J.; Selkoe, D.J. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 2002, 416, 535–539. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.M.; Klyubin, I.; Shankar, G.M.; Townsend, M.; Fadeeva, J.V.; Betts, V.; Podlisny, M.B.; Cleary, J.P.; Ashe, K.H.; Rowan, M.J.; et al. The role of cell-derived oligomers of Abeta in Alzheimer’s disease and avenues for therapeutic intervention. Biochem. Soc. Trans. 2005, 33, 1087–1090. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Olsson, T.T.; Klementieva, O.; Gouras, G.K. Prion-like seeding and nucleation of intracellular amyloid-β. Neurobiol. Dis. 2018, 113, 1–10. [Google Scholar] [CrossRef]
- Cohen, M.; Appleby, B.; Safar, J.G. Distinct prion-like strains of amyloid beta implicated in phenotypic diversity of Alzheimer’s disease. Prion 2016, 10, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Walker, L.C.; Schelle, J.; Jucker, M. The Prion-Like Properties of Amyloid-beta Assemblies: Implications for Alzheimer’s Disease. Cold Spring Harb. Perspect. Med. 2016, 6, a024398. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.B.; Leng, L.Z.; Zhang, B.; Kwong, L.; Trojanowski, J.Q.; Abel, T.; Lee, V.M. Targeting amyloid-beta peptide (Abeta) oligomers by passive immunization with a conformation-selective monoclonal antibody improves learning and memory in Abeta precursor protein (APP) transgenic mice. J. Biol. Chem. 2006, 281, 4292–4299. [Google Scholar] [CrossRef]
- Goure, W.F.; Krafft, G.A.; Jerecic, J.; Hefti, F. Targeting the proper amyloid-beta neuronal toxins: A path forward for Alzheimer’s disease immunotherapeutics. Alzheimer’s Res. Ther. 2014, 6, 42. [Google Scholar] [CrossRef]
- Schenk, D.; Barbour, R.; Dunn, W.; Gordon, G.; Grajeda, H.; Guido, T.; Hu, K.; Huang, J.; Johnson-Wood, K.; Khan, K.; et al. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature 1999, 400, 173–177. [Google Scholar] [CrossRef]
- Bard, F.; Cannon, C.; Barbour, R.; Burke, R.L.; Games, D.; Grajeda, H.; Guido, T.; Hu, K.; Huang, J.; Johnson-Wood, K.; et al. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat. Med. 2000, 6, 916–919. [Google Scholar] [CrossRef] [PubMed]
- Wilcock, D.M.; Rojiani, A.; Rosenthal, A.; Levkowitz, G.; Subbarao, S.; Alamed, J.; Wilson, D.; Wilson, N.; Freeman, M.J.; Gordon, M.N.; et al. Passive amyloid immunotherapy clears amyloid and transiently activates microglia in a transgenic mouse model of amyloid deposition. J. Neurosci. 2004, 24, 6144–6151. [Google Scholar] [CrossRef] [PubMed]
- Doody, R.S.; Thomas, R.G.; Farlow, M.; Iwatsubo, T.; Vellas, B.; Joffe, S.; Kieburtz, K.; Raman, R.; Sun, X.; Aisen, P.S.; et al. Phase 3 trials of solanezumab for mild-to-moderate Alzheimer’s disease. N. Engl. J. Med. 2014, 370, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Salloway, S.; Sperling, R.; Fox, N.C.; Blennow, K.; Klunk, W.; Raskind, M.; Sabbagh, M.; Honig, L.S.; Porsteinsson, A.P.; Ferris, S.; et al. Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N. Engl. J. Med. 2014, 370, 322–333. [Google Scholar] [CrossRef] [PubMed]
- Makin, S. The amyloid hypothesis on trial. Nature 2018, 559, S4–S7. [Google Scholar] [CrossRef]
- DiFrancesco, J.C.; Longoni, M.; Piazza, F. Anti-Abeta Autoantibodies in Amyloid Related Imaging Abnormalities (ARIA): Candidate Biomarker for Immunotherapy in Alzheimer’s Disease and Cerebral Amyloid Angiopathy. Front. Neurol. 2015, 6, 207. [Google Scholar] [CrossRef]
