3.1. Prediction Performance
In this study, effort is directed toward the determination of feature types that are beneficial for the prediction of APCs. This was performed by comparing the performance of different feature types as constructed using SVM and RF by means of 5-fold CV and jackknife test. The five basic features consisting of AAC, DPC, PCP, PseAAC and Am-PseAAC were considered as well as their combinations: AAC + PseACC, AAC + Am-PseACC, PseACC + Am-PseACC and AAC + PseACC + Am-PseACC, which were also utilized to investigate the complementarity of the five aforementioned features.
Prior to the development of the predictive model, the assessment of modelability of the benchmark dataset was performed by computing the MODI index. This index helps modelers to estimate the feasibility of obtaining robust and reliable predictive models. For the binary classification problem, if the value of MODI index is greater than 0.65, the feature is considered to be reliable for classification modelling; otherwise, the feature is not recommended for classification modelling.
As shown in
Table A1, all feature types and their combinations from the benchmark dataset met these criteria. Thus, it could be stated that our proposed features are reliable and efficient for constructing ACP predictor.
Table 2 and
Table 3 shows the performance comparison amongst different feature types with RF and SVM on the benchmark dataset via 5-fold CV and jackknife test, respectively. In addition,
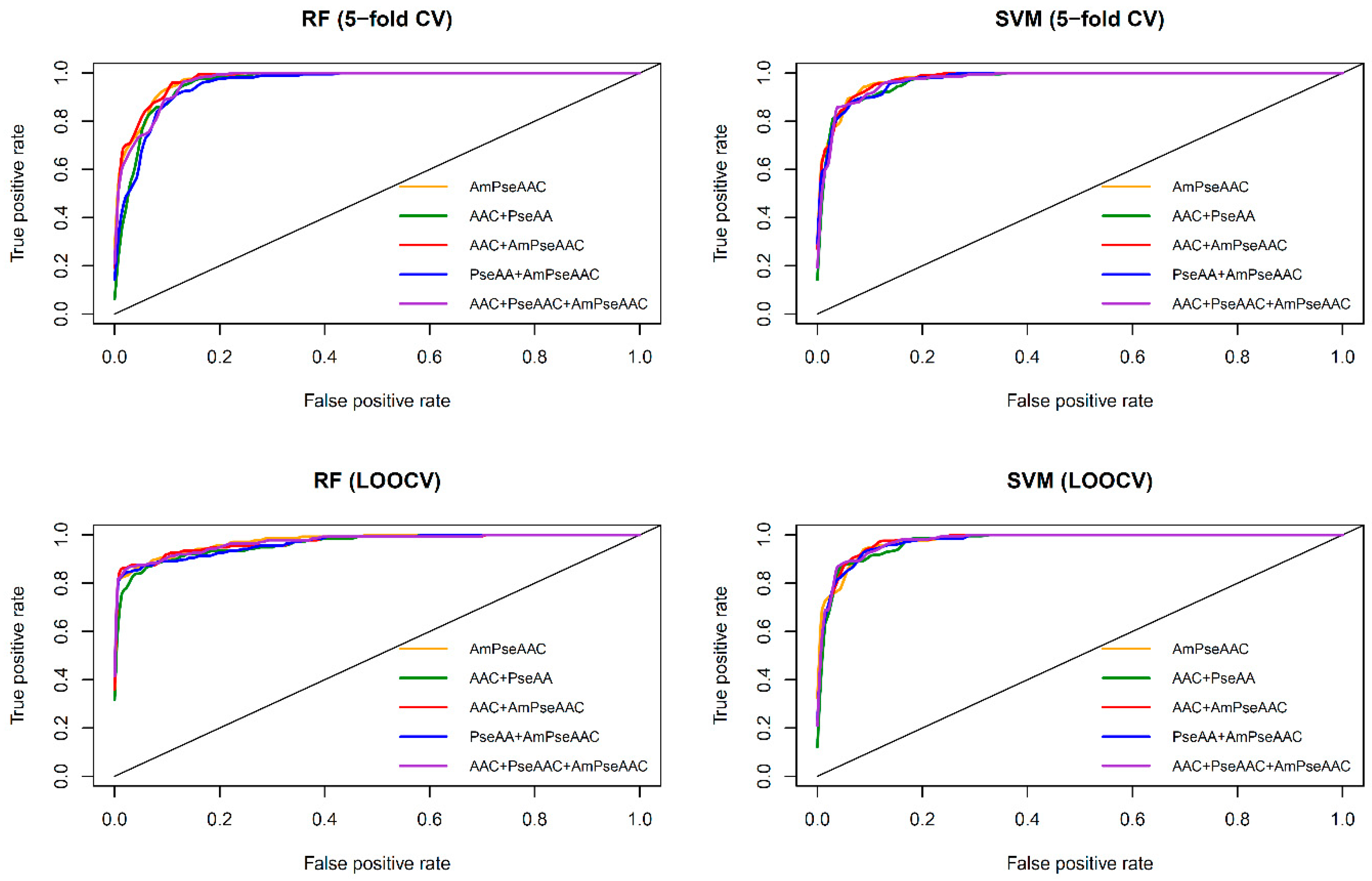
Figure 3 shows the receiver operating characteristic (
ROC) curve of the five considered feature sets obtained from RF (top and bottom left) and SVM (top and bottom right) as evaluated by 5-fold CV (top left and right) and jackknife test (bottom left and right). As seen in
Table 2 and
Table 3, SVM model with the Am-PseAAC feature afforded the highest accuracy of 95.03% as evaluated by the jackknife test. Meanwhile, RF model with PseACC feature and SVM model with AAC feature performed well with the second and third the highest accuracy of 93.28% and 92.98%, respectively. These results showed that ACC, PseACC and Am-PseAAC were effective for ACP prediction. Since using a combination of various features might yield better prediction performance, four combinations of the three effective features were also considered. The highest accuracy and
MCC of 95.61% and 0.91, respectively, was achieved by using SVM model cooperating with the combination feature of AAC and Am-PseAAC, while using the combination of the three effective features, i.e., ACC, PseACC and Am-PseAAC, performed slightly worse than the combination feature of AAC and Am-PseAAC with an accuracy and
MCC of 94.74% and 0.89, respectively. In addition, our prediction results were well consistent with previous studies [
27,
28,
29] and related studies [
33,
34,
36,
72].
