
p53 Binds Preferentially to Non-B DNA Structures Formed by the Pyrimidine-Rich Strands of GAA·TTC Trinucleotide Repeats Associated with Friedreich’s Ataxia



 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
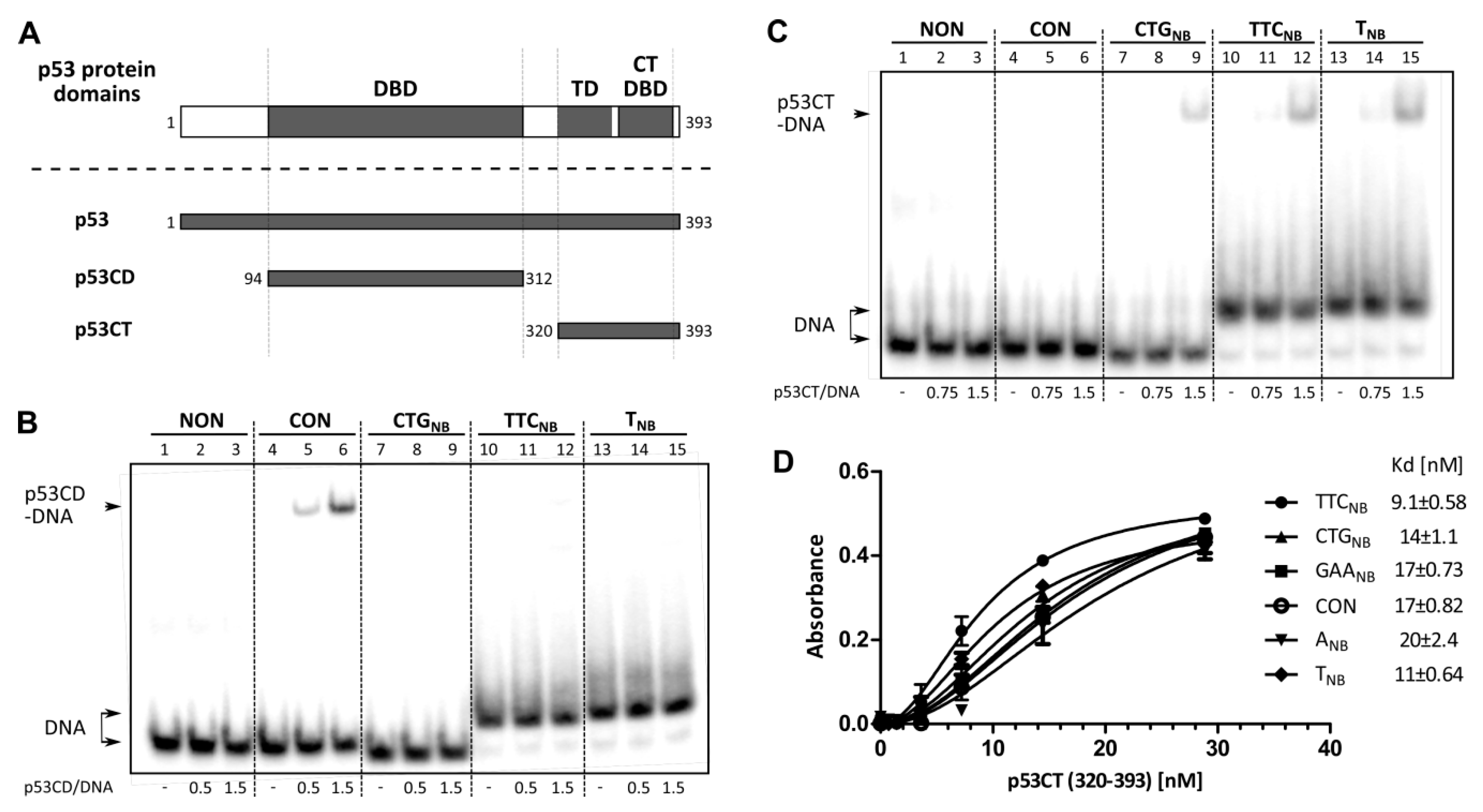
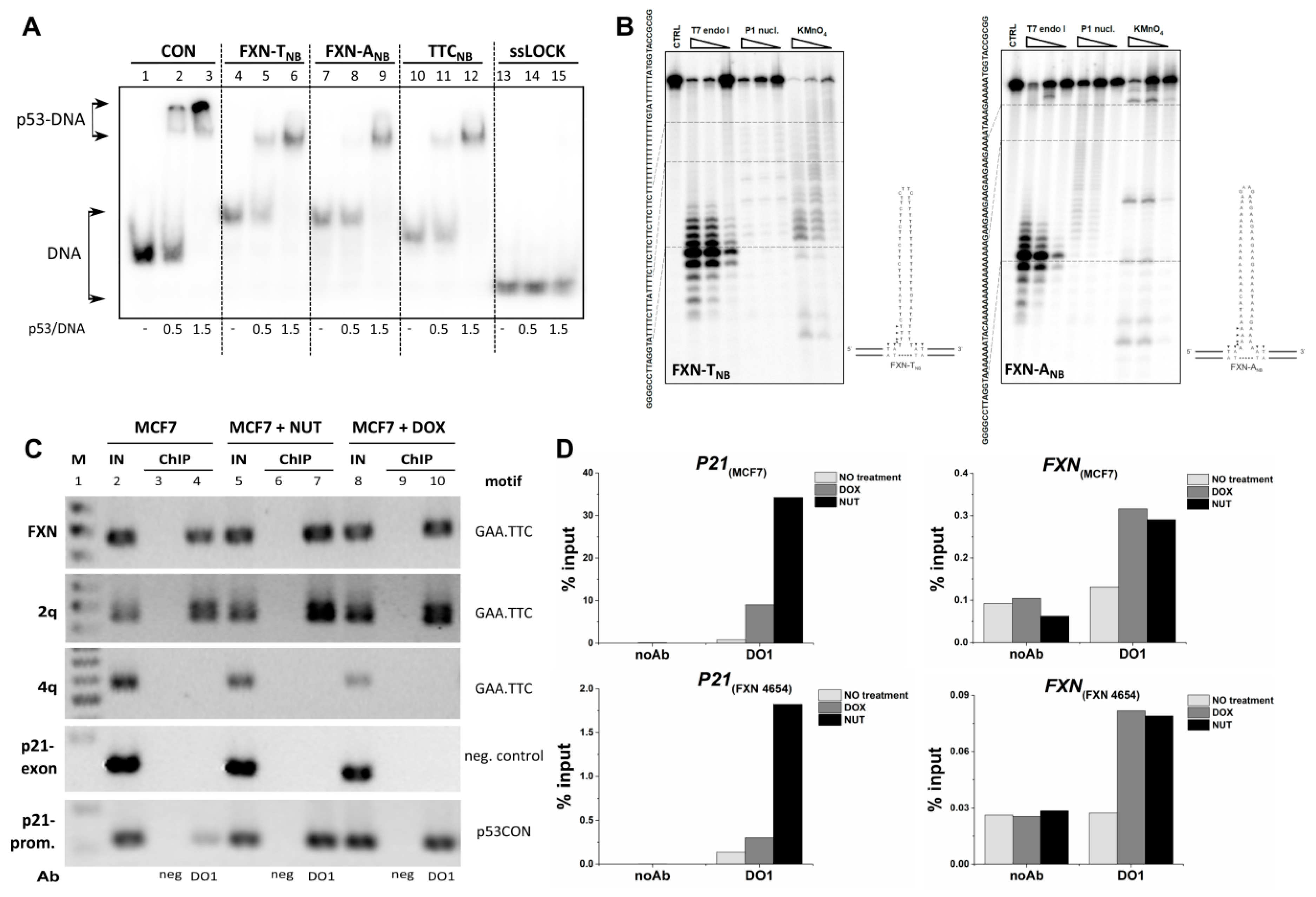
2.1. Interaction of p53 with Non-B DNA Structures Derived from TTC, GAA, CTG and CAG Trinucleotide Repeat Sequences
2.2. Role of p53 Core and C-Terminal DNA Binding Domains in TNR Non-B DNA Recognition
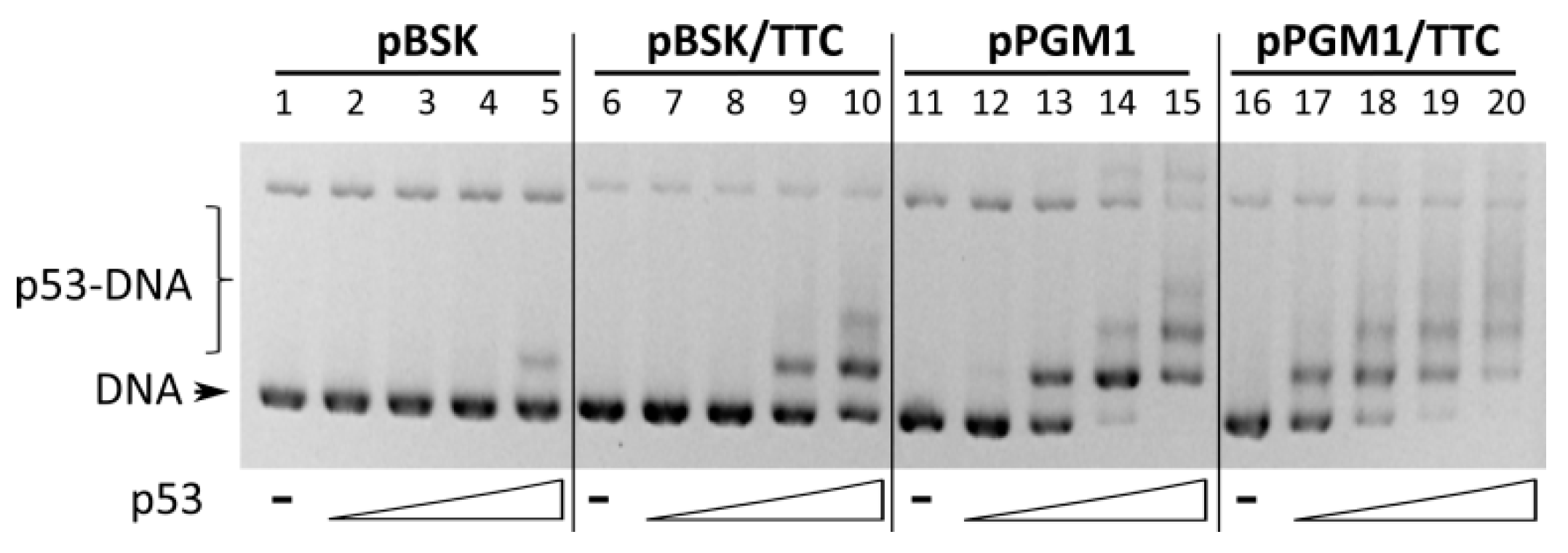
2.3. Binding of Wild-Type p53 to GAA·TTC Sequence in scDNA
2.4. Analysis of p53 Interaction with TNR Region from the First Frataxin Intron in Cells
2.5. Future Prospects of p53 and TNR Non-B DNA Structures
3. Materials and Methods
3.1. Oligonucleotides
3.2. Recombinant Plasmids
3.3. Purification of p53 Recombinant Proteins
3.4. EMSA in Polyacrylamide Gels
3.5. EMSA in Agarose Gels
3.6. ELISA
3.7. Chromatin Immunoprecipitation
3.8. Expression Analysis
3.9. Enzymatic and Chemical Footprinting
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lane, D.P. Cancer. p53, guardian of the genome. Nature 1992, 358, 15–16. [Google Scholar] [CrossRef] [PubMed]
- Laptenko, O.; Prives, C. p53: master of life, death, and the epigenome. Genes Dev. 2017, 31, 955–956. [Google Scholar] [CrossRef]
- el-Deiry, W.S.; Kern, S.E.; Pietenpol, J.A.; Kinzler, K.W.; Vogelstein, B. Definition of a consensus binding site for p53. Nat. Genet. 1992, 1, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Zotchev, S.B.; Protopopova, M.; Selivanova, G. p53 C-terminal interaction with DNA ends and gaps has opposing effect on specific DNA binding by the core. Nucleic Acids Res. 2000, 28, 4005–4012. [Google Scholar] [CrossRef][Green Version]
- Brazdova, M.; Palecek, J.; Cherny, D.I.; Billova, S.; Fojta, M.; Pecinka, P.; Vojtesek, B.; Jovin, T.M.; Palecek, E. Role of tumor suppressor p53 domains in selective binding to supercoiled DNA. Nucleic Acids Res. 2002, 30, 4966–4974. [Google Scholar] [CrossRef] [PubMed]
