
Introduction of Reversible Urethane Bonds Based on Vanillyl Alcohol for Efficient Self-Healing of Polyurethane Elastomers



Abstract
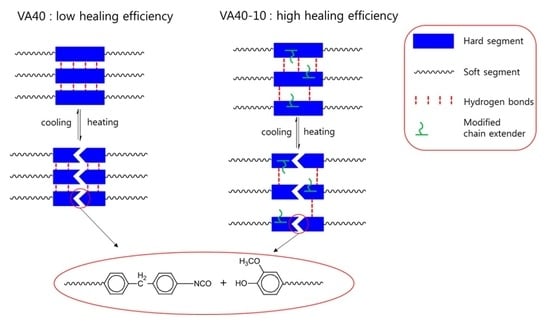
1. Introduction
2. Results and Discussion
2.1. Characterization of Vanillyl Alcohol (VA)-Based Polyurethane Elastomers (PUs)
2.2. Microdomain Structure of VA-Based PUs
2.3. Thermal Analyses of PUs Based on VA
2.4. Mechanical Properties of the VA-based PUs
2.5. Self-Healing Properties of the VA-based PUs
3. Materials and Methods
3.1. Materials
3.2. Preparation of Modified Chain Extender
3.3. Preparation of Control PU and VA-based PUs
3.4. Characterization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Hentschel, T.; Münstedt, H. Kinetics of the molar mass decrease in a polyurethane melt: A rheological study. Polymer 2001, 42, 3195–3203. [Google Scholar] [CrossRef]
- Grassie, N.; Zulfiqar, M. Thermal Degradation of the Polyurethane From 1,4-Butanediol and Methylene Bis(4-Phenyl Isocyanate). J. Polym. Sci. Polym. Chem. Ed. 1978, 16, 1563–1574. [Google Scholar] [CrossRef]
- Montaudo, G.; Puglisi, C.; Scamporrino, E.; Vitalini, D. Mechanism of thermal degradation of polyurethanes. Effect of ammonium polyphosphate. Macromolecules 1984, 17, 1605–1614. [Google Scholar] [CrossRef]
- Zia, K.M.; Bhatti, H.N.; Ahmad Bhatti, I. Methods for polyurethane and polyurethane composites, recycling and recovery: A review. React. Funct. Polym. 2007, 67, 675–692. [Google Scholar] [CrossRef]
- Wegener, G.; Brandt, M.; Duda, L.; Hofmann, J.; Klesczewski, B.; Koch, D.; Kumpf, R.J.; Orzesek, H.; Pirkl, H.G.; Six, C.; et al. Trends in industrial catalysis in the polyurethane industry. Appl. Catal. A Gen. 2001, 221, 303–335. [Google Scholar] [CrossRef]
- Janik, H.; Marzec, M. A review: Fabrication of porous polyurethane scaffolds. Mater. Sci. Eng. C 2015, 48, 586–591. [Google Scholar] [CrossRef]
- Zdrahala, R.J.; Zdrahala, I.J. Biomedical applications of polyurethanes: A review of past promises, present realities, and a vibrant future. J. Biomater. Appl. 1999, 14, 67–90. [Google Scholar] [CrossRef]
- Samadzadeh, M.; Boura, S.H.; Peikari, M.; Kasiriha, S.M.; Ashrafi, A. A review on self-healing coatings based on micro/nanocapsules. Prog. Org. Coatings 2010, 68, 159–164. [Google Scholar] [CrossRef]
- Li, X.; Gao, Y.; Boott, C.E.; Winnik, M.A.; Manners, I. Non-covalent synthesis of supermicelles with complex architectures using spatially confined hydrogen-bonding interactions. Nat. Commun. 2015, 6, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Syrett, J.A.; Becer, C.R.; Haddleton, D.M. Self-healing and self-mendable polymers. Polym. Chem. 2010, 1, 978–987. [Google Scholar] [CrossRef]
- Zhang, M.; Xu, D.; Yan, X.; Chen, J.; Dong, S.; Zheng, B.; Huang, F. Self-healing supramolecular gels formed by crown ether based host-guest interactions. Angew. Chemie–Int. Ed. 2012, 51, 7011–7015. [Google Scholar] [CrossRef]
- Cook, T.R.; Zheng, Y.R.; Stang, P.J. Metal-organic frameworks and self-assembled supramolecular coordination complexes: Comparing and contrasting the design, synthesis, and functionality of metal-organic materials. Chem. Rev. 2013, 113, 734–777. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Mahmood, N.; Beiner, M.; Binder, W.H. Self-Healing Materials from V- and H-Shaped Supramolecular Architectures. Angew. Chemie–Int. Ed. 2015, 54, 10188–10192. [Google Scholar] [CrossRef]
