Small Multitarget Molecules Incorporating the Enone Moiety


 ,
,  and
and 
Abstract
1. Introduction
2. Results and Discussion
2.1. Chemistry
2.2. Physicochemical Studies
2.2.1. Experimental Determination of Lipophilicity as RM Values
2.2.2. In Silico Determination of Lipophilicity Values as cLogP
N = 11, r = 0.854, q2 = 0.592, s = 0.257, F1, 9 = 24.266, α = 0.01
2.2.3. Molecular Properties Prediction—Lipinski “Rule of Five”
2.3. Biological Assays
3. Experimental Section
3.1. Materials and Instruments
3.2. Chemistry General Procedure
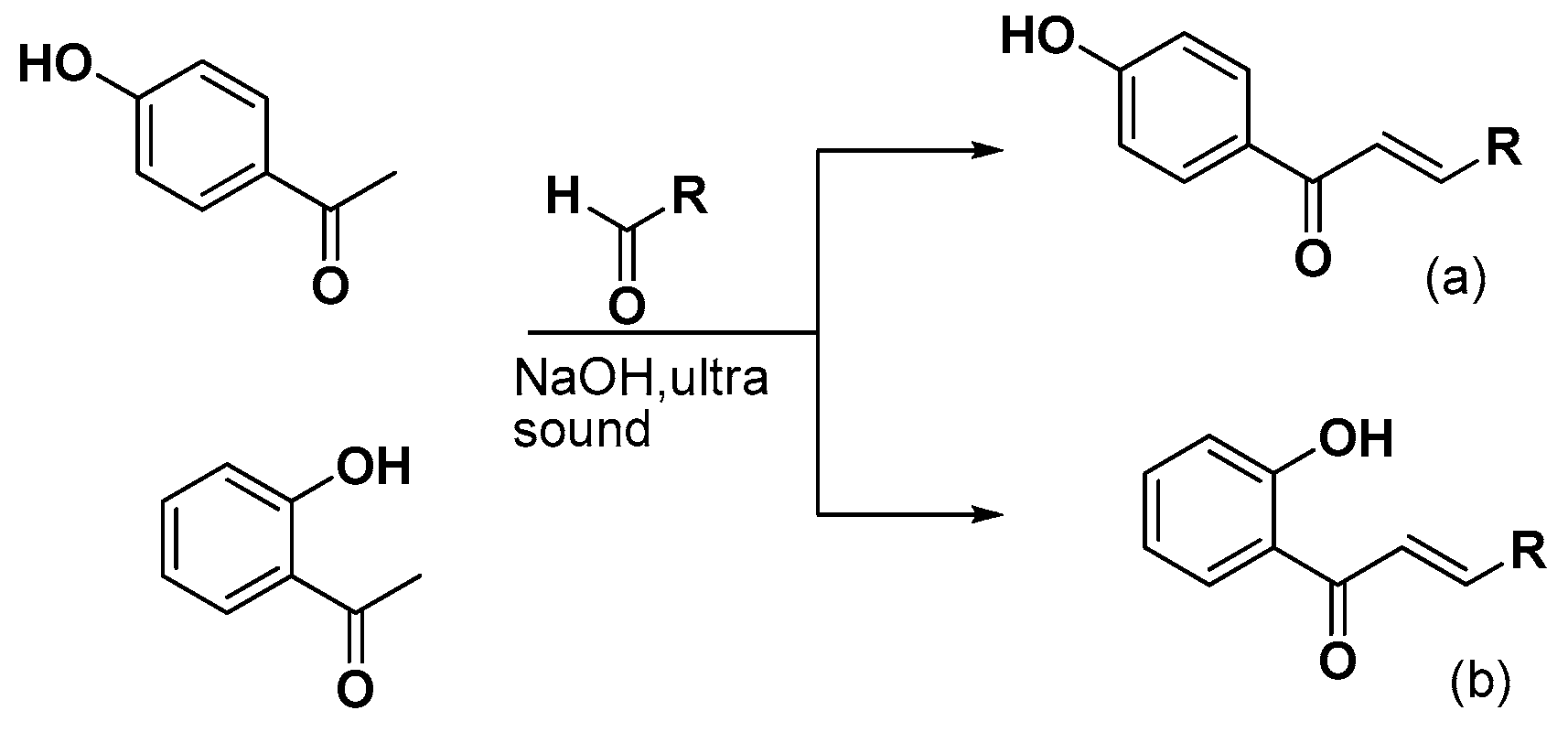
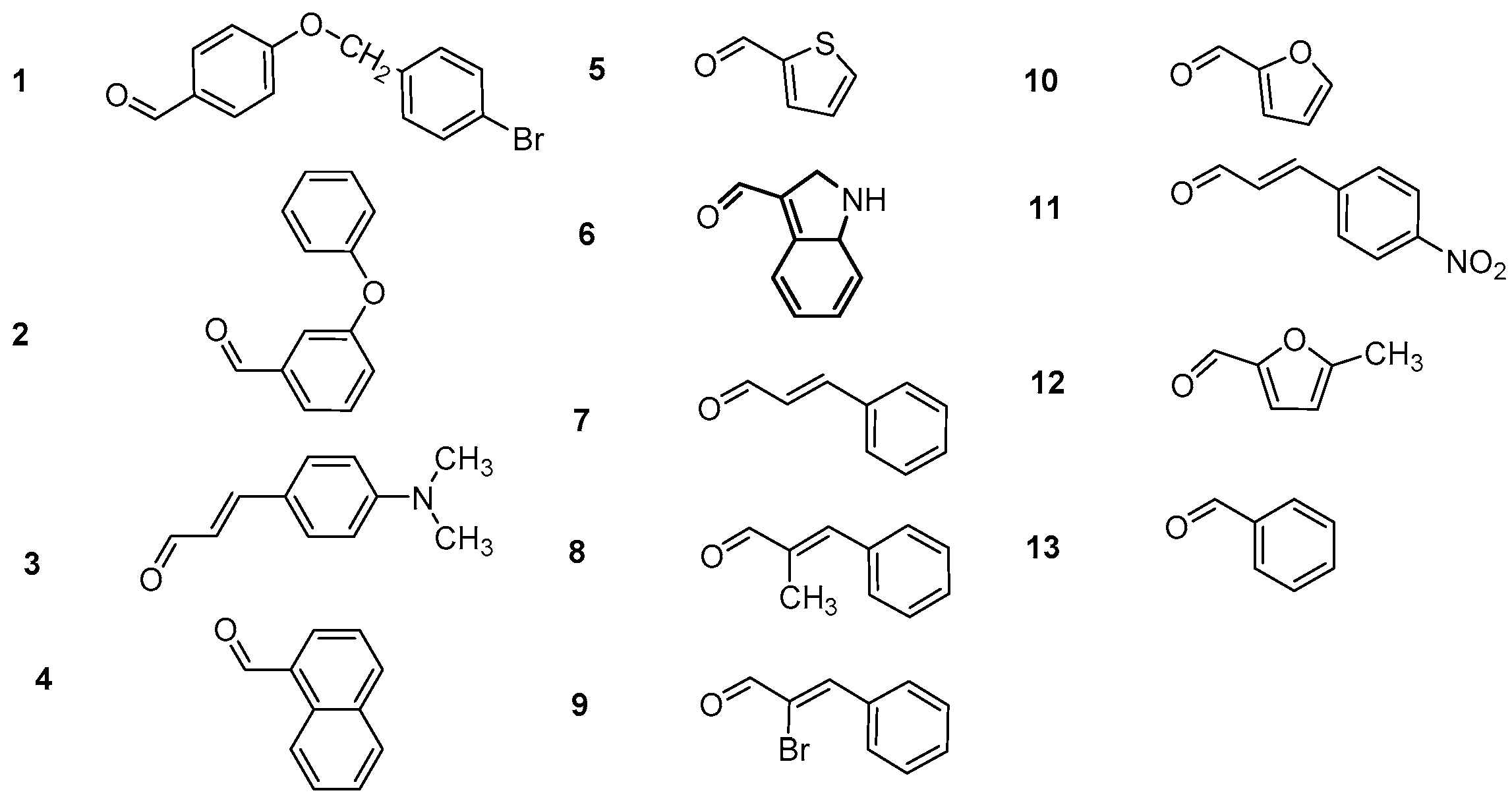
3.2.1. Synthesis of 4-Hydroxy-Chalcones (a1–13)
3.2.2. Synthesis of 2-Hydroxy-Chalcones (b1–4, b7–9, b11, b13)
3.2.3. Synthesis of bis-Ethers (di–dvii)
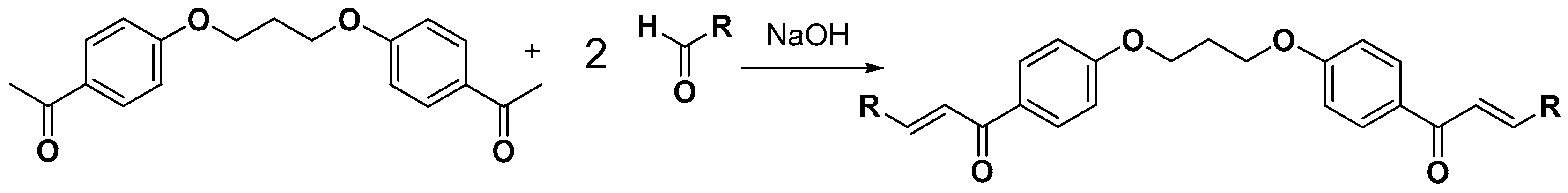
3.2.4. Synthesis of bis-Etherified Double Chalcones (c1–13)
3.2.5. Synthesis of bis-Etherified Double Chalcones of (E)-3-(4-(Dimethylamino) Phenyl-Acryl-Aldehyde (ei, eiii–evii)
3.3. Physicochemical Studies
3.3.1. Molecular Properties Prediction-Lipinski “Rule of Five”
3.3.2. Determination of RM Values
3.4. Biological In Vitro Assays
3.4.1. Determination of the Reducing Activity Using the Stable Radical 1,1-Diphenyl-Picrylhydrazyl (DPPH)
3.4.2. Inhibition of Linoleic Acid Lipid Peroxidation
3.4.3. In Vitro Lipid Peroxidation Assay
3.4.4. ABTS•+—Decolorization Assay for Antioxidant Activity
3.4.5. Soybean Lipoxygenase Inhibition Study In Vitro
3.4.6. Human h-15-LOX-1 Screening UV Assay [77]
3.4.7. Inhibition of Acetyl-Cholinesterase
3.4.8. Evaluation of the Cytotoxicity
3.4.9. Circular Dichroism Studies
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AA | Arachidonic Acid |
| AAPH | 2,2′-Azobis-(2-amidinopropane) hydrochloride |
| DPPH | 2,2-Diphenyl-1-picrylhydrazyl radical |
| LOX | Lipoxygenase |
| NDGA | Nordihydroguaiaretic acid |
| RPTLC | Reverse-phase thin layer chromatography |
| TCA | Trichloroacetic acid |
References
- Di Carlo, G.; Mascolo, N.; Izzo, A.A.; Capasso, F. Flavonoids: Old and new aspects of a class of natural therapeutic drugs. Life Sci. 1999, 65, 337–353. [Google Scholar] [CrossRef]
- Gupta, D.; Jaina, D.K.; Trivedi, P. Recent advances in chalcones as antiinfective agents. Int. J. Chem. Sci. 2010, 8, 649–654. [Google Scholar]
- Rahman, M.A. Chalcone: A valuable insight into the recent advances and potential pharmacological activities. Chem. Sci. J. 2011, 29, 1–16. [Google Scholar] [CrossRef]
- Sinha, S.; Medhi, B.; Sehga, R. Chalcones as an emerging lead molecule for antimalarial therapy: A review. J. Mod. Med. Chem. 2013, 1, 64–77. [Google Scholar]
- Yadav, V.R.; Prasad, S.; Sung, B.; Aggarwal, B.B. The role of chalcones in suppression of NF-κB-mediated inflammation and cancer. Int. Immunopharmacol. 2011, 11, 295–309. [Google Scholar] [CrossRef] [PubMed]
- Nasir, S.; Bukhari, A.; Jasamai, M.; Jantan, I. Synthesis and biological evaluation of chalcone derivatives (mini review). Med. Chem. 2012, 12, 1394–1403. [Google Scholar]
- Nowakowska, Z. A review of anti-infective and anti-inflammatory chalcones. Eur. J. Med. Chem. 2007, 42, 125–137. [Google Scholar] [CrossRef] [PubMed]
- Katsori, A.M.; Hadjipavlou-Litina, D. Recent progress in therapeutic applications of chalcones. Expert Opin. Ther. Pat. 2011, 21, 1575–1596. [Google Scholar] [CrossRef]
- Guida, A.; Lhouty, M.H.; Tichit, D.; Figueras, F.; Geneste, P. Hydrotalcites as base catalysts. Kinetics of Claisen-Schmidt condensation, intramolecular condensation of acetonylacetone and synthesis of chalcone. Appl. Catal. A 1997, 164, 251–264. [Google Scholar] [CrossRef]
- Romanelli, G.; Pasquale, G.; Sathicq, A.; Thomas, H.; Autino, J.; Vazquez, P. Synthesis of chalcones catalyzed by aminopropylated silica sol–gel under solvent-free conditions. J. Mol. Catal. A Chem. 2011, 340, 24–32. [Google Scholar] [CrossRef]