- Adamik, M.; Kejnovska, I.; Bazantova, P.; Petr, M.; Renciuk, D.; Vorlickova, M.; Brazdova, M. p53 binds human telomeric G-quadruplex in vitro. Biochimie 2016, 83–91. [Google Scholar] [CrossRef]
- Quante, T.; Otto, B.; Brazdova, M.; Kejnovska, I.; Deppert, W.; Tolstonog, G.V. Mutant p53 is a transcriptional co-factor that binds to G-rich regulatory regions of active genes and generates transcriptional plasticity. Cell Cycle 2012, 11, 3290–3303. [Google Scholar] [CrossRef] [PubMed]
- Brazdova, M.; Tichy, V.; Helma, R.; Bazantova, P.; Polaskova, A.; Krejci, A.; Petr, M.; Navratilova, L.; Ticha, O.; Nejedly, K.; et al. p53 Specifically Binds Triplex DNA In Vitro and in Cells. PLoS ONE 2016, 11, e0167439. [Google Scholar] [CrossRef]
- Petr, M.; Helma, R.; Polaskova, A.; Krejci, A.; Dvorakova, Z.; Kejnovska, I.; Navratilova, L.; Adamik, M.; Vorlickova, M.; Brazdova, M. Wild-type p53 binds to MYC promoter G-quadruplex. Biosci. Rep. 2016, 36. [Google Scholar] [CrossRef]
- Bacolla, A.; Wojciechowska, M.; Kosmider, B.; Larson, J.E.; Wells, R.D. The involvement of non-B DNA structures in gross chromosomal rearrangements. DNA Repair 2006, 5, 1161–1170. [Google Scholar] [CrossRef] [PubMed]
- Kozlowski, P.; de Mezer, M.; Krzyzosiak, W.J. Trinucleotide repeats in human genome and exome. Nucleic Acids Res. 2010, 38, 4027–4039. [Google Scholar] [CrossRef]
- Pearson, C.E.; Sinden, R.R. Trinucleotide repeat DNA structures: dynamic mutations from dynamic DNA. Curr. Opin. Struct. Biol. 1998, 8, 321–330. [Google Scholar] [CrossRef]
- Sinden, R.R.; Pytlos-Sinden, M.J.; Potaman, V.N. Slipped strand DNA structures. Front. Biosci. 2007, 12, 4788–4799. [Google Scholar] [CrossRef]
- Zhao, J.; Bacolla, A.; Wang, G.; Vasquez, K.M. Non-B DNA structure-induced genetic instability and evolution. CMLS 2010, 67, 43–62. [Google Scholar] [CrossRef]
- Schmidt, M.H.; Pearson, C.E. Disease-associated repeat instability and mismatch repair. DNA Repair 2016, 38, 117–126. [Google Scholar] [CrossRef]
