Carbon Dot-Mediated Capillary Electrophoresis Separations of Metallated and Demetallated Forms of Transferrin Protein



Abstract
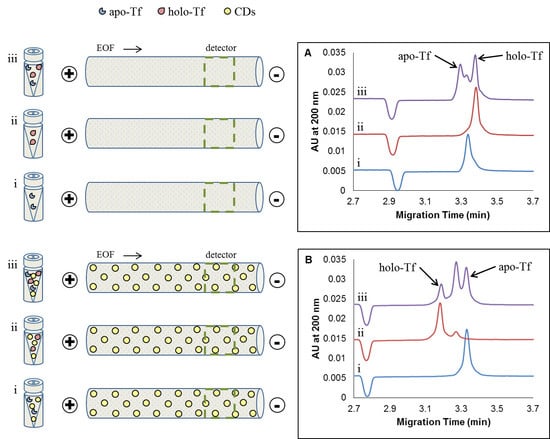
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
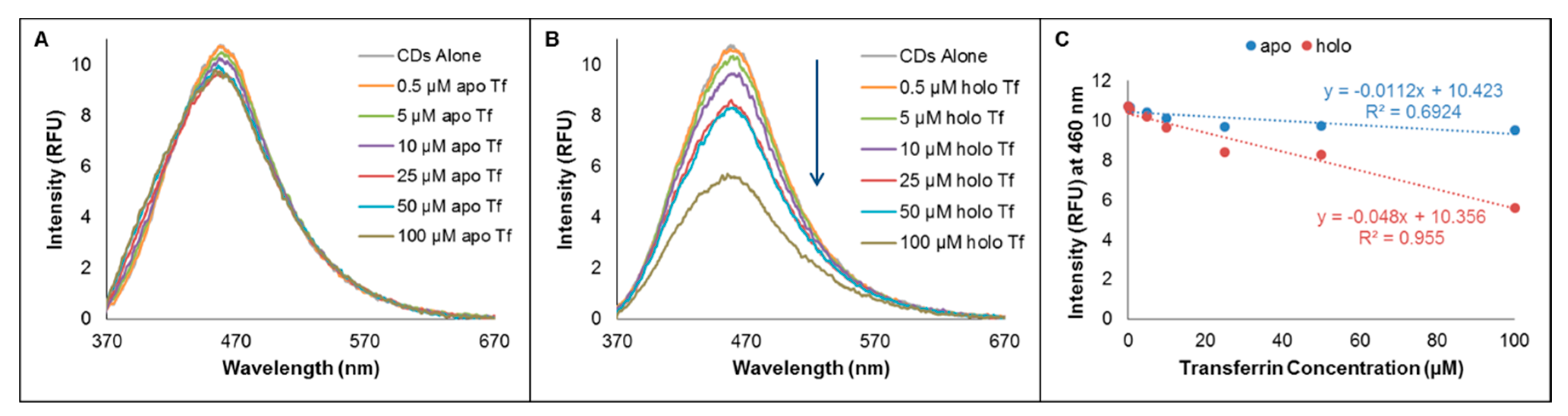
2.1. Probing Interactions Between CDs and Tf by Fluorimetry
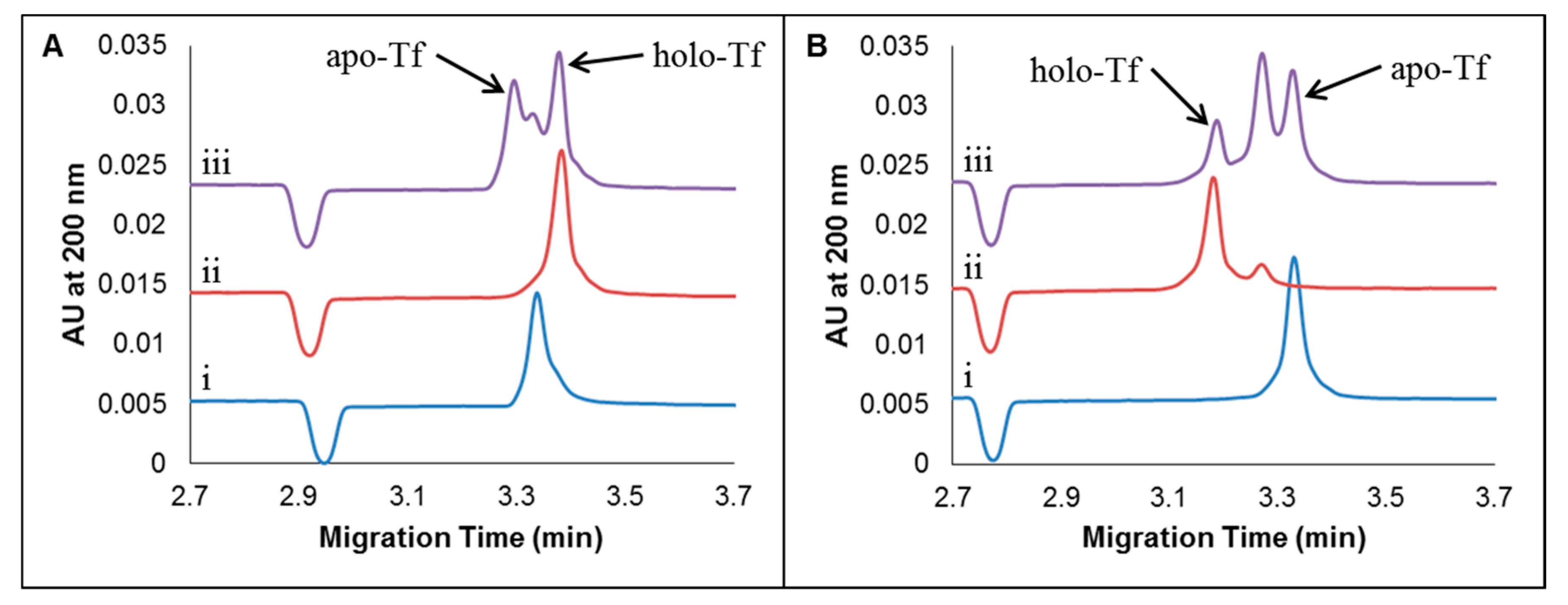
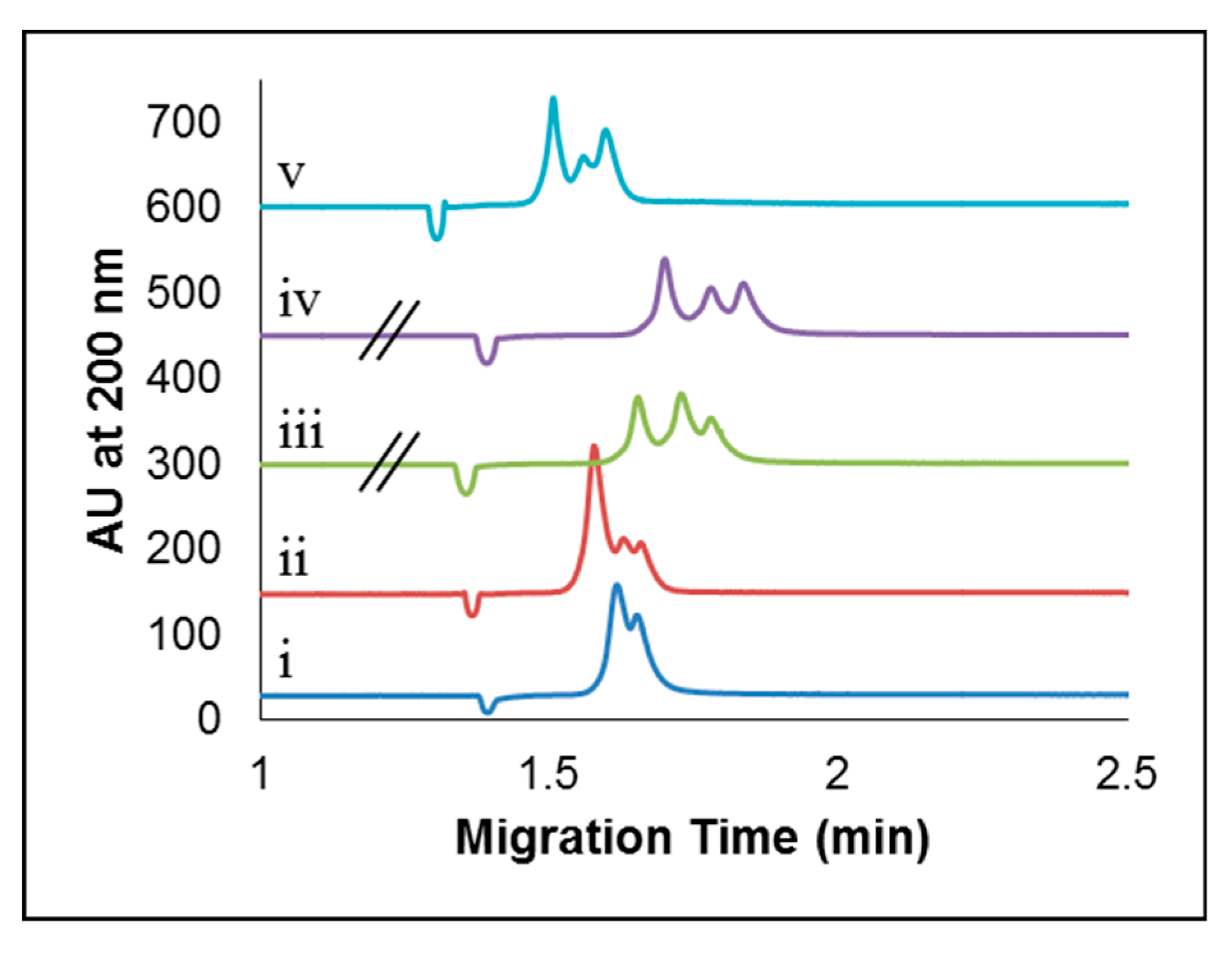
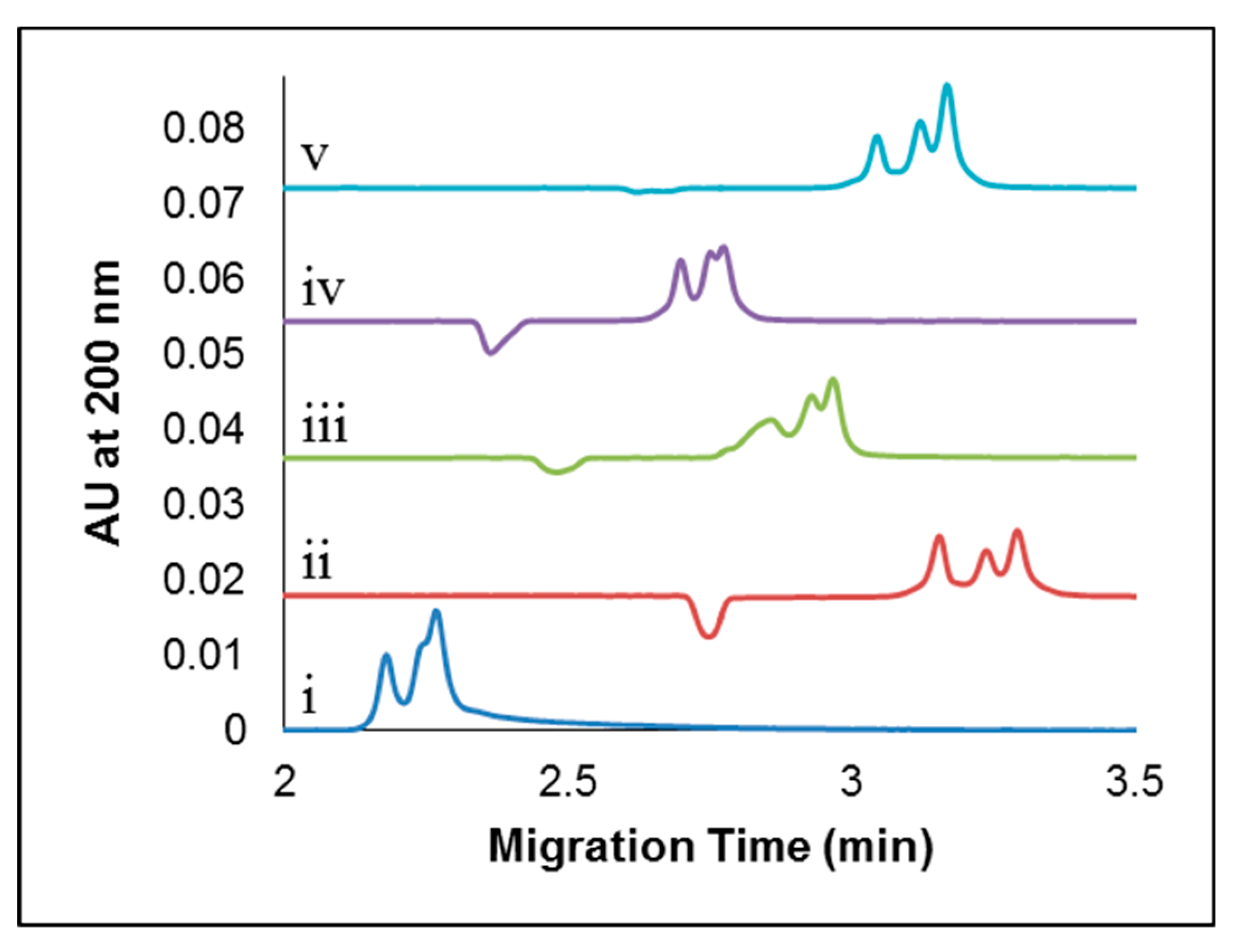
2.2. CE Method Development and Optimization for the Separation of Apo-Tf and Holo-Tf
2.2.1. Studying the Effects of Sample Preparation: Diluent and Sample Additives
2.2.2. Effect of Concentration of Added CDs
2.2.3. Separation Buffer Composition: Background Electrolyte, pH, and Concentration Effects
2.2.4. Optimizing Capillary Inside Diameter and Temperature
3. Materials and Methods
3.1. Reagents
3.2. Carbon Dots
3.3. Separation Buffer and Sample Preparation
3.4. Instrumentation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Shan, D.; Hsieh, J.-T.; Bai, X.; Yang, J. Citrate-based fluorescent biomaterials. Adv. Healthc. Mater. 2018, 7. [Google Scholar] [CrossRef]
- Zhao, P.; Zhu, L. Dispersibility of carbon dots in aqueous and/or organic solvents. Chem. Commun. 2018, 54, 5401–5406. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Bi, Q.; Zhang, X.; Wang, L.; Zhang, X.; Dong, S.; Zhao, L. Graphene quantum dots as additives in capillary electrophoresis for separation cinnamic acid and its derivatives. Anal. Biochem. 2016, 500, 38–44. [Google Scholar] [CrossRef]
- Miao, P.; Han, K.; Tang, Y.; Wang, B.; Lin, T.; Cheng, W. Recent advances in carbon nanodots: Synthesis, properties and biomedical applications. Nanoscale 2015, 7, 1586–1595. [Google Scholar] [CrossRef]