- Eddarir, S.; Cotelle, N.; Bakkour, Y.; Rolando, C. An efficient synthesis of chalcones based on the Suzuki reaction. Tetrahedron Lett. 2003, 44, 5359–5363. [Google Scholar] [CrossRef]
- Dhar, D.N. Chemistry of Chalcones and Related Compounds; John Wiley & Sons, Inc.: New York, NY, USA, 1981; p. 213. [Google Scholar]
- Pihlaja, R.; Haaparanta-Solin, M.; Rinne, O.M. The anti-inflammatory effects of lipoxygenase and cyclo-oxygenase inhibitors in inflammation-induced human fetal glia cells and the Aβ degradation capacity of human fetal astrocytes in an ex vivo assay. Front. Neurosci. 2017, 11, 299. [Google Scholar] [CrossRef] [PubMed]
- Czapski, G.A.; Czubowicz, K.; Strosznajder, J.B.; Strosznajder, R.P. The lipoxygenases: Their regulation and implication in Alzheimer’s disease. Neurochem. Res. 2016, 41, 243–257. [Google Scholar] [CrossRef] [PubMed]
- Chinnici, C.M.; Yao, Y.; Ding, T.; Funk, C.D.; Praticò, D. Absence of 12/15 lipoxygenase reduces brain oxidative stress in apolipoprotein e-deficient mice. Am. J. Pathol. 2005, 167, 1371–1377. [Google Scholar] [CrossRef]
- Piomelli, D. Eicosanoids in synaptic transmission. Crit. Rev. Neurobiol. 1994, 8, 65–83. [Google Scholar] [PubMed]
- Yao, Y.; Clark, C.M.; Trojanowski, J.Q.; Lee, V.M.; Praticò, D. Elevation of 12/15 lipoxygenase products in AD and mild cognitive impairment. Ann. Neurol. 2005, 58, 623–626. [Google Scholar] [CrossRef] [PubMed]
- Brash, A.R. Lipoxygenases: Occurrence, functions, catalysis, and acquisition of substrate. J. Biol. Chem. 1999, 274, 23679–23682. [Google Scholar] [CrossRef] [PubMed]
- Ka, M.H.; Choi, E.H.; Chun, H.S.; Lee, K.G. Antioxidative activity of volatile extracts isolated from Angelica tenuissimae roots, peppermint leaves, pine needles, and sweet flag leaves. J. Agric. Food Chem. 2005, 53, 4124–4129. [Google Scholar] [CrossRef] [PubMed]
- Molina-Jimenez, M.F.; Sanchez-Reus, M.I.; Andres, D.; Cascales, M.; Benedi, J. Neuroprotective effect of fraxetin and myricatin against rotenone-induced apoptosis in neuroblastoma cells. Brain Res. 2004, 1009, 9–16. [Google Scholar] [CrossRef]
- Lukiw, W.J.; Cui, J.G.; Marcheselli, V.L.; Bodker, M.; Botkjaer, A.; Gotlinger, K.; Serhan, C.N.; Bazan, N.G. A role for docosahexaenoic acid-derived neuroprotectin D1 in neural cell survival and Alzheimer disease. J. Clin. Investig. 2005, 115, 2774–2783. [Google Scholar] [CrossRef]
- Firuzi, O.; Zhuo, J.; Chinnici, C.M.; Wisniewski, T.; Praticò, D. 5-Lipoxygenase gene disruption reduces amyloid-β pathology in a mouse model of Alzheimer’s disease. FASEB J. 2008, 22, 1169–1178. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Li, J.; Follett, P.L.; Zhang, Y.; Cotanche, D.A.; Jensen, F.E.; Volpe, J.J.; Rosenberg, P.A. 12-Lipoxygenase plays a key role in cell death caused by glutathione depletion and arachidonic acid in rat oligodendrocytes. Eur. J. Neurosci. 2004, 20, 2049–2058. [Google Scholar] [CrossRef] [PubMed]
- Czubowicz, K.; Czapski, G.A.; Cieślik, M.; Strosznajder, R.P. Lipoxygenase inhibitors protect brain cortex macromolecules against oxidation evoked by nitrosative stress. Folia Neuropathol. 2010, 48, 283–292. [Google Scholar]
- Csermely, P.; Agoston, V.; Pongor, S. The efficiency of multi-target drugs: The network approach might help drug design. Trends Pharmacol. Sci. 2005, 26, 178–182. [Google Scholar] [CrossRef] [PubMed]
- Anighoro, A.; Bajorath, J.; Rastelli, G. Polypharmacology: Challenges and opportunities in drug discovery. J. Med. Chem. 2014, 57, 7874–7887. [Google Scholar] [CrossRef]
- Geldenhuys, W.J.; Van der Schyf, C.J. Rationally designed multitargeted agents against neurodegenerative diseases. Curr. Med. Chem. 2013, 20, 1662–1672. [Google Scholar] [CrossRef]