- Sinden, R.R.; Potaman, V.N.; Oussatcheva, E.A.; Pearson, C.E.; Lyubchenko, Y.L.; Shlyakhtenko, L.S. Triplet repeat DNA structures and human genetic disease: dynamic mutations from dynamic DNA. J. Biosci. 2002, 27, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Andrew, S.E.; Goldberg, Y.P.; Kremer, B.; Telenius, H.; Theilmann, J.; Adam, S.; Starr, E.; Squitieri, F.; Lin, B.; Kalchman, M.A.; et al. The relationship between trinucleotide (CAG) repeat length and clinical features of Huntington’s disease. Nat. Genet. 1993, 4, 398–403. [Google Scholar] [CrossRef] [PubMed]
- Walter, K.; Warnecke, G.; Bowater, R.; Deppert, W.; Kim, E. tumor suppressor p53 binds with high affinity to CTG.CAG trinucleotide repeats and induces topological alterations in mismatched duplexes. J. Biol. Chem. 2005, 280, 42497–42507. [Google Scholar] [CrossRef]
- Cobb, A.M.; Jackson, B.R.; Kim, E.; Bond, P.L.; Bowater, R.P. Sequence-specific and DNA structure-dependent interactions of Escherichia coli MutS and human p53 with DNA. Anal. Biochem. 2013, 442, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Bacolla, A.; Wells, R.D. Non-B DNA conformations as determinants of mutagenesis and human disease. Mol. Carcinogenes. 2009, 48, 273–285. [Google Scholar] [CrossRef]
- Campuzano, V.; Montermini, L.; Molto, M.D.; Pianese, L.; Cossee, M.; Cavalcanti, F.; Monros, E.; Rodius, F.; Duclos, F.; Monticelli, A.; et al. Friedreich’s ataxia: Autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 1996, 271, 1423–1427. [Google Scholar] [CrossRef] [PubMed]
- Rajeswari, M.R. DNA triplex structures in neurodegenerative disorder, Friedreich’s ataxia. J. Biosci. 2012, 37, 519–532. [Google Scholar] [CrossRef]
- Heidenfelder, B.L.; Makhov, A.M.; Topal, M.D. Hairpin formation in Friedreich’s ataxia triplet repeat expansion. J. Biol. Chem. 2003, 278, 2425–2431. [Google Scholar] [CrossRef]
- LeProust, E.M.; Pearson, C.E.; Sinden, R.R.; Gao, X. Unexpected formation of parallel duplex in GAA and TTC trinucleotide repeats of Friedreich’s ataxia. J. Mol. Biol. 2000, 302, 1063–1080. [Google Scholar] [CrossRef]