- Ju, J.; Chen, W. Synthesis of highly fluorescent nitrogen-doped graphene quantum dots for sensitive, label-free detection of Fe (III) in aqueous media. Biosens. Bioelectron. 2014, 58, 219–225. [Google Scholar] [CrossRef]
- Dong, Y.; Shao, J.; Chen, C.; Li, H.; Wang, R.; Chi, Y.; Lin, X.; Chen, G. Blue luminescent graphene quantum dots and graphene oxide prepared by tuning the carbonization degree of citric acid. Carbon 2012, 50, 4738–4743. [Google Scholar] [CrossRef]
- Qu, S.; Wang, X.; Lu, Q.; Liu, X.; Wang, L. A biocompatible fluorescent ink based on water-soluble luminescent carbon nanodots. Angew. Chem. Int. Ed. 2012, 51, 12215–12218. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Zhang, H.; Lai, J.; Peng, X.; Hu, Y.; Gu, W.; Ye, L. Carbon dots with red-shifted photoluminescence by fluorine doping for optical bio-imaging. Carbon 2018, 128, 78–85. [Google Scholar] [CrossRef]
- Bhaisare, M.L.; Talib, A.; Khan, M.S.; Pandey, S.; Wu, H.-F. Synthesis of fluorescent carbon dots via microwave carbonization of citric acid in presence of tetraoctylammonium ion, and their application to cellular bioimaging. Microchim. Acta 2015, 182, 2173–2181. [Google Scholar] [CrossRef]
- Goh, E.J.; Kim, K.S.; Kim, Y.R.; Jung, H.S.; Beack, S.; Kong, W.H.; Scarcelli, G.; Yun, S.H.; Hahn, S.K. Bioimaging of hyaluronic acid derivatives using nanosized carbon dots. Biomacromolecules 2012, 13, 2554–2561. [Google Scholar] [CrossRef] [PubMed]
- Zholobak, N.M.; Popov, A.L.; Shcherbakov, A.B.; Popova, N.R.; Guzyk, M.M.; Antonovich, V.P.; Yegorova, A.V.; Scrypynets, Y.V.; Leonenko, I.I.; Baranchikov, A.Y.; et al. Facile fabrication of luminescent organic dots by thermolysis of citric acid in urea melt, and their use for cell staining and polyelectrolyte microcapsule labelling. Beilstein J. Nanotechnol. 2016, 7, 1905–1917. [Google Scholar] [CrossRef]