- Zhu, K.; Song, Q.; Chen, H.; Hu, P. Thermally assisted self-healing polyurethane containing carboxyl groups. J. Appl. Polym. Sci. 2018, 135, 1–7. [Google Scholar] [CrossRef]
- Karami, Z.; Zohuriaan-Mehr, M.J.; Rostami, A. Bio-based thermo-healable non-isocyanate polyurethane DA network in comparison with its epoxy counterpart. J. CO2 Util. 2017, 18, 294–302. [Google Scholar] [CrossRef]
- Yu, F.; Cao, X.; Du, J.; Wang, G.; Chen, X. Multifunctional Hydrogel with Good Structure Integrity, Self-Healing, and Tissue-Adhesive Property Formed by Combining Diels-Alder Click Reaction and Acylhydrazone Bond. ACS Appl. Mater. Interfaces 2015, 7, 24023–24031. [Google Scholar] [CrossRef] [PubMed]
- Ying, H.; Zhang, Y.; Cheng, J. Dynamic urea bond for the design of reversible and self-healing polymers. Nat. Commun. 2014, 5, 1–9. [Google Scholar] [CrossRef]
- Cao, S.; Li, S.; Li, M.; Xu, L.; Ding, H.; Xia, J.; Zhang, M.; Huang, K. A thermal self-healing polyurethane thermoset based on phenolic urethane. Polym. J. 2017, 49, 775–781. [Google Scholar] [CrossRef]
- Pérez-San Vicente, A.; Peroglio, M.; Ernst, M.; Casuso, P.; Loinaz, I.; Grande, H.J.; Alini, M.; Eglin, D.; Dupin, D. Self-Healing Dynamic Hydrogel as Injectable Shock-Absorbing Artificial Nucleus Pulposus. Biomacromolecules 2017, 18, 2360–2370. [Google Scholar] [CrossRef] [PubMed]
- Pepels, M.; Filot, I.; Klumperman, B.; Goossens, H. Self-healing systems based on disulfide-thiol exchange reactions. Polym. Chem. 2013, 4, 4955–4965. [Google Scholar] [CrossRef]
- Gao, W.; Bie, M.; Liu, F.; Chang, P.; Quan, Y. Self-Healable and Reprocessable Polysulfide Sealants Prepared from Liquid Polysulfide Oligomer and Epoxy Resin. ACS Appl. Mater. Interfaces 2017, 9, 15798–15808. [Google Scholar] [CrossRef]
- Li, G.; Zhang, H.; Daniel, F.; Weizheng, F.; Xia, H.; Zhao, Y. A composite material with room temperature shape processabillity and optical repair. J. Mater. Chem. C 2016, 4, 5932–5939. [Google Scholar] [CrossRef]
- Habault, D.; Hongji Zhang, H.; Zhao, Y. Light-triggered self-healing and shape-memory polymers. Chem. Soc. Rev. 2013, 42, 7244–7256. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.M.; Rong, M.Z.; Zhang, M.Q. Sunlight driven self-healing, reshaping and recycling of a robust, transparent and yellowing resistant polymer. J. Mater. Chem. A 2016, 4, 10683–10690. [Google Scholar] [CrossRef]
- Kim, S.M.; Jeon, H.; Shin, S.H.; Park, S.A.; Jegal, J.; Hwang, S.Y.; Oh, D.X.; Park, J. Superior Toughness and Fast Self-Healing at Room Temperature Engineered by Transparent Elastomers. Adv. Mater. 2018, 30, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Yanagisawa, Y.; Nan, Y.; Okuro, K.; Aida, T. Mechanically robust, readily repairable polymers via tailored noncovalent cross-linking. Science 2018, 359, 72–76. [Google Scholar] [CrossRef] [PubMed]
- Capelot, M.; Montarnal, D.; Tournilhac, F.; Leibler, L. Metal-catalyzed transesterification for healing and assembling of thermosets. J. Am. Chem. Soc. 2012, 134, 7664–7667. [Google Scholar] [CrossRef] [PubMed]
- Denissen, W.; Rivero, G.; Nicolaÿ, R.; Leibler, L.; Winne, J.M.; Du Prez, F.E. Vinylogous urethane vitrimers. Adv. Funct. Mater. 2015, 25, 2451–2457. [Google Scholar] [CrossRef]