- Cavalli, A.; Bolognesi, M.L.; Minarini, A.; Rosini, M.; Tumiatti, V.; Recanatini, M.; Melchiorre, C. Multi-target-directed ligands to combat neurodegenerative diseases. J. Med. Chem. 2008, 51, 347–372. [Google Scholar] [CrossRef]
- Liargkova, T.; Hadjipavlou-Litina, D.J.; Koukoulitsa, C.; Voulgari, E.; Avgoustakis, C. Simple chalcones and bis-chalcones ethers as possible pleiotropic agents. J. Enzyme Inhib. Med. Chem. 2015, 31, 302–313. [Google Scholar] [CrossRef]
- Chavan, B.B.; Gadekar, A.S.; Mehta, P.P.; Vawhal, P.K.; Kolsure, A.K.; Chabukswar, A.R. Synthesis and medicinal significance of chalcones—A review. Asian J. Biomed. Pharm. Sci. 2015, 6, 1–7. [Google Scholar]
- Yazdan, S.K.; Vidya, S.D.; Afzal, B.S. Chemical and biological potentials of chalcones: A review. Org. Med. Chem. 2015, 1, 1–8. [Google Scholar] [CrossRef]
- Ng, H.L.; Ma, X.; Chew, E.H.; Chui, W.K. Design, synthesis, and biological evaluation of coupled bioactive scaffolds as potential anticancer agents for dual targeting of dihydrofolate reductase and thioredoxin reductase. J. Med. Chem. 2017, 60, 1734–1745. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.J.; Jiang, S.M.; Chen, Z.H.; Ye, B.J.; Piao, H.R. Synthesis and anti-bacterial activity of some heterocyclic chalcone derivatives bearing thiofuran, furan, and quinoline moieties. Arch. Pharm. Chem. Life Sci. 2011, 344, 689–695. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Luo, X.; Zou, Z.; Zhang, X.; Huang, F.; Li, R.; Liao, S.; Liu, Y. Synthesis and evaluation of hydroxychalcones as multifunctional non-purine xanthine oxidase inhibitors for the treatment of hyperuricemia. Bioorg. Med. Chem. Lett. 2017, 27, 3602–3606. [Google Scholar] [CrossRef] [PubMed]
- Mohd Faudzia, S.M.; Leonga, S.W.; Abas, F.; Mohd Aluwid, M.F.F.; Rullahd, K.; Lamd, K.W.; Ahmade, S.; Thamf, C.L.; Shaaria, K.; Lajis, N.H. Synthesis, biological evaluation and QSAR studies of diarylpentanoid analogues as potential nitric oxide inhibitors. MedChemComm 2015, 6, 1069–1080. [Google Scholar] [CrossRef]
- Hwang, T.L.; Yeh, S.H.; Leu, Y.L.; Chern, C.Y.; Hsu, H.C. Inhibition of superoxide anion and elastase release in human neutrophils by 30-isopropoxychalcone via a cAMP-dependent pathway. Br. J. Pharm. 2006, 148, 78–87. [Google Scholar] [CrossRef]
- Rekker, R. Hydrophobic Fragmental Constant, Its Derivation and Application: A Means of Characterizing Membrane Systems; Elsevier Eds. Scientific Co.: New York, NY, USA, 1977. [Google Scholar]
- Leśniewska, M.A.; Gdaniec, Z.; Muszalska, I. Calculation procedures and HPLC method for analysis of the lipophilicity of acyclovir esters. Drug Dev. Ind. Pharm. 2015, 41, 663–669. [Google Scholar] [CrossRef]
- Główka, F.; Romański, M.; Siemiątkowska, A. Determination of partition coefficients n-octanol/water for treosulfan and its epoxy-transformers: An example of a negative correlation between lipophilicity of unionized compounds and their retention in reversed-phase chromatography. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2013, 923, 92–97. [Google Scholar] [CrossRef]
- Bakht, M.A.; Alajm, M.F.; Alam, P.; Alam, A.; Alam, P.; Ajarba, T.M. Theoretical and experimental study on lipophilicity and wound healing activity of ginger compounds. Asian Pac. J. Trop. Biomed. 2014, 4, 329–333. [Google Scholar] [CrossRef]
- BioByte. Available online: www.biobyte.com (accessed on 7 November 2018).
- Sakuratani, Y.; Kasai, K.; Noguchi, Y.; Yamada, J. Comparison of predictivities of log P calculation models based on experimental data for 134 simple organic compounds. QSAR Comb. Sci. 2006, 26, 109–116. [Google Scholar] [CrossRef]
- Canavan, N. FDA and drug companies alike want ADME-tox testing performed earlier and earlier in a drug’s life cycle. Drug Discov. Dev. 2007, 10, 34–36. [Google Scholar]
- Molinspiration Cheminformatics. Available online: www.molinspiration.com (accessed on 20 April 2018).