Furthermore, observations pertaining to the performance comparisons from
Table 2 and
Table 3 and
Figure 3, it can be briefly summarized as follows.
Table 2 and
Table 3 shows that both RF and SVM models afforded improved prediction performances when using the Am-PseACC feature while the best prediction performance on the benchmark dataset via both 5-fold CV and jackknife test were achieved by the SVM model trained using the combination of AAC and Am-PseAAC features.
For the convenience of subsequent descriptions, we will refer to this method as ACPred as it represents the best model that will be used for further comparisons with other tools.
3.2. Comparison with Other Methods
To indicate the effectiveness of the proposed method, we compared the performance of our selected model (named ACPred) with other popular ACP predictors. Since Hajisharifi et al. [
24] were the first group to establish the benchmark dataset, and iACP [
26], iACP-GAEnsC [
28] and TargetACP [
31] were state-of-the-art ACP predictors that provided prediction results as assessed by the jackknife test, therefore these predictors were used for performance comparisons.
Table 4 lists the performance comparisons of ACPred with the three ACP predictors. As noticed from
Table 4, prediction results were obtained from two different experimental designs: (i) the prediction model was performed on the benchmark dataset as derived from the work of [
24] as well as those of Hajisharifi et al.’s method, iACP, iACP-GAEnsC, TargetACP and ACPred, (ii) the prediction model was built using the balanced dataset including TargetACP and ACPred. The reported results from aforementioned methods as summarized in
Table 4 was obtained directly from the work of iACP-GAEnsC [
28].
As for performance comparisons on the benchmark dataset, it was found that ACPred was comparable with that of iACP-GAEnsC and TargetACP as indicated by four statistical parameters. Moreover, ACPred yielded a greater prediction performance than the method proposed by Hajisharifi et al. and iACP. To the best of our knowledge, iACP-GAEnsC is the only method that utilizes the synthetic minority oversampling technique (SMOTE) [
73] technique for constructing a balanced dataset from the original benchmark dataset [
24]. From the perspectives of machine learning, it is not fair to directly compare prediction results between TargetACP and ACPred because TargetACP was trained and tested on the balanced dataset consisting of 205 ACPs and 205 non-ACPs. In order to perform a fair comparison, the herein proposed ACPred method was applied on the balanced dataset (called ACPred-modified), which was generated by means of the SMOTE technique. Herein, the oversampling method was used to add synthetic samples for the minority class (i.e., ACPs) presented in the benchmark dataset by setting the parameters of the number of nearest neighbours (
k), the number of extra cases (oversampling) to add to the minority class (perc.over) and the number of cases to reduce (undersampling) from the majority class (perc.under) were set to 9, 50 and 300, respectively. As seen in
Table 4, ACPred-modified was found to outperform Hajisharifi et al.’s method, iACP and iACP-GAEnsC with an improvement of 2%–4% and 3%–7% on
Ac and
MCC, respectively. However, it was observed that ACPred-modified performed slightly worse than TargetACP by approximately 2%–3% where ACPred-modified and TargetACP provided 97.56%
Ac/0.95
MCC and 98.78%
Ac/0.97
MCC, respectively. Nevertheless, TargetACP was constructed with a sophisticated design thus, it is not easy to identify and assess which features offer the most contribution to the prediction improvement. On the other hand, ACPred was designed in a systematic manner for prediction and characterization of anticancer activities of peptides. In addition, a user-friendly web server ACPred was developed to facilitate high-throughput prediction of ACP. Therefore, the proposed ACPred model could become a practical tool for predicting and interpreting the anticancer activity of peptides, or at least as a complementary tool to existing methods in the field.
Thinking to the future, some possible improvements that could enhance the prediction performance for predicting anticancer activity of peptides are described hereafter. Firstly, it is worthy to explore the separation of peptides according to their sequence lengths (e.g., peptides having 4, 5 or 6 amino acids, peptides having 7–12 amino acids, and peptides having more than 12 amino acids) followed by the development of separate predictive models for each of the five sequence range [
36,
74]. Secondly, extract 5, 10 and 15 amino acids from the N or C terminus for the development of predictive QSAR models, which has successfully been demonstrated by the group of P.S. Raghava [
23] Thirdly, explore the modelability of the dataset using the approach of Golbraikh et al. [
44] such that robust models could be developed. Fourthly, the currently employed non-anticancer peptides [
24] are not based on true experimentally-determined inactive peptides but are random peptides obtained from the UniProt database therefore, if such true inactive peptides could be determined they may aid in the development of robust models.