- Ketter, N.; Brashear, H.R.; Bogert, J.; Di, J.; Miaux, Y.; Gass, A.; Purcell, D.D.; Barkhof, F.; Arrighi, H.M. Central Review of Amyloid-Related Imaging Abnormalities in Two Phase III Clinical Trials of Bapineuzumab in Mild-To-Moderate Alzheimer’s Disease Patients. J. Alzheimer’s Dis. 2017, 57, 557–573. [Google Scholar] [CrossRef]
- Tornquist, M.; Michaels, T.C.T.; Sanagavarapu, K.; Yang, X.; Meisl, G.; Cohen, S.I.A.; Knowles, T.P.J.; Linse, S. Secondary nucleation in amyloid formation. Chem. Commun. (Camb.) 2018, 54, 8667–8684. [Google Scholar] [CrossRef]
- Meisl, G.; Yang, X.; Hellstrand, E.; Frohm, B.; Kirkegaard, J.B.; Cohen, S.I.; Dobson, C.M.; Linse, S.; Knowles, T.P. Differences in nucleation behavior underlie the contrasting aggregation kinetics of the Abeta40 and Abeta42 peptides. Proc. Natl. Acad. Sci. USA 2014, 111, 9384–9389. [Google Scholar] [CrossRef]
- Novo, M.; Freire, S.; Al-Soufi, W. Critical aggregation concentration for the formation of early Amyloid-beta (1-42) oligomers. Sci. Rep. 2018, 8, 1783. [Google Scholar] [CrossRef]
- Wolff, M.; Zhang-Haagen, B.; Decker, C.; Barz, B.; Schneider, M.; Biehl, R.; Radulescu, A.; Strodel, B.; Willbold, D.; Nagel-Steger, L. Abeta42 pentamers/hexamers are the smallest detectable oligomers in solution. Sci. Rep. 2017, 7, 2493. [Google Scholar] [CrossRef] [PubMed]
- Gremer, L.; Scholzel, D.; Schenk, C.; Reinartz, E.; Labahn, J.; Ravelli, R.B.G.; Tusche, M.; Lopez-Iglesias, C.; Hoyer, W.; Heise, H.; et al. Fibril structure of amyloid-beta(1-42) by cryo-electron microscopy. Science 2017, 358, 116–119. [Google Scholar] [CrossRef] [PubMed]
- Stöhr, J.; Condello, C.; Watts, J.C.; Bloch, L.; Oehler, A.; Nick, M.; DeArmond, S.J.; Giles, K.; DeGrado, W.F.; Prusiner, S.B. Distinct synthetic Aβ prion strains producing different amyloid deposits in bigenic mice. Proc. Natl. Acad. Sci. USA 2014, 111, 10329–10334. [Google Scholar] [CrossRef] [PubMed]
- Watts, J.C.; Condello, C.; Stöhr, J.; Oehler, A.; Lee, J.; DeArmond, S.J.; Lannfelt, L.; Ingelsson, M.; Giles, K.; Prusiner, S.B. Serial propagation of distinct strains of Aβ prions from Alzheimer’s disease patients. Proc. Natl. Acad. Sci. USA 2014, 111, 10323–10328. [Google Scholar] [CrossRef] [PubMed]
- Mullard, A. BACE failures lower AD expectations, again. Nat. Rev. Drug Discov. 2018, 17, 385. [Google Scholar] [CrossRef] [PubMed]
- Gold, M. Phase II clinical trials of anti-amyloid beta antibodies: When is enough, enough? Alzheimer’s Dement. 2017, 3, 402–409. [Google Scholar] [CrossRef]
- Koenigsknecht-Talboo, J.; Meyer-Luehmann, M.; Parsadanian, M.; Garcia-Alloza, M.; Finn, M.B.; Hyman, B.T.; Bacskai, B.J.; Holtzman, D.M. Rapid microglial response around amyloid pathology after systemic anti-Abeta antibody administration in PDAPP mice. J. Neurosci. 2008, 28, 14156–14164. [Google Scholar] [CrossRef] [PubMed]
- Weyand, C.M.; Goronzy, J.J. Aging of the Immune System. Mechanisms and Therapeutic Targets. Ann. Am. Thorac. Soc. 2016, 13 (Suppl. 5), S422–S428. [Google Scholar] [CrossRef]
- Miller-Vedam, L.; Ghaemmaghami, S. Strain specificity and drug resistance in anti-prion therapy. Curr. Top. Med. Chem. 2013, 13, 2397–2406. [Google Scholar] [CrossRef]
- Cummings, J.; Lee, G.; Ritter, A.; Zhong, K. Alzheimer’s disease drug development pipeline: 2018. Alzheimer’s Dement. 2018, 4, 195–214. [Google Scholar] [CrossRef]
- van Groen, T.; Schemmert, S.; Brener, O.; Gremer, L.; Ziehm, T.; Tusche, M.; Nagel-Steger, L. The Abeta oligomer eliminating D-enantiomeric peptide RD2 improves cognition without changing plaque pathology. Sci. Rep. 2017, 7, 16275. [Google Scholar] [CrossRef] [PubMed]