- Lipinski, C.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Rishton, G.M.; LaBonte, K.; Williams, A.J.; Kassam, K.; Kolovanov, E. Computational approaches to the prediction of bloodbrain barrier permeability: A comparative analysis of central nervous system drugs versus secretase inhibitors for Alzheimer’s disease. Curr. Opin. Drug. Discov. Devel. 2006, 9, 303–313. [Google Scholar] [PubMed]
- Crooks, S.W.; Stockley, R.A. Leukotriene B4. Int. J. Biochem. Cell Biol. 1998, 30, 173–178. [Google Scholar] [CrossRef]
- Chu, J.; Praticò, D. 5-Lipoxygenase as an endogenous modulator of amyloid beta formation in vivo. Ann. Neurol. 2011, 69, 34–46. [Google Scholar] [CrossRef] [PubMed]
- Praticò, D. Oxidative stress hypothesis in Alzheimer’s disease: A reappraisal. Trends Pharmacol. Sci. 2008, 29, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Pratico, D.; Zhukareva, V.; Yao, Y.; Uryu, K.; Funk, C.D.; Lawson, J.A.; Trojanowski, J.Q.; Lee, V.M. 12/15 lipoxygenase is increased in Alzheimer’s disease. Possible involvement in Brain oxidative stress. Am. J. Pathol. 2004, 164, 1655–1662. [Google Scholar] [CrossRef]
- Chu, J.; Li, J.G.; Ceballos-Diaz, C.; Golde, T.; Praticò, D. The influence of 5-lipoxygenase on Alzheimer’s disease-related tau pathology: In vivo and in vitro evidence. Biol. Psychiatry 2013, 74, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Skrzypczak-Jankun, E.; Zhou, K.; Jankun, J. Inhibition of lipoxygenase by (−)- epigallocatechin gallate: X-ray analysis at 2.1 A reveals degradation of EGCG and shows soybean LOX-3 complex with EGC instead. Int. J. Mol. Med. 2003, 12, 415–420. [Google Scholar] [CrossRef]
- Skrzypczak-Jankun, E.; Chorostowska-Wynimko, J.; Selman, S.H.; Jankun, J. Lipoxygenases—A challenging problem in enzyme inhibition and drug development. Curr. Enzyme Inhib. 2007, 3, 119–132. [Google Scholar] [CrossRef]
- Gleason, M.M.; Rojas, C.J.; Learn, K.S.; Perrone, M.; Bilder, G.E. Characterization and inhibition of 15-lipoxygenase in human monocytes–Comparison with soybean 15-lipoxygenase. Am. J. Physiol. 1995, 258, 1301–1307. [Google Scholar] [CrossRef]
- Lapenna, D.; Ciofani, G.; Pierdomenico, S.D.; Giamberardino, M.A.; Cuccurullo, F. Dihydrolipoic acid inhibits 15-lipoxygenase-dependent lipid peroxidation. Free Rad. Biol. Med. 2003, 35, 1203–1209. [Google Scholar] [CrossRef]
- Nuhn, P.; Büge, A.; Köhler, T.; Lettau, H.; Schneider, R. Trends bei der entwicklung von lipoxygenase-hemmern. Pharmazie 1991, 46, 81–88. [Google Scholar] [PubMed]
- Maccarrone, M.; van Aarie, P.G.M.; Veldink, G.A.; Vliegenthart, J.F.G. In vitro oxygenation of soybean biomembranes by lipoxygenase-2. Biochim. Biophys. Acta 1994, 1190, 164–169. [Google Scholar] [CrossRef]
- Pontiki, E.; Hadjipavlou-Litina, D. Lipoxygenase inhibitors: A comparative QSAR study review and evaluation of new QSARs. Med. Res. Rev. 2008, 28, 39–117. [Google Scholar] [CrossRef] [PubMed]
- Steinhilber, D.; Hofmann, B. Recent advances in the search for novel 5-lipoxygenase inhibitors. Basic Clin. Pharmacol. Toxicol. 2014, 114, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Sendobry, S.M.; Cornicelli, J.A.; Welch, K.; Bocan, T.; Tait, B.; Trivedi, B.K.; Colbry, N.; Dyer, R.D.; Feinmark, S.J.; Daugherty, A. Attenuation of diet-induced atherosclerosis in rabbits with a highly selective 15-lipoxygenase inhibitor lacking significant antioxidant properties. Br. J. Pharmacol. 1997, 120, 1199–1206. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.E.; Cho, J.K.; Marcus, J.; Long, C.; Ryu, H.W.; Kim, J.H.; Kim, H.J.; Yuk, H.J.; Kim, D.W.; Hun, K. Inhibitory evaluation of sulfonamide chalcones on β-secretase and acylcholinesterase. Park Mol. 2013, 18, 140–153. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P.; LeVineIII, H. Alzheimer’s disease and the β-amyloid peptide. J. Alzheimers Dis. 2010, 19, 311. [Google Scholar] [CrossRef] [PubMed]
- Barten, D.M.; Albright, C.F. Therapeutic strategies for Alzheimer’s disease. Mol. Neurobiol. 2008, 37, 171–186. [Google Scholar] [CrossRef]