- Zheng, M.; Liu, S.; Li, J.; Qu, D.; Zhao, H.; Guan, X.; Hu, X.; Xie, Z.; Jing, X.; Sun, Z. Integrating oxaliplatin with highly luminescent carbon dots: An unprecedented theranostic agent for personalized medicine. Adv. Mater. 2014, 26, 3554–3560. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, A.; Iqbal, K.; Xu, L.; Li, B.; Gong, D.; Liu, X.; Guo, Y.; Liu, W.; Qin, W.; Guo, H. Heterogeneous synthesis of nitrogen-doped carbon dots prepared via anhydrous citric acid and melamine for selective and sensitive turn on-off-on detection of Hg (II), glutathione and its cellular imaging. Sens. Actuators B Chem. 2018, 255, 1130–1138. [Google Scholar]
- Zhang, Q.; Zhang, C.; Li, Z.; Ge, J.; Li, C.; Dong, C.; Shuang, S. Nitrogen-doped carbon dots as fluorescent probe for detection of curcumin based on the inner filter effect. RSC Adv. 2015, 5, 95054–95060. [Google Scholar] [CrossRef]
- Wang, Y.; Gao, D.; Chen, Y.; Hu, G.; Liu, H.; Jiang, Y. Development of N, S-doped carbon dots as a novel matrix for the analysis of small molecules by negative ion MALDI-TOF MS. RSC Adv. 2016, 6, 79043–79049. [Google Scholar] [CrossRef]
- Duan, L.-P.; Ding, G.-S.; Tang, A.-N. Preparation of chitosan-modified silica nanoparticles and their applications in the separation of auxins by capillary electrophoresis. J. Sep. Sci. 2015, 38, 3976–3982. [Google Scholar] [CrossRef]