- Mariappan, S.V.; Catasti, P.; Silks, L.A.; Bradbury, E.M.; Gupta, G. The high-resolution structure of the triplex formed by the GAA/TTC triplet repeat associated with Friedreich’s ataxia. J. Mol. Biol. 1999, 285, 2035–2052. [Google Scholar] [CrossRef] [PubMed]
- Mitas, M. Trinucleotide repeats associated with human disease. Nucleic Acids Res. 1997, 25, 2245–2254. [Google Scholar] [CrossRef]
- Potaman, V.N.; Oussatcheva, E.A.; Lyubchenko, Y.L.; Shlyakhtenko, L.S.; Bidichandani, S.I.; Ashizawa, T.; Sinden, R.R. Length-dependent structure formation in Friedreich ataxia (GAA)n*(TTC)n repeats at neutral pH. Nucleic Acids Res. 2004, 32, 1224–1231. [Google Scholar] [CrossRef]
- Sawamoto, M.; Imai, T.; Umeda, M.; Fukuda, K.; Kataoka, T.; Taketani, S. The p53-dependent expression of frataxin controls 5-aminolevulinic acid-induced accumulation of protoporphyrin IX and photo-damage in cancerous cells. Photochem. Photobiol. 2013, 89, 163–172. [Google Scholar] [CrossRef]
- Shimizu, R.; Lan, N.N.; Tai, T.T.; Adachi, Y.; Kawazoe, A.; Mu, A.; Taketani, S. p53 directly regulates the transcription of the human frataxin gene and its lack of regulation in tumor cells decreases the utilization of mitochondrial iron. Gene 2014, 551, 79–85. [Google Scholar] [CrossRef]
- Mouli, S.; Nanayakkara, G.; AlAlasmari, A.; Eldoumani, H.; Fu, X.; Berlin, A.; Lohani, M.; Nie, B.; Arnold, R.D.; Kavazis, A.; et al. The role of frataxin in doxorubicin-mediated cardiac hypertrophy. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, 844–859. [Google Scholar] [CrossRef]
- Lupoli, F.; Vannocci, T.; Longo, G.; Niccolai, N.; Pastore, A. The role of oxidative stress in Friedreich’s ataxia. FEBS Lett. 2018, 592, 718–727. [Google Scholar] [CrossRef] [PubMed]
- Marmolino, D.; Acquaviva, F. Friedreich’s Ataxia: From the (GAA)n repeat mediated silencing to new promising molecules for therapy. Cerebellum 2009, 8, 245–259. [Google Scholar] [CrossRef] [PubMed]