- Fezoui, Y.; Teplow, D. Kinetic studies of amyloid β-protein fibril assembly. J. Biol. Chem. 2002, 277, 36948–36954. [Google Scholar] [CrossRef]
- Habchi, J.; Chia, S.; Limbocker, R.; Mannini, B.; Ahn, M.; Perni, M.; Hansson, O.; Arosio, P.; Kumita, J.R.; Challa, P.K.; et al. Systematic development of small molecules to inhibit specific microscopic steps of Aβ42 aggregation in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2017, 114, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Matis, I.; Delivoria, D.C.; Mavroidi, B.; Papaevgeniou, N.; Panoutsou, S.; Bellou, S.; Papavasileiou, K.D.; Linardaki, Z.I.; Stavropoulou, A.V.; Vekrellis, K.; et al. An integrated bacterial system for the discovery of chemical rescuers of disease-associated protein misfolding. Nat. Biomed. Eng. 2017, 1, 838–852. [Google Scholar] [CrossRef]
- Mete, E.; Comez, B.; Gul, H.I.; Gulcin, I.; Supuran, C.T. Synthesis and carbonic anhydrase inhibitory activities of new thienyl-substituted pyrazoline benzenesulfonamides. J. Enzyme Inhib. Med. Chem. 2016, 31, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Dimmock, J.R.; Kandepu, N.M.; Hetherington, M.; Quail, J.W.; Pugazhenthi, U.; Sudom, A.M.; Chamankhah, M.; Rose, P.; Pass, E.; Allen, T.M.; et al. Cytotoxic activities of mannich bases of chalcones and related compounds. J. Med. Chem. 1998, 41, 1014–1026. [Google Scholar] [CrossRef] [PubMed]
- Cassidy, J.E.; Witcher, W.J. The polarographic characteristics of furfurylideneacetophenone and some of its para derivatives. J. Phys. Chem. 1959, 63, 1824–1826. [Google Scholar] [CrossRef]
- Amin, A.N.; El-Khouly, M.E.; Subbaiyan, N.K.; Zandler, M.E.; Supur, M.; Fukuzumi, S.; D’Souza, F. Syntheses, electrochemistry, and photodynamics of ferrocene_azadipyrromethane donor_acceptor dyads and triads. J. Phys. Chem. 2011, 115, 9810–9819. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, C.; Tharmaraj, P. Cobalt(II), nickel(II) and copper(II) complexes of 1-(2-hydroxyphenyl)-3-(1-naphthalenyl)-2-propen-1-one and 1-(1-hydroxy-2-naphthalenyl)-3 (1-naphthalenyl)-2-propen-1-one. Syn. React. Inorg. Metalorg. Chem. 2006, 20, 151–166. [Google Scholar] [CrossRef]
- Silva, A.M.S.; Pinto, D.C.G.A.; Travares, H.R.; Cavaleiro, J.A.S.; Jimeno, M.L.; Elguero, J. Novel (E)- and (Z)-2-styrylchromones from (E,E)-2′-hydroxycinnamylideneacetophenones—xanthones from daylight photooxidative cyclization of (E)-2-styrylchromones. Eur. J. Org. Chem. 1998, 9, 2031–2038. [Google Scholar] [CrossRef]
- Hofmann, E.; Webster, J.; Do, T.; Kline, R.; Snider, L.; Hauser, Q.; Higginbottom, G.; Campbell, A.; Ma, L.; Paula, S. Hydroxylated chalcones with dual properties: Xanthine oxidase inhibitors and radical scavengers. Bioorg. Med. Chem. 2016, 24, 578–587. [Google Scholar]
- Sodani, R.S.; Choudhary, P.C.; Sharma, H.O.; Verma, B.L. Syntheses and reactions of 4′-[(ω-bromoalkyl) oxy]-and 4′, 4′′′-(polymethylenedoxy)-bis substituted chalcones. J. Chem. 2010, 7, 763–769. [Google Scholar] [CrossRef]
- Prabhakar, K.R.; Veerapur, V.P.; Bansal, P.; Vipan, P.K.P.; Reddy, K.M.; Barik, A.; Reddy, B.K.D.; Reddanna, P.; Priyadarsinib, K.I.; Unnikrishnana, M.K. Identification and evaluation of antioxidant, analgesic/anti-inflammatory activity of the most active ninhydrin–phenol adducts synthesized. Bioorg. Med. Chem. 2006, 14, 7113–7120. [Google Scholar] [CrossRef] [PubMed]
- Pontiki, E.; Hadjipavlou-Litina, D.; Litinas, K.; Nicolotti, O.; Carotti, A. Design, synthesis and pharmacobiological evaluation of novel acrylic acid derivatives acting as lipoxygenase and cyclooxygenase-1 inhibitors with antioxidant and anti-inflammatory activities. Eur. J. Med. Chem. 2011, 46, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Eleftheriadis, N.; Thee, S.; Biesebeek, J.Y.; Van Der Wouden, P.; Baas, B.J.; Dekker, F.J. Identification of 6-benzyloxysalicylates as a novel class of inhibitors of 15-lipoxygenase-1. Eur. J. Med. Chem. 2015, 94, 265–275. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the synthesized compounds are available from the authors. |