Aside from the aforementioned methodological improvements that have been witnessed from the literature in the development of anticancer peptide classifiers, however, the underlying origin of anticancer activity of investigated peptides as rationalized by peptide features is an area that deserves more attention. Current predictive models used for the prediction of anticancer activity of peptides are primarily based on sequence order-independent descriptors that does not consider the order of amino acids in a peptide sequence (e.g., AAC and DPC), which hampers the identification of motifs from peptides. Such limitation has been addressed by the group of KC Chou [
41,
75,
76] in which they developed PseAAC and Am-PseAAC descriptors that take into account sequence order information based on physicochemical properties. However, the interpretability of these descriptors is limited and thus, existing approaches for rationalizing biological interpretation from these predictive models are resorted back to the sequence order-independent descriptors such as AAC and DPC. Analysis of the literature revealed that interpretable descriptors such as the
z-scale descriptors [
77,
78] have been successful for predicting robust and interpretable models, however the flaw of this approach is that the peptides used in the development of the model should be of equal length so as to conduct sequence alignment. Methodological tweaks addressing such limitation have been made via the use of autocorrelation such that sequence alignment is not needed and would allow the models to be built on peptides of varied lengths. In spite of this, this method is not applicable in this study field in which the prepared dataset removes peptides having high similarity of greater than 90% therefore, rendering the removal of a large number of peptides that is normally needed when using
z-scale descriptors. As such, there is ample room for the discovery of new and interpretable descriptors for the development of predictive models of peptides, which is an important area for further driving the field of anticancer peptide prediction.
3.3. Biological Space
The identification of feature importance from AAC and DPC descriptors can provide a better understanding on the biochemical and biophysical properties of anticancer activities of peptides. Previously, AAC and DPC features have been analyzed as to further gain insights on how to characterize therapeutic peptides [
29,
34,
36,
47] and protein functions [
33,
51,
79]. In this study, the value of MDGI was adopted to rank and estimate the importance of each AAC and DPC feature. Such information is derived from the analysis of the benchmark dataset that consists of 138 ACPs and 205 non-ACPs.
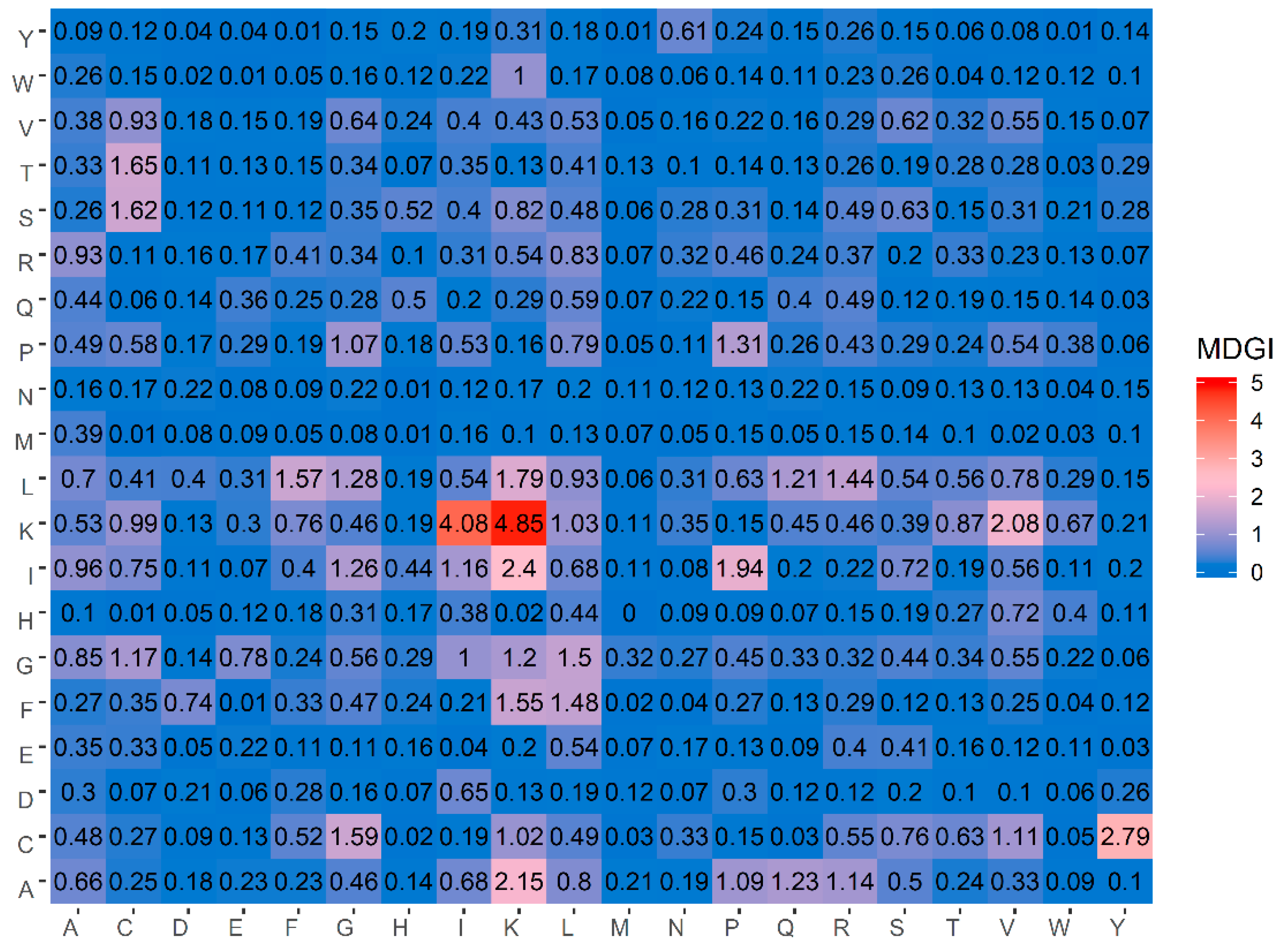
Table 5 lists the percentage values of the 20 amino acids for both ACPs and non-ACPs along with amino acid composition difference between the two classes as well as their MDGI values. In addition, a heatmap showing the feature importance for DPC features is shown in
Figure 4. From
Table 5, it can be observed that the ten informative amino acids with the highest MDGI values are Lys, Cys, Gln, Arg, Ile, Leu, His, Pro, Glu and Gly (29.54, 19.71, 15.89, 11.42, 10.13, 7.84, 7.80, 7.37, 6.88 and 6.56, respectively). Meanwhile,
Figure 4 shows that the 20 top-ranked dipeptides according to their MDGI value are KK, KI, CY, IK, AK, KV, IP, LK, TC, SC, CG, LF, FK, GL, FL, LR, PP, LG, IG and AQ. Interestingly, among the 5 top-ranked informative amino acids, 3 of these are polar (i.e., Lys, Gln and Arg), while 2 are non-polar (i.e., Cys and Ile) and hydrophobic residues. In addition, Lys and Arg are also positively-charged residues, which may support the anti-cancer cell penetrating properties of peptides (i.e., that is required for targeting tumors with specificity and low toxicity) [