- Fojta, M.; Kubicarova, T.; Vojtesek, B.; Palecek, E. Effect of p53 protein redox states on binding to supercoiled and linear DNA. J. Biol. Chem. 1999, 274, 25749–25755. [Google Scholar] [CrossRef]
- Bacolla, A.; Tainer, J.A.; Vasquez, K.M.; Cooper, D.N. Translocation and deletion breakpoints in cancer genomes are associated with potential non-B DNA-forming sequences. Nucleic Acids Res. 2016, 44, 5673–5688. [Google Scholar] [CrossRef] [PubMed]
- Neil, A.J.; Kim, J.C.; Mirkin, S.M. Precarious maintenance of simple DNA repeats in eukaryotes. BioEssays 2017, 39. [Google Scholar] [CrossRef] [PubMed]
- Hampp, S.; Kiessling, T.; Buechle, K.; Mansilla, S.F.; Thomale, J.; Rall, M.; Ahn, J.; Pospiech, H.; Gottifredi, V.; Wiesmuller, L. DNA damage tolerance pathway involving DNA polymerase iota and the tumor suppressor p53 regulates DNA replication fork progression. Proc. Natl. Acad. Sci. USA 2016, 113, 4311–4319. [Google Scholar] [CrossRef]
- Kim, E.; Deppert, W. The complex interactions of p53 with target DNA: We learn as we go. Biochem. Cell Biol. 2003, 81, 141–150. [Google Scholar] [CrossRef]
- Sakamoto, N.; Larson, J.E.; Iyer, R.R.; Montermini, L.; Pandolfo, M.; Wells, R.D. GGA*TCC-interrupted triplets in long GAA*TTC repeats inhibit the formation of triplex and sticky DNA structures, alleviate transcription inhibition, and reduce genetic instabilities. J. Biol. Chem. 2001, 276, 27178–27187. [Google Scholar] [CrossRef]
- Singh, S.K.; Qiao, Z.; Song, L.; Jani, V.; Rice, W.; Eng, E.; Coleman, R.A.; Liu, W.L. Structural visualization of the p53/RNA polymerase II assembly. Genes Dev. 2016, 30, 2527–2537. [Google Scholar] [CrossRef]
- Brazdova, M.; Navratilova, L.; Tichy, V.; Nemcova, K.; Lexa, M.; Hrstka, R.; Pecinka, P.; Adamik, M.; Vojtesek, B.; Palecek, E.; et al. Preferential binding of hot spot mutant p53 proteins to supercoiled DNA in vitro and in cells. PLoS ONE 2013, 8, e59567. [Google Scholar] [CrossRef]