{kind=link}
{kind=link}
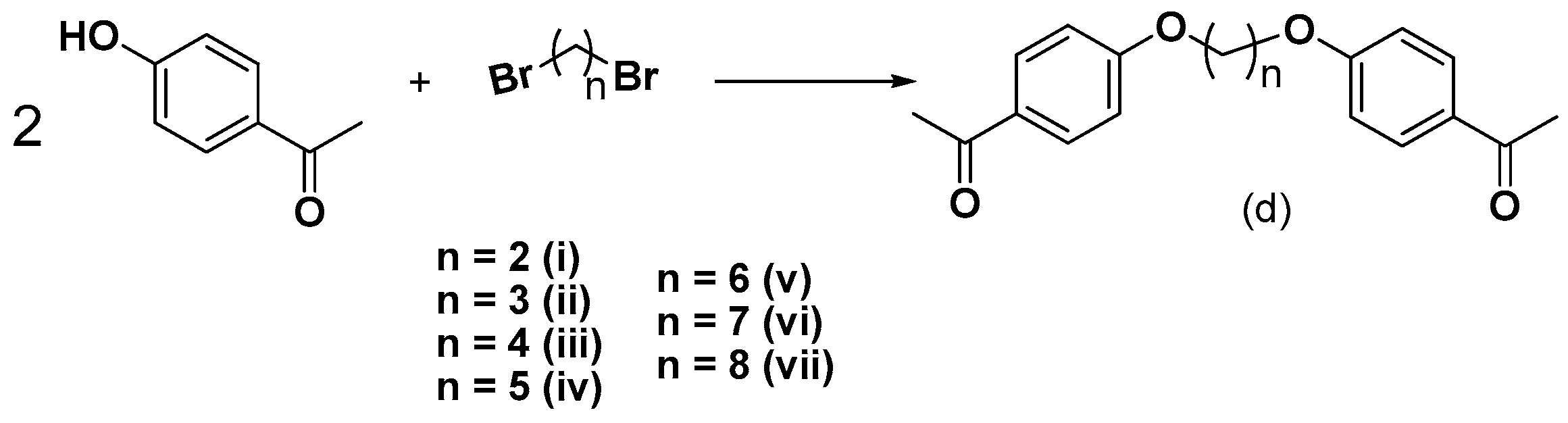{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | mi Log P a | TPSA b | n Atoms | n c O, N | n d OH, NH | n Violations | Nrotb e | mol. Wt f | Volume g | h LogBB |
|---|---|---|---|---|---|---|---|---|---|---|
| a1 * | 5.79 | 46.53 | 26 | 3 | 1 | 1 | 6 | 409.28 | 324.95 | 0.55275 |
| a2 * | 5.06 | 46.53 | 24 | 3 | 1 | 1 | 5 | 316.36 | 290.26 | 0.483 |
| a3 * | 3.95 | 40.54 | 22 | 3 | 1 | 0 | 5 | 293.37 | 283.19 | 0.3135 |
| a4 * | 4.31 | 37.3 | 21 | 2 | 1 | 0 | 3 | 274.32 | 253.86 | 0.43115 |
| a5 | 3.05 | 37.3 | 16 | 2 | 1 | 0 | 3 | 230.04 | 200.58 | 0.1940 |
| a6 | 2.43 | 49.33 | 20 | 3 | 2 | 0 | 3 | 265.31 | 245.06 | −0.03325 |
| a7 | 3.85 | 37.3 | 19 | 2 | 1 | 0 | 4 | 250.3 | 237.29 | 0.31955 |
| a8 | 4.4 | 37.3 | 20 | 2 | 1 | 0 | 4 | 264.32 | 253.85 | 0.38155 |
| a9 | 4.32 | 37.3 | 20 | 2 | 1 | 0 | 4 | 329.19 | 255.17 | 0.3955 |
| a10 | 2.41 | 50.44 | 16 | 3 | 1 | 0 | 3 | 214.22 | 191.44 | −0.01035 |
| a11 | 3.81 | 83.12 | 22 | 5 | 1 | 0 | 5 | 295.29 | 260.62 | −0.17895 |
| a12 | 3.32 | 33.37 | 15 | 2 | 1 | 0 | 2 | 200.24 | 189.02 | 0.23795 |
| a13 | 3.33 | 37.3 | 17 | 2 | 1 | 0 | 3 | 224.26 | 209.87 | 0.2498 |
| b1 | 6.21 | 46.53 | 26 | 3 | 1 | 1 | 6 | 409.28 | 324.95 | 0.62405 |
| b2 | 5.48 | 46.53 | 24 | 3 | 1 | 1 | 5 | 316.36 | 290.26 | 0.5543 |
| b3 | 4.37 | 40.54 | 22 | 3 | 1 | 0 | 5 | 293.37 | 283.19 | 0.3848 |
| b4 | 4.73 | 37.3 | 21 | 2 | 1 | 0 | 3 | 274.32 | 253.86 | 0.50245 |
| b7 | 4.27 | 37.3 | 19 | 2 | 1 | 0 | 4 | 250.3 | 237.29 | 0.39085 |
| b8 | 4.82 | 37.3 | 20 | 2 | 1 | 0 | 4 | 264.32 | 253.85 | 0.45285 |
| b9 | 4.74 | 37.3 | 20 | 2 | 1 | 0 | 4 | 239.19 | 255.17 | 0.4668 |
| b11 | 4.23 | 83.12 | 22 | 5 | 1 | 0 | 5 | 295.29 | 260.62 | −0.10765 |
| b13 | 3.75 | 37.3 | 17 | 2 | 1 | 0 | 3 | 224.26 | 209.87 | 0.3211 |
| di | 3.47 | 52.61 | 22 | 4 | 0 | 0 | 7 | 298.34 | 278.11 | 0.15095 |
| Dii * | 3.74 | 52.61 | 23 | 4 | 0 | 0 | 8 | 312.37 | 294.92 | 0.2083 |
| diii | 4.01 | 52.61 | 24 | 4 | 0 | 0 | 9 | 326.39 | 311.72 | 0.25325 |
| div | 4.52 | 52.61 | 25 | 4 | 0 | 0 | 10 | 340.42 | 328.52 | 0.33385 |
| dv | 5.03 | 52.61 | 26 | 4 | 0 | 1 | 11 | 354.45 | 345.32 | 0.416 |
| dvi | 5.53 | 52.61 | 27 | 4 | 0 | 1 | 12 | 368.74 | 362.12 | 0.50435 |
| dvii | 6.04 | 52.61 | 28 | 4 | 0 | 1 | 13 | 382.5 | 378.93 | 0.5803 |
| c1 * | 9.72 | 71.08 | 55 | 6 | 0 | 2 | 18 | 858.62 | 689.61 | 1.2791 |
| c2 * | 9.42 | 71.08 | 51 | 6 | 0 | 2 | 16 | 672.78 | 620.23 | 1.1396 |
| c3 * | 8.66 | 59.09 | 47 | 6 | 0 | 2 | 16 | 626.8 | 606.09 | 0.8007 |
| c4 * | 8.98 | 52.61 | 45 | 4 | 0 | 2 | 12 | 588.7 | 547.43 | 1.03755 |
| c5 | 7.13 | 52.61 | 35 | 4 | 0 | 2 | 12 | 500.64 | 440.87 | 0.5648 |
| c6 | 5.82 | 76.66 | 43 | 6 | 2 | 2 | 12 | 570.69 | 529.83 | 0.2561 |
| c7 | 8.55 | 52.61 | 41 | 4 | 0 | 2 | 14 | 540.66 | 514.28 | 0.81435 |
| c8 | 9.04 | 52.61 | 43 | 4 | 0 | 2 | 14 | 568.71 | 547.4 | 0.93835 |
| c9 | 8.98 | 52.61 | 43 | 4 | 0 | 2 | 14 | 698.45 | 550.05 | 0.9678 |
| c10 | 5.85 | 78.89 | 35 | 6 | 0 | 1 | 12 | 468.5 | 422.58 | 0.1563 |
| c11 | 8.5 | 144.26 | 47 | 10 | 0 | 2 | 16 | 630.65 | 560.95 | -0.1812 |
| c12 | 6.29 | 78.89 | 37 | 6 | 0 | 1 | 12 | 496.56 | 455.7 | 0.3113 |
| c13 | 7.69 | 52.61 | 37 | 4 | 0 | 1 | 12 | 488.58 | 459.44 | 0.67485 |
| ei | 8.51 | 59.09 | 46 | 6 | 0 | 2 | 15 | 612.77 | 589.29 | 0.74335 |
| eiii | 8.79 | 59.09 | 48 | 6 | 0 | 2 | 17 | 640.82 | 622.89 | 0.93565 |
| eiv | 9 | 59.09 | 49 | 6 | 0 | 2 | 18 | 654.85 | 639.69 | 1.0178 |
| ev | 9.17 | 59.09 | 50 | 6 | 0 | 2 | 19 | 668.88 | 656.5 | 1.00995 |
| evi | 9.31 | 59.09 | 51 | 6 | 0 | 2 | 20 | 682.9 | 673.3 | 1.0921 |
| evii | 9.44 | 59.09 | 52 | 6 | 0 | 2 | 21 | 696.93 | 690.1 | 1.17425 |
| tacrine | 3.05 | 38.91 | 15 | 2 | 2 | 0 | 0 | 198.27 | 191.53 | 1.053785 |