80]. Although the percentage compositions differentiating ACPs from non-ACPs as presented in
Table 5 highlight that ACPs a have higher percentage of polar residues and lower percentage of basic residues as compared to non-ACPs, cell surface binding and internalization are critical for specific targeting of cells with anti-cancer activities. According to the membrane of cancer cells presenting anionic molecules such as phosphatidyl serine (PS), heparin sulfate and O-glycosylated mucins, the ACPs that contain positive charge are critical for endocytosis and selectively killing cancer cells. While hydrogen bonds that appear on ACPs were designed for improving the solubilization of hydrophobic molecules on ACPs. However, positively charged amino acids alone are not enough to completely neutralize cancer cells but rather aid in rapid internalization of cancer cells for selective killing with low toxicity to normal cells. Moreover, hydrogen bonded interactions to encapsulate anti-cancer drugs has been utilized for increasing the oral efficiency and drug-controlled release. Therefore, only hydrogen bond on ACPs are not adequate to resist cancer cells. Thus, the combination between crucial amino acids and physicochemical properties could provide a synergistic effect that enhances the efficiency of ACPs [
81]. As such, results from the analysis of the top ranked informative AAC are discussed below.
To roughly analyze the characteristics of ACPs, a few studies have shed light on important amino acids and dipeptides commonly found in ACPs by using simple composition analysis approaches without the use of experimental methods. For instance, Tyagi et al. [
23] reported results of residue preference at 10 N-terminus and 10 C-terminus by using the sequence logos. Their analysis revealed that Leu and Lys were typically found at the N-terminus while Cys, Leu and Lys were typically found at the C-terminus. Chen et al. [
26] also confirmed that amino acids including Lys, Ile, Cys, Glu, and Gly were abundant in ACPs as compared to non-ACPs. Recently, Manavalan et al. [
29] revealed that the three top-ranked informative dipeptides consisted of KK, AK and KL. As can be noticed in
Table 5 and
Figure 4, our analysis results derived from the MDGI values of AAC and DPC were quite consistent with the three aforementioned studies.
As mentioned above, amongst the five top-ranked informative amino acids, Lys had the highest MDGI value of 29.54. Such feature with the highest MDGI is considered to be the most important factor governing the anticancer activity. As a basic residue, Lys is highly conserved in the composition of therapeutic peptides as it enhances the formation of electrostatic interactions between the peptide and the plasma membrane owing to its cationicity [
82]. The role of cationicity in ACPs have been previously investigated by the study of Gopal et al. [
83]. The authors modified a synthetic AMP known as HPA3NT3 (FKRLKKLFKKIWNWK), which was derived from Helicobacter pylori by substituting Arg and Asn with Lys at positions 3 and 13, respectively. Gopal et al. reported that the Lys-modified peptide increased the peptide selectivity toward negatively-charged membrane surface, which could allow for endocytosis and specificity of the APC in killing cancer cells. In addition, Wang et al. [
84] has shown that the cell-penetrating ability is critical for specifically targeting cationic cancer cells. The authors observed that a Lys-rich peptide, L-K6 (IKKILSKIKKLLK-NH2) could internalize into MCF-7 cells without significant cell surface disruption. Interestingly, the authors also found that the internalized L-K6 could induce MCF-7 cell death without significant cytoskeleton disruption and mitochondrial impairment. Moreover, these findings indicated that the negatively-charged phosphatidylserine (PS) reported to be abundantly exposed on cancer cell surfaces [