- Laptenko, O.; Shiff, I.; Freed-Pastor, W.; Zupnick, A.; Mattia, M.; Freulich, E.; Shamir, I.; Kadouri, N.; Kahan, T.; Manfredi, J.; et al. The p53 C terminus controls site-specific DNA binding and promotes structural changes within the central DNA binding domain. Mol. Cell 2015, 57, 1034–1046. [Google Scholar] [CrossRef]
- Clark, R.M.; Bhaskar, S.S.; Miyahara, M.; Dalgliesh, G.L.; Bidichandani, S.I. Expansion of GAA trinucleotide repeats in mammals. Genomics 2006, 87, 57–67. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Helma, R.; Bažantová, P.; Petr, M.; Adámik, M.; Renčiuk, D.; Tichý, V.; Pastuchová, A.; Soldánová, Z.; Pečinka, P.; Bowater, R.P.; et al. p53 Binds Preferentially to Non-B DNA Structures Formed by the Pyrimidine-Rich Strands of GAA·TTC Trinucleotide Repeats Associated with Friedreich’s Ataxia. Molecules 2019, 24, 2078. https://doi.org/10.3390/molecules24112078
Helma R, Bažantová P, Petr M, Adámik M, Renčiuk D, Tichý V, Pastuchová A, Soldánová Z, Pečinka P, Bowater RP, et al. p53 Binds Preferentially to Non-B DNA Structures Formed by the Pyrimidine-Rich Strands of GAA·TTC Trinucleotide Repeats Associated with Friedreich’s Ataxia. Molecules. 2019; 24(11):2078. https://doi.org/10.3390/molecules24112078
Chicago/Turabian StyleHelma, Robert, Pavla Bažantová, Marek Petr, Matej Adámik, Daniel Renčiuk, Vlastimil Tichý, Alena Pastuchová, Zuzana Soldánová, Petr Pečinka, Richard P. Bowater, and et al. 2019. "p53 Binds Preferentially to Non-B DNA Structures Formed by the Pyrimidine-Rich Strands of GAA·TTC Trinucleotide Repeats Associated with Friedreich’s Ataxia" Molecules 24, no. 11: 2078. https://doi.org/10.3390/molecules24112078
APA StyleHelma, R., Bažantová, P., Petr, M., Adámik, M., Renčiuk, D., Tichý, V., Pastuchová, A., Soldánová, Z., Pečinka, P., Bowater, R. P., Fojta, M., & Brázdová, M. (2019). p53 Binds Preferentially to Non-B DNA Structures Formed by the Pyrimidine-Rich Strands of GAA·TTC Trinucleotide Repeats Associated with Friedreich’s Ataxia. Molecules, 24(11), 2078. https://doi.org/10.3390/molecules24112078