| NDGA | 3.48 | 80.91 | 22 | 4 | 4 | 0 | 5 | 302.37 | 287.90 | 1.6613 |
| Compound | RA% 100 μM 20 min | RA% 100 μM 60 min | ABTS•+ Inhb. % @ 100 μM | AAPH% @ 100 μM | % Inhb. of Liposomes LP @ 100 μM |
|---|---|---|---|---|---|
| a1 * | 1 | 6 | No | * | nt |
| a2 * | 3 | 4 | No | * | nt |
| a3 * | 18 | 17 | 12 | * | nt |
| a4 * | 7 | 3 | No | * | nt |
| a5 | 17 | 3 | No | 23 | nt |
| a6 | 15 | 2 | No | 45 | nt |
| a7 | 19 | 6 | 12 | 31 | nt |
| a8 | 6 | 2 | No | 32 | nt |
| a9 | 10 | 1 | No | 33 | nt |
| a10 | 23 | 10 | 2 | 24 | nt |
| a11 | 7 | 3 | 14 | 55 | nt |
| a12 | 24 | 9 | 6 | 45 | nt |
| a13 | 7 | 3 | No | 38 | nt |
| b1 | 46 | 0 | 40 | 89 | nt |
| b2 | 45 | 15 | No | 44 | nt |
| b3 | 72 | 15 | 96 | 100 | nt |
| b4 | 30 | 20 | No | 82 | nt |
| b7 | 13 | 0 | 5 | 29 | nt |
| b8 | 17 | 3 | 12 | 25 | nt |
| b9 | 20 | 8 | No | 17 | nt |
| b11 | 4 | 2 | No | 31 | nt |
| b13 | 10 | 0 | No | 15 | nt |
| di | No | 22 | nt | ||
| Dii * | No | no | No | 20 | nt |
| diii | No | 29 | nt | ||
| div | No | 30 | nt | ||
| dv | No | 18 | nt | ||
| dvi | No | 12 | nt | ||
| dvii | No | 27 | nt | ||
| c1 * | 36 | 55 | no | 42 | nt |
| c2 * | No | 8 | no | 22 | 2 |
| c3 * | 17 | 14 | 99 | 70 | 81 |
| c4 * | 19 | 50 | no | 95 | 3 |
| c5 | No | no | No | 46 | nt |
| c6 | No | No | No | 100 | nt |
| c7 | No | no | No | 82 | nt |
| c8 | No | no | No | 75 | nt |
| c9 | No | no | No | 32 | nt |
| c10 | No | no | 11 | 77 | nt |
| c11 | No | no | No | 68 | nt |
| c12 | No | no | 17 | 68 | 74 |
| c13 | No | no | No | 71 | nt |
| ei | No | no | 98 | 71 | 4 |
| eiii | No | no | No | 47 | nt |
| eiv | No | no | 73 | 72 | 77 |
| ev | No | no | 25 | 14 | nt |
| evi | No | no | 79 | 64 | 64 |
| evii | No | no | 88 | 6 | nt |
| Trolox | Nt | nt | 96 | 93 | 69 |
| Tacrine | Nt | nt | 88 | nt | nt |
| NDGA | 81 | 83 | nt | nt | nt |
| Compound | % LOX Inhb. @ 100 μM/IC50 μM | % AChE Inhb. @ 100 μM/IC50 μM | RM ± (SD) ¥ | cLogP |
|---|---|---|---|---|
| a1 * | 14 | 7 | 0.34 | 5.51 |
| a2 * | 10 | 26 | 0.36 | 5.06 |
| a3 * | 56/100 μM | 50/100 μM | −0.69 | 3.58 |
| a4 * | 44 | 25 | −0.26 | 4.13 |
| a5 | 9 | 38 | −0.62 | 2.60 |
| a6 | 60 | Νο | −0.65 | 1.91 |
| a7 | 4 | 63/87 μM | −0.64 | 3.41 |
| a8 | 14 | 57/100 μM | −0.68 | 3.81 |
| a9 | 16 | 48 | −0.82 | 3.90 |
| a10 | 23 | 27 | −0.06 | 2.13 |
| a11 | 36 | 56/100 μM | −1.09 | 3.15 |
| a12 | 2 | 42 | −0.81 | 2.63 |
| a13 | 23 | 25 | −0.73 | 2.96 |
| b1 | 89/56 μM | 89/100 μM | −0.91 | 5.97 |
| b2 | 44 | 44 | 0.04 | 5.52 |
| b3 | 100/57 μM | 100 # | −0.66 | 4.04 |
| b4 | 82/65 μM | 82 # | −0.03 | 4.59 |
| b7 | 29 | 29 | −0.85 | 3.87 |
| b8 | 25 | 25 | −0.79 | 4.27 |
| b9 | 16 | 16 | −0.37 | 4.36 |
| b11 | 31 | 31 | −0.91 | 3.61 |
| b13 | 14 | 14 | 0.43 | 3.42 |
| di | 12 | 22 | 0.67 | 3.31 |
| Dii * | no | 20 | −0.23 | 3.68 |
| diii | 41 | 29 | −0.37 | 3.97 |
| div | 17 | 29 | 0.23 | 4.49 |
| dv | 7 | 18 | −0.53 | 5.02 |
| dvi | 17 | 12 | 0.32 | 5.59 |
| dvii | 5 | 27 | −0.36 | 6.08 |
| c1 * | 41 | 23 | −0.68 | 10.88 |
| c2 * | 100/55 μM | 71/49 μM | −0.89 | 7.92 |
| c3 * | 93/56 μM | 95/52 μM | 0.77 | 9.03 |
| c4 * | 100/55 μM | 58/100 μM | 0.43 | 5.98 |
| c5 | 32 | 51/100 μM | 0.43 | 5.98 |
| c6 | 98/54 μM | Νο | −0.97 | 5.54 |
| c7 | 41 | 94/56 μM | −0.77 | 7.59 |
| c8 | 26.5 | 100/58 μM | −0.85 | 8.39 |
| c9 | 24.5 | 85/74 μM | −0.86 | 8.58 |
| c10 | 57/85 μM | 69/76 μM | 0.7 | 5.04 |
| c11 | 95/50 μM | 100/52 μM | −0.88 | 7.08 |
| c12 | 52/96 μM | 100/48 μM | −0.96 | 6.04 |
| c13 | 76/65 μM | 50.5/100 μM | 0.34 | 6.69 |
| ei | 77/62.5 μM | 71/57.5 μM | 0.56 | 7.55 |
| eiii | 27 | 47 | −0.07 | 8.21 |
| eiv | 23 | 72/76 μM | 0.13 | 8.74 |
| ev | 7 | 14 | Nd | 9.27 |
| evi | 65/76 μM | 64/62 μM | Nd | 9.79 |
| evii | 12 | 6/85 μM | Nd | 10.4 |
| Tacrine | 98/0.03 μM | |||
| NDGA | 93/0.5 μM |
| Compound | 1 μM Average (% pi) ± σ | 10 μM Average (% pi) ± σ | 20 μM Average (% pi) ± σ | 50 μM Average (% pi) ± σ | 100 μM Average (% pi) ± σ |
|---|---|---|---|---|---|