85,
86] might contribute to the preferential binding of L-K6 to MCF-7 cancer cells. Furthermore, defensins are a large subfamily of natural cationic human AMP showing anticancer properties. Their roles in the immunomodulation of the innate and adaptive immune system have extensively been studied in previous works [
87,
88]. Defensins are categorized as either α-defensins or β-defensins, depending on their sequence homology and Cys connectivity (
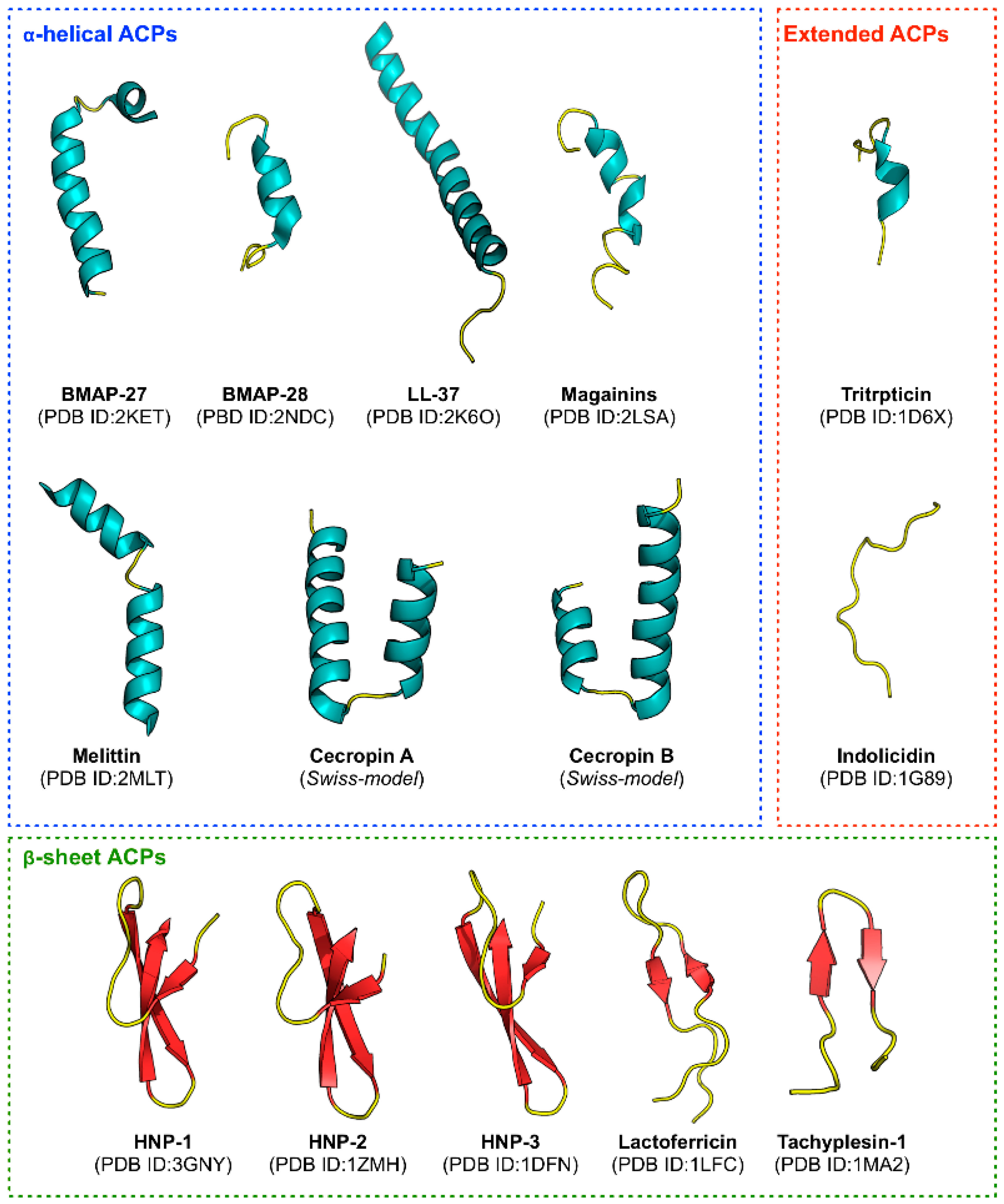
Figure 1), they are found to be rich in cationic residues (e.g., Lys and Arg) that enhances their immunomodulatory effects [
89]. Papo et al. [
90] constructed a short 15 amino acid peptide consisting of
d- or
l- amino acids (
d-K
6L
9; LKLLKKLLKKLLKLL-NH2) and observed that
d-K
6L
9 specifically targets and lyses cancer cells as well as inhibiting the growth of primary and metastatic tumors in in vivo models. The ability of
d-K
6L
9 in selectively targeting cancer cells can be attributed to its cationicity whereby the peptide can bind to highly enriched surface-exposed PS in cancer cells as compared to non-cancer cells [
91]. Similarly, Arg, which is a positively-charged residue that differs from Lys by its side chain, was shown to be the fourth informative amino acid form our analysis thus, reiterating the importance of the cationic property of APCs.
The second most important amino acid was Cys that afforded an MDGI value of 19.71. Cys is a highly reactive amino acid in which it contains a thiol group that is oxidized to form a dimer thus creating a disulfide bridge between two spatially adjacent Cys residues. The significance of disulfide bridges are seen in their strong, hydrophobic nature which are extremely important for the overall structural fold of the peptide, while also increasing the peptide stability against proteolytic degradation [
92]. Almost a decade ago, GO-201 ([R]
9CQCRRKNYGQLDIFP) achieved in facilitating the inhibition of mucin-1 cytoplasmic domain (MUC1-CD) [
93]. Thus far, many studies have established the function of the GO-201 peptide whereby its anticancer activity was demonstrated both in vitro and in vivo against human breast cancer, prostate cancer, chronic myelogenous leukemia and pancreatic cancer [
93,
94,
95,
96]. From these aforementioned studies, the researchers were able to determine key motifs responsible for the anticancer activity of GO-201 as CxC, thus reiterating the crucial role of Cys disulfide bridges [
97]. Furthermore, Tyuryaeva et al. [
98] explored the molecular mechanism of Cys-induced cytotoxicity and the resulting apoptosis of cancer cells by designing and testing peptide sequences containing the key CxC ([R]
9KCGCFF; named DIL) or CxxC ([R]
9FF
CPH
CYQ; named DOL) motifs. The results obtained from this study suggests that the disulfide oxidoreductase is responsible for the effective cytotoxtic action of Cys. Therefore, it can be inferred that peptides containing the CxC or CxxC motifs form disulfide bridges that pilot a partial loss of protein function and the rapid onset of apoptosis. It can therefore be anticipated that such peptide sequences could help to create a potentially valuable new class of ACPs. Similarly, Schroeder et al. [
99] ascertained a novel peptide GKAKCCK having pronounced antimicrobial activity. The authors discovered that the efficiency of this peptide was critically dependent on the two continuous cysteines and that upon mutating these residues to Ala or Ser, its activity was completely abrogated. Hence, given the well-established connection between antimicrobial and anticancer peptides [
13,
100], it is likely that the GKAKCCK peptide is able to induce antitumorigenic effects.