- Gong, Z.-S.; Duan, L.-P.; Tang, A.-N. Amino-functionalized silica nanoparticles for improved enantiomeric separation in capillary electrophoresis using carboxymethyl-beta-cyclodextrin (CM-beta-CD) as a chiral selector. Microchim. Acta 2015, 182, 1297–1304. [Google Scholar] [CrossRef]
- Manuel Jimenez-Soto, J.; Moliner-Martinez, Y.; Cardenas, S.; Valcarcel, M. Evaluation of the performance of singlewalled carbon nanohorns in capillary electrophoresis. Electrophoresis 2010, 31, 1681–1688. [Google Scholar] [CrossRef]
- Benitez-Martinez, S.; Simonet, B.M.; Valcarcel, M. Graphene nanoparticles as pseudostationary phase for the electrokinetic separation of nonsteroidal anti-inflammatory drugs. Electrophoresis 2013, 34, 2561–2567. [Google Scholar] [CrossRef]
- Cao, J.; Qu, H.; Cheng, Y. Separation of flavonoids and phenolic acids in complex natural products by microemulsion electrokinetic chromatography using surfactant-coated and carboxylic single-wall carbon nanotubes as additives. Electrophoresis 2010, 31, 1689–1696. [Google Scholar] [CrossRef]
- Neiman, B.; Grushka, E.; Lev, O. Use of gold nanoparticles to enhance capillary electrophoresis. Anal. Chem. 2001, 73, 5220–5227. [Google Scholar] [CrossRef] [PubMed]
- Viberg, P.; Jornten-Karlsson, M.; Petersson, P.; Spegel, P.; Nilsson, S. Nanoparticles as pseudostationary phase in capillary electrochrornatography/ESI-MS. Anal. Chem. 2002, 74, 4595–4601. [Google Scholar] [CrossRef]
- Zhao, T.; Zhou, G.; Wu, Y.; Liu, X.; Wang, F. Gold nanomaterials based pseudostationary phases in capillary electrophoresis: A brand-new attempt at chondroitin sulfate isomers separation. Electrophoresis 2015, 36, 588–595. [Google Scholar] [CrossRef]
- Harris, D.C. Chromatographic methods and capillary electrophoresis. In Quantitative Chemical Analysis; W.H. Freeman and Co.: New York, NY, USA, 2003; pp. 654–672. ISBN 0-7167-4464-3. [Google Scholar]