| a1 * | 1 ± 0.6 | 5 ± 2.8 | 5.5 ± 0.71 | 32 ± 4.9 | 32 ± 4.9 |
| a2 * | 1 ± 0.5 | 3.5 ± 0.7 | 27.5 ± 8.9 | 63 ± 11.2 | 63 ± 11.2 |
| a3 * | 14 ± 3.5 | 15 ± 0.8 | 19 ± 1.4 | 14 ± 0.7 | 14 ± 0.7 |
| a4 * | 8.5 ± 2.1 | 12 ± 4.2 | 29.5 ± 0.6 | 51 ± 9.9 | 51 ± 9.9 |
| a5 | 11.36 ± 0.62 | Νο ± No | 28.32 ± 2.91 | 36.15 ± 2.33 | 36.15 ± 2.33 |
| a6 | 1.71 ± 2.08 | 1.66 ± 0.59 | 3.25 ± 2.6 | 6.67 ± 1.35 | 6.67 ± 1.35 |
| a7 | 6.85 ± 3.12 | 9.27 ± 1.9 | 9.24 ± 6.49 | 60.95 ± 0.01 | 60.95 ± 0.01 |
| a8 | 1.39 ± 0.1 | 2.95 ± 1.23 | 1.84 ± 2.05 | 8.15 ± 0.34 | 8.15 ± 0.34 |
| a9 | 0.55 ± 0.09 | 0.59 ± 0.83 | 2.7 ± 0.68 | 7.59 ± 0.13 | 7.59 ± 0.13 |
| a10 | 3.03 ± 0.88 | 2.25 ± 0.38 | 2.39 ± 1.08 | 17.15 ± 0.32 | 17.15 ± 0.32 |
| a11 | 3.13 ± σ 2.43 | 6.26 ± 3 | 18.81 ± 2.57 | 67.84 ± 6.45 | 67.84 ± 6.45 |
| a12 | 0.88 ± 0.31 | 3.25± 2.11 | 8.12 ± 0.32 | 63.54 ± 6.16 | 63.54 ± 6.16 |
| di | 4.94 ± 1.02 | 9.05 ± 0.58 | 16.78 ± 5.61 | 33.22 ± 9.65 | 33.22 ± 9.65 |
| dii * | 9 ± 4.5 | 8.5 ± 4.24 | 13 ± 4.5 | 42 ± 3.5 | 42 ± 3.5 |
| diii | 14.06 ± 1.36 | 20 ± 3.12 | 20.76 ± 3.51 | 32.14 ± 6.99 | 32.14 ± 6.99 |
| div | 4.89 ± 0.51 | 8.56 ± 3.97 | 11.86 ± 2.98 | 17.05 ± 1.46 | 17.05 ±1.46 |
| dv | 4.47 ± 0.59 | 6.83 ± 2.46 | 8.05 ± 3.58 | 9.69 ± 0.57 | 9.69 ± 0.57 |
| dvi | 0.37 ± 0.52 | 1.92 ± 1.02 | 1.79 ± 1.53 | 2.55 ± 0.5 | 2.55 ± 0.5 |
| c1 * | 38.5 ± 6.9 | 49.5 ± 7.8 | 53.5 ± 0.71 | 74 ± 5.6 | 74 ± 5.6 |
| c2 * | 17 ± 1.4 | 29 ± 4.2 | 43 ± 2.8 | 55 ± 4.2 | 55 ± 4.2 |
| c3 * | 81 ± 0.8 | 90 ± 1 | 100 ± 0 | 100 ± 0 | 100 ± 0 |
| c4 * | 29.5 ± 3.5 | 42 ± 4.6 | 46 ± 5.6 | 67 ± 9.2 | 67 ± 9.2 |
| c5 | 0.32 ± 0.45 | 0.97 ± 0.2 | 3.02 ± 1.36 | 7.26 ± 0.3 | 7.26 ± 0.3 |
| c6 | 1.12 ± 1.15 | 2.2 ± 1.66 | 1.16 ± 0.81 | 3.03 ± 0.02 | 3.03 ± 0.02 |
| c7 | 22.5 ± 11.64 | 28.73 ± 11.78 | 25.4 ± 8.42 | 28.84 ± 5.62 | 28.84 ± 5.62 |
| c8 | 35.72 ± 10.23 | 29.05 ± 9.62 | 35.69 ± 5.85 | 38.7 ± 4.96 | 38.7 ± 4.96 |
| c9 | 12.5 ± 10.31 | 15.88 ± 6.31 | 28.76 ± 2.74 | 29.53 ± 6.87 | 29.53 ± 6.87 |
| c10 | 18.10 ± 11.82 | 13.48 ± 10.36 | 25.93 ± 9.73 | 32.11 ± 9.38 | 32.11 ± 9.38 |
| c11 | 6.4 ± 0.4 | 19.07 ± 7.23 | 34.63 ± 0.31 | 64.53 ± 3.07 | 64.53 ± 3.07 |
| c12 | 13.22 ± 0.64 | 31.31 ± 4.84 | 53.79 ± 16.95 | 84.03 ± 2.25 | 84.03 ± 2.25 |
| Compound | % Soybean LOX Inhb. @ 100 μM/IC50 μM | % AChE Inhb. @ 100 μM/IC50 μM | h-15-LOX-1 Inhb. IC50 μM | AAPH% @ 100 μM | % Inhb. of Liposomes LP @ 100 μM |
|---|---|---|---|---|---|
| b3 | 100/57 μM | 100 # | |||
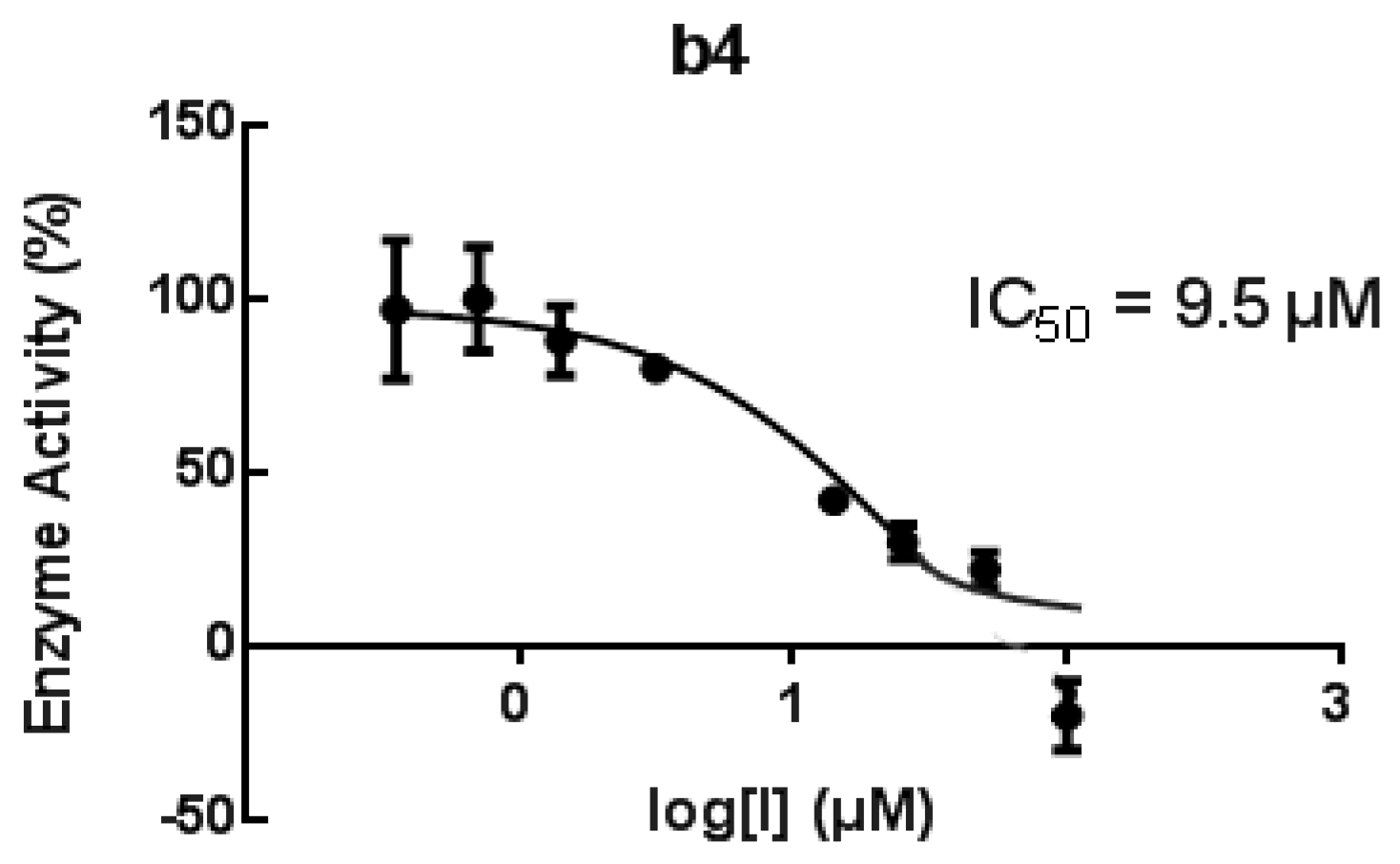
| b4 | 82/65 μM | 82 # | 9.5 μM | 82 | |
| c2 * | 100/55 μM | 71/49 μM | |||
| c3 * | 93/56 μM | 95/52 μM | 70 | 81 | |
| c4 * | 100/55 μM | 58/100 μM | 95 | ||
| c11 | 95/50 μM | 100/52 μM | (2nd potent at 50 μM) | 68 | |
| c12 | 52/96 μM | 100/48 μM | 68 | 74 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liargkova, T.; Eleftheriadis, N.; Dekker, F.; Voulgari, E.; Avgoustakis, C.; Sagnou, M.; Mavroidi, B.; Pelecanou, M.; Hadjipavlou-Litina, D. Small Multitarget Molecules Incorporating the Enone Moiety. Molecules 2019, 24, 199. https://doi.org/10.3390/molecules24010199
Liargkova T, Eleftheriadis N, Dekker F, Voulgari E, Avgoustakis C, Sagnou M, Mavroidi B, Pelecanou M, Hadjipavlou-Litina D. Small Multitarget Molecules Incorporating the Enone Moiety. Molecules. 2019; 24(1):199. https://doi.org/10.3390/molecules24010199
Chicago/Turabian StyleLiargkova, Thalia, Nikolaos Eleftheriadis, Frank Dekker, Efstathia Voulgari, Constantinos Avgoustakis, Marina Sagnou, Barbara Mavroidi, Maria Pelecanou, and Dimitra Hadjipavlou-Litina. 2019. "Small Multitarget Molecules Incorporating the Enone Moiety" Molecules 24, no. 1: 199. https://doi.org/10.3390/molecules24010199
APA StyleLiargkova, T., Eleftheriadis, N., Dekker, F., Voulgari, E., Avgoustakis, C., Sagnou, M., Mavroidi, B., Pelecanou, M., & Hadjipavlou-Litina, D. (2019). Small Multitarget Molecules Incorporating the Enone Moiety. Molecules, 24(1), 199. https://doi.org/10.3390/molecules24010199