3.4. Mechanistic Interpretation of Informative PCP
The physicochemical properties of amino acids play an essential role as effective features for identifying and characterizing protein or peptide functions from their primary sequences [
33,
34,
36,
51,
79]. It is well known that PCPs [
39], such as molecular volume, exposure or accessible surface, polarity (hydrophobicity/hydrophilicity), charge/pK, hydrogen-bonding potential and so forth are correlated with the structure and function of the amino acid sequence [
101]. Herein, we have obtained the ten top-ranked informative PCPs corresponding to their MDGI values as shown in
Table 6.
The most important PCP is seen to be ‘hydrophobicity’ with an MDGI value of 1.51, which corresponds to the AAindex ARGP820101. In addition, from the analysis, 4 of the 10 top-ranked informative PCPs are related to α-helix properties of peptides consisting of ARGP820102, BEGF750101, ISOY800106 and CHOP780201 with corresponding MDGI values of 1.40, 0.92, 0.64 and 0.61, respectively. Furthermore, 3 of 10 top-ranked informative PCPs including the AAindices of BEGF750102, BEGF750103 and CHOP780101 are related to β-sheet properties with MDGI values of 1.00, 0.87 and 0.83, respectively. Consequently, it could be inferred that the hydrophobicity of a peptide along with its structural orientation (i.e., α-helix or β-sheet) are significant in determining the anticancer properties of peptides. The important PCPs of ACPs are analyzed and discussed below.
3.4.1. Hydrophobic Residues on α-Helical Structure Enhances Cell Penetrating Properties of Anticancer Peptides
It is commonly known that the hydrophobicity property is critical for the 3D structure of a protein. Many descriptors have been developed for measuring the hydrophobicity of proteins, depending on the presence of hydrophobic residues at specific locations in the peptide sequence. These indices are usually based on the 3D crystal structure of proteins coupled with the physicochemical properties of their side chains [
102,
103]. For instance, Argos et al. [
104] utilized the hydrophobic index as measured by Nozaki and Tanford [
105] for the 20 canonical amino acids for determining the role of hydrophobicity on the structure of amino acids. As mentioned above, the property of ARGP820101 with an MDGI value of 1.51 pertained to the hydrophobicity property. This result suggests that the presence of hydrophobic amino acid residues on the α-helix might be important for governing the anticancer activity of peptides. Moreover, the property of ARGP820103 (the fourth most important PCPs) is also associated with hydrophobicity property. Many previous studies have described the need for symbiosis between hydrophobicity and cationicity of peptides in order to enhance its cell penetrating property and its ability to reach target cell as to exert its effects [
106,
107,
108,
109]. In addition, researchers have made efforts to assess the influence of hydrophobicity on anticancer effects. For instance, Huang et al. [
108] systematically altered the hydrophobicity of a 26-residue α-helical peptide (peptide
P; KWKSFLKTFKSAKKTVLHTALKAISS) by replacing Ala residues with the more hydrophobic Leu residues as to increase its hydrophobicity or by changing Leu residues to Ala residues as to decrease its hydrophobicity. On the basis of these results, the authors observed a correlation between higher hydrophobic variants whereby the Leu-substituted peptides (A12L and A20L) exhibited greater anticancer activity against cancer cells with higher IC
50 values than peptide P. Thus, peptides with higher hydrophobicity are assumed to penetrate deeper into the hydrophobic core of the cell membrane, thereby causing pores or channels on the cancer cell membrane and exhibiting greater anticancer activity. From these results, it can be inferred that the presence of hydrophobic residues on the α-helical structure of ACPs can enhance its anticancer properties.
3.4.2. Peptides Forming an Amphipathic α-Helix Contributes to Anticancer Activity
Interestingly, four of the ten top-ranked informative PCPs consisting of ARGP820102, BEGF750101, ISOY800106 and CHOP780201 described helical properties of peptides such as helical potential [
104], inner helix [
110], frequency of helix end [
111] and frequency of α-helix [
112], respectively, as shown in
Table 6. This supports the notion that the helical structure plays a principal role in governing the anticancer property of peptides. In addition, the α-helical conformation as adopted by many ACPs in biological membranes, is now regarded as a key determinant of anticancer activity [
113]. BMAP-27 (GRFKRFRKKFKKLFKKLSPVIPLLHL) and BMAP-28 (GGLRSLGRKILRAWKKY GPIIVPIIRI) are well-known peptides derived from bovine sources having a cationic NH
2 terminal that forms an amphipathic α-helix. They exert their cytotoxic activity against human leukemia cells by enhancing membrane permeability, which allows for an influx of Ca++ ions, thereby leading to apoptosis via DNA fragmentation [
114]. In addition, the structures of Cecropin A (KWKLFKKIEKVGQNIRDGIIKAGPAVAVVGQATQIAK) and Cecropin B (KWKVFKKIEKMGRNIRNGIVKAGPAIAVLGEAKAL), which were first derived from insects, primarily consisted of two α-helix [
115,
116] while its amphipathic N-terminal (i.e., that is capable of interacting with anionic membrane components) is responsible for mediating the cytotoxic activity against cancer cells. On the other hand, the C-terminal is hydrophobic and it is postulated to facilitate peptide entry into the membrane thereby enabling oligomerization to occur, which subsequently leads to leakage of cell components due to pore formation and eventual cell death [
117]. In addition, Srisailam et al. [
118] designed a custom lytic peptide by modifying the cecropin B (CB) to contain two identical hydrophobic segments on both the N and the C terminals, thus disrupting its normal α-helical structure. The resulting peptide CB-3, was unable to effectively lyse cell membranes on cancer cells as well as bacterial cells as compared to the control peptide, therefore highlighting the significance of the peptide structure on its function. Interestingly, Moore et al. [
119] uncovered that CB exhibited anticancer activities when tested against colon adenocarcinoma cells in vivo and cytotoxicity against multidrug-resistant breast and ovarian cancer cell lines in vitro.