- Montarnal, D.; Capelot, M.; Tournilhac, F.; Leibler, L. Silica-Like Malleable Materials from. Science 2011, 334, 965–968. [Google Scholar] [CrossRef] [PubMed]
- Koberstein, J.T.; Galambos, A.F.; Leung, L.M. Compression-molded polyurethane block copolymers. 1. Microdomain morphology and thermomechanical properties. Macromolecules 1992, 25, 6195–6204. [Google Scholar] [CrossRef]
- Saiani, A.; Rochas, C.; Eeckhaut, G.; Daunch, W.A.; Leenslag, J.-W.; Higgins, J.S. Origin of Multiple Melting Endotherms in a High Hard Block Content Polyurethane. 2. Structural Investigation. Macromolecules 2004, 37, 1411–1421. [Google Scholar] [CrossRef]
- Leung, L.M.; Koberstein, J.T. Small-angle scattering analysis of hard-microdomain structure and microphase mixing in polyurethane elastomers. J. Polym. Sci. 1985, 23, 1883–1913. [Google Scholar] [CrossRef]
- Kojio, K.; Matsuo, K.; Motokucho, S.; Yoshinaga, K.; Shimodaira, Y.; Kimura, K. Simultaneous small-angle X-ray scattering/wide-angle X-ray diffraction study of the microdomain structure of polyurethane elastomers during mechanical deformation. Polym. J. 2011, 43, 692–699. [Google Scholar] [CrossRef]
- Zoran, S.P.; Zoltan, Z.; Joseph, H.F.; William, J.M. Thermal degradation of segmented polyurethanes. J. Appl. Polym. Sci. 1994, 51, 1087–1095. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available or not from the authors. |











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Code | Molecular Weight | |||
|---|---|---|---|---|
| Mn | Mw | Mz | Mw/Mn | |
| Control PU | 18,000 | 31,000 | 47,000 | 1.78 |
| VA10 | 22,000 | 93,000 | 495,000 | 4.19 |
| VA20 | 28,000 | 69,000 | 156,000 | 2.45 |
| VA30 | 15,000 | 33,000 | 60,000 | 2.24 |
| VA40 | 12,000 | 26,000 | 46,000 | 2.10 |
| VA40-5 | 24,000 | 72,000 | 214,000 | 3.00 |
| VA40-10 | 27,000 | 84,000 | 258,000 | 3.09 |
| Sample Code | Composition (Molar Ratio) | |||
|---|---|---|---|---|
| PTMEG1000 | VA | MDIz | BDO m-CE | |
| Control PU | 1 | - | 2 | 1 |
| VA10 | 0.9 | 0.1 | 2 | 1 |
| VA20 | 0.8 | 0.2 | 2 | 1 |
| VA30 | 0.7 | 0.3 | 2 | 1 |
| VA40 | 0.6 | 0.4 | 2 | 1 |
| VA40-5 | 0.6 | 0.4 | 2 | 0.95 0.05 |
| VA40-10 | 0.6 | 0.4 | 2 | 0.90 0.10 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, D.-W.; Kim, H.-N.; Lee, D.-S. Introduction of Reversible Urethane Bonds Based on Vanillyl Alcohol for Efficient Self-Healing of Polyurethane Elastomers. Molecules 2019, 24, 2201. https://doi.org/10.3390/molecules24122201
Lee D-W, Kim H-N, Lee D-S. Introduction of Reversible Urethane Bonds Based on Vanillyl Alcohol for Efficient Self-Healing of Polyurethane Elastomers. Molecules. 2019; 24(12):2201. https://doi.org/10.3390/molecules24122201
Chicago/Turabian StyleLee, Dae-Woo, Han-Na Kim, and Dai-Soo Lee. 2019. "Introduction of Reversible Urethane Bonds Based on Vanillyl Alcohol for Efficient Self-Healing of Polyurethane Elastomers" Molecules 24, no. 12: 2201. https://doi.org/10.3390/molecules24122201
APA StyleLee, D.-W., Kim, H.-N., & Lee, D.-S. (2019). Introduction of Reversible Urethane Bonds Based on Vanillyl Alcohol for Efficient Self-Healing of Polyurethane Elastomers. Molecules, 24(12), 2201. https://doi.org/10.3390/molecules24122201