- Kinsel, G.R. Miscellaneous separation methods. In Fundamentals of Analytical Chemistry 8th EDITION; Skoog, D.A., West, D.M., Holler, F.J., Crouch, S.R., Eds.; Cengage Learning: Boston, MA, USA, 2003. [Google Scholar]
- Nowak, P.; Spiewak, K.; Brindell, M.; Wozniakiewicz, M.; Stochel, G.; Koscielniak, P. Separation of iron-free and iron-saturated forms of transferrin and lactoferrin via capillary electrophoresis performed in fused-silica and neutral capillaries. J. Chromatogr. A 2013, 1321, 127–132. [Google Scholar] [CrossRef]
- Yue, C.-Y.; Ding, G.-S.; Liu, F.-J.; Tang, A.-N. Water-compatible surface molecularly imprinted silica nanoparticles as pseudostationary phase in electrokinetic chromatography for the enantioseparation of tryptophan. J. Chromatogr. A 2013, 1311, 176–182. [Google Scholar] [CrossRef]
- Turiel, E.; Martin-Esteban, A. Molecular imprinting technology in capillary electrochromatography. J. Sep. Sci. 2005, 28, 719–728. [Google Scholar] [CrossRef] [PubMed]
- Ren, L.; Kim, H.K.; Zhong, W. Capillary electrophoresis-assisted identification of peroxyl radical generated by single-walled carbon nanotubes in a cell-free system. Anal. Chem. 2009, 81, 5510–5516. [Google Scholar] [CrossRef]
- Lippard, S.J.; Berg, J.M. Principles of Bioinorganic Chemistry; University Science Books: Sausalito, CA, USA, 1994; ISBN 0-935702-72-5. [Google Scholar]
- Li, H.Y.; Qian, Z.M. Transferrin/transferrin receptor-mediated drug delivery. Med. Res. Rev. 2002, 22, 225–250. [Google Scholar] [CrossRef]