Similarly, Magainins isolated from the skin of
Xenopus laevis is comprised of a α-helical secondary structure with separate cationic and hydrophobic faces. Magainin-2 (GIGKFLHSAKKFGKAFVGEIMNS) causes lysis of both hematopoietic and solid tumor cell lines as while observing selective cytotoxic activity against several human bladder cell lines [
120]. In addition, it was perceived using florescence spectrometry that magainins could lyse tumor cells by forming ion-conducting α-helical channels in the cancer cell membrane [
121]. Melittin (GIGAVLKVLTTGLPALISWIKRKRQQ) is another well-known ACP that is derived from the venom of European honeybee (
Apis mellifera) [
122]. The N-terminal region of melittin is largely hydrophobic whereas the C-terminal contains positively-charged amino acid residues. Melittin form channels in lipid bilayers and is lytic against both cancer cells and normal cells [
123]. Self-association of amphipathic α-helical monomers of melittin is largely suggested to be responsible for perturbations of membrane integrity thereby leading to cellular lysis via the barrel stave mechanism [
124]. Furthermore, the only human cathelicidin-derived cationic ACP to date, LL-37 (LLGDFFRKSKEKIGKEFKRIVQRIKDFLRNLVPRTES) is present throughout the body and assumes a α-helical conformation that is comprised of both cationic and hydrophobic faces oriented in a parallel manner with lipid membranes. As such these peptides are suggested to engage in a carpet-like mechanism for its cytotoxicity [
125]. However, unlike other ACPs, LL-37 is toxic to eukaryotic cells at slightly higher concentrations and thus lacks the selectivity of an ACP. Nevertheless, Okumura et al. [
126] showed that a C-terminal fragment of LL-37 (hCAP18) was able to induce mitochondrial depolarization and apoptosis in human oral squamous carcinoma cells but not in healthy human gingival fibroblasts and human keratinocyte cells. In summary, it is evident that the amphipathicity associated with certain α-helical structures is pivotal for the elicitation of peptide anticancer activities.
3.4.3. Peptides Forming β-Sheet Are Vital to Peptide Structure and Contribute to Anticancer Activity
As seen in
Table 6, 3 out of the 20 top-ranked informative PCPs pertain to β-sheet structure (i.e., BEGF750102, BEGF750103 and CHOP78010) which correspond to properties such as conformational parameter of β-structure, conformational parameter of β-turn and normalized frequency of β-turn [
110], respectively. Although, most of the well-known APCs are α-helical structures, some β-sheet ACPs have also been extensively studied. Many studies showed that defensins contain a group of closely related Cys-Arg rich cationic peptides. Out of these, human α- and β-defensins remain the most studied, comprising of six conserved Cys residues that form three intramolecular disulfide bridges between the N-terminal and the C-terminal regions of the peptide [
127,
128]. In the studies of [
129,
130], human neutrophil peptides (HNPs) -1 (ACYCRIPACIAGERRYGTCIYQGRLWAFCC), -2 (CYCRIPACIAGERRYGTCIYQGRLWAFCC) and -3 (DCYCRIPACIAGERRYGTCIY QGRLWAFCC) were shown to possess cytotoxic effects against several human and mouse tumor cells such as human B-lymphoma cells, human oral squamous carcinoma cells, and mouse teratocarcinoma cells. HNPs were also shown to kill cancer cells through membrane binding mediated by electrostatic interactions, followed by rapid collapse of the membrane potential and loss of membrane integrity [
130,
131]. In addition, Mader et al. [
100,
132] reported that bovine lactoferricin (LfcinB; FKCRRWQWRMKKLGAPSITCVRRAF) isolated from cow’s milk, represents a cationic ACP with amphipathic β-sheet configuration displaying anticancer activity against leukemia cells and various other carcinomas [
100,
132]. Moreover, LfcinB not only exerts its action by inducing apoptosis through direct disruption of the mitochondrial membrane, but is also capable of lysing the membrane itself, depending on the cancer cell type [
133,
134].
Furthermore, isolated from the hemocytes of horseshoe crab (
Tachypleus tridentatus), the ACP tachyplesin I (KWCFRVCYRGICYRRCR) is arranged in two anti-parallel β-sheets that are held in place by two disulfide bonds [
135]. Chen et al. [
136] discovered that tachyplesin I killed cancer cells in a unique way by binding to hyaluronan on human prostate carcinoma cells, as well as to the C1q complement component in human serum, leading to the activation of the classical immune complement pathway. As a result, complement-mediated lysis of tachyplesin I-coated cancer cells was achieved. In a separate study, Adamia et al. [
137] observed that the binding activity of tachyplesin I to C1q was dependent on its secondary structure which consists of two disulfide bonds formed by Cys residues at positions 3–16 and 7–12. Previously it has also been demonstrated that, the deletion of Cys residues from tachyplesin I altered its β-sheet structure to that of a linear form thus, disrupting its activity [
138]. Therefore, from the aforementioned studies, it can be postulated that β-sheet is vital for maintaining the stability of peptide structures. As noticed in
Table 5, our result of informative amino acids is consistent with the above-mentioned studies, where Cys is the second most important amino acid having a MDGI value of 19.71.
Nevertheless, further research is necessary to determine the key amino acids that could transform non-ACPs into ACPs and thus, allow the generation of a library of potentially novel ACPs. The design of effective ACPs from non-ACPs could possibly be based on the improvement of physicochemical properties, like hydrophobicity, positive charge, replacing the amino acid used or adding a functional motif with better activity towards the targeted cancer cells. In that regard, Tada et al. [
139] revealed that the replacement of amino acid His to Arg in epidermal growth factor receptor (EGFR) binding peptide increased its anti-tumor activity via stronger binding affinity to the EGFR on cancer cells. Furthermore, the utilization of a motif containing the amino acids Trp, Met and Trp (WMW) was able to sensitize apoptosis and reduce the migratory effect on cancer cells which in turn could synergize the anti-cancer activity of peptides [
140]. Therefore, it is of paramount importance to explore this area further.