- Schemmert, S.; Schartmann, E.; Zafiu, C.; Kass, B.; Hartwig, S.; Lehr, S.; Bannach, O.; Langen, K.J.; Shah, N.J.; Kutzsche, J.; et al. Abeta Oligomer Elimination Restores Cognition in Transgenic Alzheimer’s Mice with Full-blown Pathology. Mol. Neurobiol. 2018. [Google Scholar] [CrossRef]
- Kutzsche, J.; Schemmert, S.; Tusche, M.; Neddens, J.; Rabl, R.; Jurgens, D.; Brener, O.; Willuweit, A. Large-Scale Oral Treatment Study with the Four Most Promising D3-Derivatives for the Treatment of Alzheimer’s Disease. Molecules 2017, 22, 1693. [Google Scholar] [CrossRef] [PubMed]
- Schemmert, S.; Schartmann, E.; Honold, D.; Zafiu, C.; Ziehm, T.; Langen, K.J.; Shah, N.J.; Kutzsche, J.; Willuweit, A.; Willbold, D. Deceleration of the neurodegenerative phenotype in pyroglutamate-Abeta accumulating transgenic mice by oral treatment with the Abeta oligomer eliminating compound RD2. Neurobiol. Dis. 2019, 124, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Purro, S.A.; Nicoll, A.J.; Collinge, J. Prion Protein as a Toxic Acceptor of Amyloid-beta Oligomers. Biol. Psychiatry 2018, 83, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Yadav, S.P.; Surewicz, W.K. Interaction between human prion protein and amyloid-beta (Abeta) oligomers: Role OF N-terminal residues. J. Biol. Chem. 2010, 285, 26377–26383. [Google Scholar] [CrossRef] [PubMed]
- Rosener, N.S.; Gremer, L.; Reinartz, E.; Konig, A.; Brener, O.; Heise, H.; Hoyer, W.; Neudecker, P.; Willbold, D. A d-enantiomeric peptide interferes with heteroassociation of amyloid-beta oligomers and prion protein. J. Biol. Chem. 2018, 293, 15748–15764. [Google Scholar] [CrossRef] [PubMed]
- Leithold, L.H.; Jiang, N.; Post, J.; Ziehm, T.; Schartmann, E.; Kutzsche, J.; Shah, N.J.; Breitkreutz, J.; Langen, K.J.; Willuweit, A.; et al. Pharmacokinetic Properties of a Novel D-Peptide Developed to be Therapeutically Active Against Toxic beta-Amyloid Oligomers. Pharm. Res. 2016, 33, 328–336. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Funke, S.A.; Willbold, D. Transport of Alzheimer disease amyloid-beta-binding D-amino acid peptides across an in vitro blood-brain barrier model. Rejuvenation Res. 2010, 13, 210–213. [Google Scholar] [CrossRef] [PubMed]
- Drin, G.; Rousselle, C.; Scherrmann, J.M.; Rees, A.R.; Temsamani, J. Peptide delivery to the brain via adsorptive-mediated endocytosis: Advances with SynB vectors. AAPS PharmSci. 2002, 4, E26. [Google Scholar] [CrossRef] [PubMed]
- Elfgen, A.; Hupert, M.; Bochinsky, K.; Tusche, M.; González de San Román Martin, E.; Gering, I.; Sacchi, S.; Pollegioni, L.; Huesgen, P.F.; Hartmann, R.; et al. Metabolic resistance of the D-peptide RD2 developed for direct elimination of amyloid-β oligomers. Sci. Rep. 2019, 9, 5715. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Willbold, D.; Kutzsche, J. Do We Need Anti-Prion Compounds to Treat Alzheimer’s Disease? Molecules 2019, 24, 2237. https://doi.org/10.3390/molecules24122237
Willbold D, Kutzsche J. Do We Need Anti-Prion Compounds to Treat Alzheimer’s Disease? Molecules. 2019; 24(12):2237. https://doi.org/10.3390/molecules24122237
Chicago/Turabian StyleWillbold, Dieter, and Janine Kutzsche. 2019. "Do We Need Anti-Prion Compounds to Treat Alzheimer’s Disease?" Molecules 24, no. 12: 2237. https://doi.org/10.3390/molecules24122237
APA StyleWillbold, D., & Kutzsche, J. (2019). Do We Need Anti-Prion Compounds to Treat Alzheimer’s Disease? Molecules, 24(12), 2237. https://doi.org/10.3390/molecules24122237