- Ali, S.A.; Joao, H.C.; Hammerschmid, F.; Eder, J.; Steinkasserer, A. Transferrin Trojan horses as a rational approach for the biological delivery of therapeutic peptide domains. J. Biol. Chem. 1999, 274, 24066–24073. [Google Scholar] [CrossRef] [PubMed]
- Singh, M. Transferrin as a targeting ligand for liposomes and anticancer drugs. Curr. Pharm. Des. 1999, 5, 443–451. [Google Scholar]
- Qian, Z.M.; Li, H.Y.; Sun, H.Z.; Ho, K. Targeted drug delivery via the transferrin receptor-mediated endocytosis pathway. Pharmacol. Rev. 2002, 54, 561–587. [Google Scholar] [CrossRef]
- Du, W.; Fan, Y.; Zheng, N.; He, B.; Yuan, L.; Zhang, H.; Wang, X.; Wang, J.; Zhang, X.; Zhang, Q. Transferrin receptor specific nanocarriers conjugated with functional 7peptide for oral drug delivery. Biomaterials 2013, 34, 794–806. [Google Scholar] [CrossRef]
- Camp, E.R.; Wang, C.; Little, E.C.; Watson, P.M.; Pirollo, K.F.; Rait, A.; Cole, D.J.; Chang, E.H.; Watson, D.K. Transferrin receptor targeting nanomedicine delivering wild-type p53 gene sensitizes pancreatic cancer to gemcitabine therapy. Cancer Gene Ther. 2013, 20, 222–228. [Google Scholar] [CrossRef]
- Martinez, A.; Suarez, J.; Shand, T.; Magliozzo, R.S.; Sanchez-Delgado, R.A. Interactions of arene-Ru(II)-chloroquine complexes of known antimalarial and antitumor activity with human serum albumin (HSA) and transferrin. J. Inorg. Biochem. 2011, 105, 39–45. [Google Scholar] [CrossRef]
- Tortorella, S.; Karagiannis, T.C. Transferrin receptor-mediated endocytosis: A useful target for cancer therapy. J. Membr. Biol. 2014, 247, 291–307. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, A.; Chatterjee, S.; Khorwal, V.; Mukherjee, T.K. Luminescence turn-on/off sensing of biological iron by carbon dots in transferrin. Phys. Chem. Chem. Phys. 2016, 18, 5148–5158. [Google Scholar] [CrossRef]