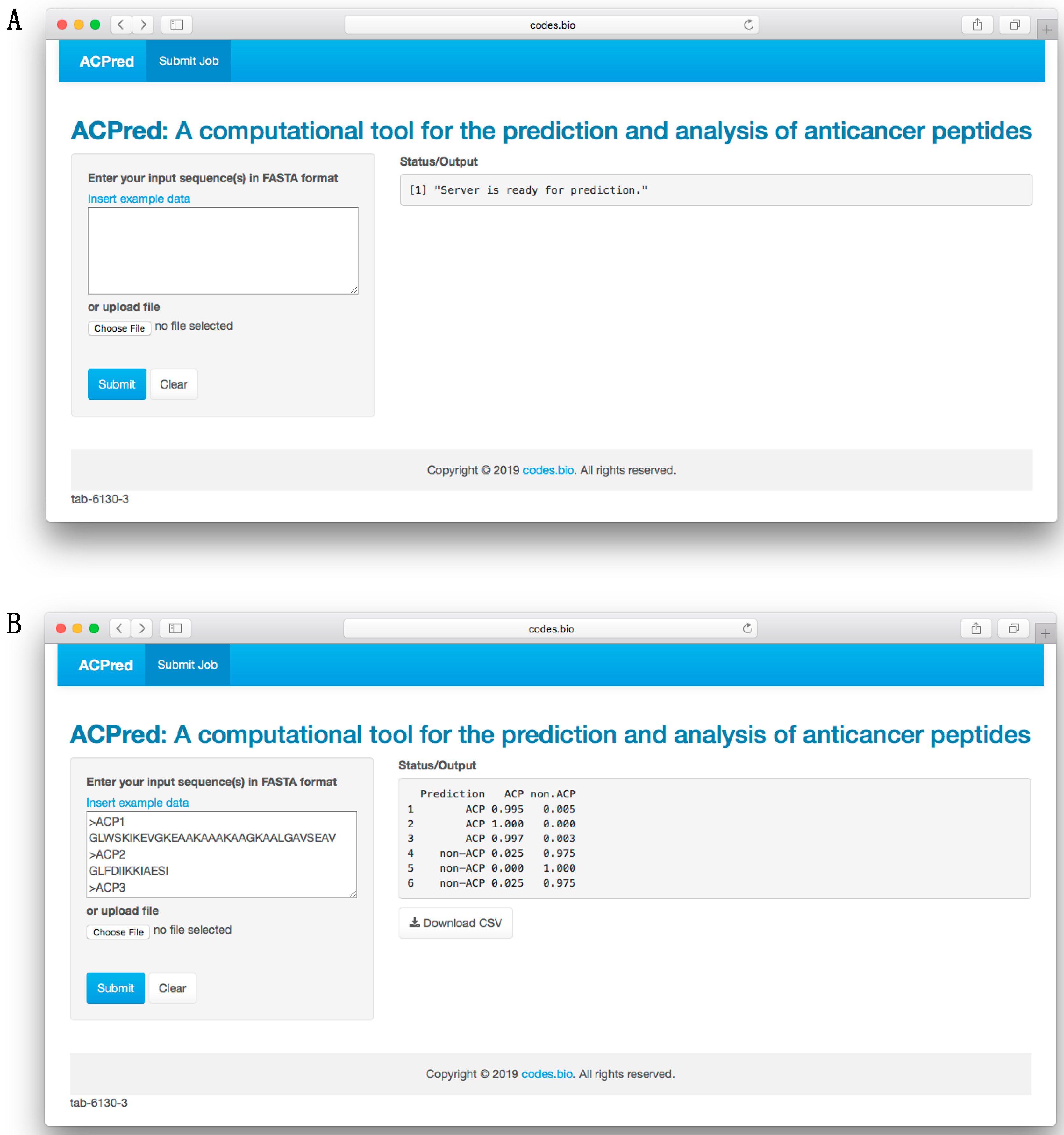
3.6. ACPred Web Server
In order to make the prediction model presented herein a practical tool that can be widely used by the scientific community, we have constructed a web server called the ACPred using the best model as described in previous sections. The web interface has been established using the Shiny package under the R programming environment. The web server is freely accessible at
http://codes.bio/acpred/. Screenshots of the ACPred web server is shown in
Figure 5 in which panel A shows the web server prior to submission of input data and panel B shown the web server after the prediction has been made.
Briefly, a step-by-step guide on using the web server is given below:
Step 2. Either enter the query peptide sequence into the Input box or upload the sequence file by clicking on the “Choose file” button (i.e., found below the “Enter your input sequence(s) in FASTA format heading”).
Step 3. Click on the “Submit” button to initiate the prediction process.
Step 4. Prediction results are automatically displayed in the grey box found below the “Status/Output” heading. Typically, it takes a few seconds for the server to process the task. Users can also download the prediction results as a CSV file by pressing on the “Download CSV button”.
Additionally, users can also run a local copy of ACPred on their own computer by using the following one-line code in an R environment:
shiny::runGitHub(‘acpred-webserver’, ‘chaninlab’)
However, prior to running the aforementioned code, it is recommended that users first install the prerequisite R packages. This can be performed by using the following code:
install.packages(c(‘shiny’, ‘shinyjs’, ‘shinythemes’, ‘protr’, ‘seqinr’, ‘caret’, ‘markdown’))

 ,
, 



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