- Nowak, P.; Spiewak, K.; Nowak, J.; Brindell, M.; Wozniakiewicz, M.; Stochel, G.; Koscielniak, P. Selective separation of ferric and non-ferric forms of human transferrin by capillary micellar electrokinetic chromatography. J. Chromatogr. A 2014, 1341, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Meng, Q.; Wang, L.; Zhang, J.; Song, Y.; Jin, H.; Zhang, K.; Sun, H.; Wang, H.; Yang, B. Highly photoluminescent carbon dots for multicolor patterning, sensors, and bioimaging. Angew. Chem. Int. Ed. 2013, 52, 3953–3957. [Google Scholar] [CrossRef] [PubMed]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sirkisoon, L.R.; Makamba, H.C.; Saito, S.; Colyer, C.L. Carbon Dot-Mediated Capillary Electrophoresis Separations of Metallated and Demetallated Forms of Transferrin Protein. Molecules 2019, 24, 1916. https://doi.org/10.3390/molecules24101916
Sirkisoon LR, Makamba HC, Saito S, Colyer CL. Carbon Dot-Mediated Capillary Electrophoresis Separations of Metallated and Demetallated Forms of Transferrin Protein. Molecules. 2019; 24(10):1916. https://doi.org/10.3390/molecules24101916
Chicago/Turabian StyleSirkisoon, Leona R., Honest C. Makamba, Shingo Saito, and Christa L. Colyer. 2019. "Carbon Dot-Mediated Capillary Electrophoresis Separations of Metallated and Demetallated Forms of Transferrin Protein" Molecules 24, no. 10: 1916. https://doi.org/10.3390/molecules24101916
APA StyleSirkisoon, L. R., Makamba, H. C., Saito, S., & Colyer, C. L. (2019). Carbon Dot-Mediated Capillary Electrophoresis Separations of Metallated and Demetallated Forms of Transferrin Protein. Molecules, 24(10), 1916. https://doi.org/10.3390/molecules24101916