Amino Acids in the Development of Prodrugs

 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
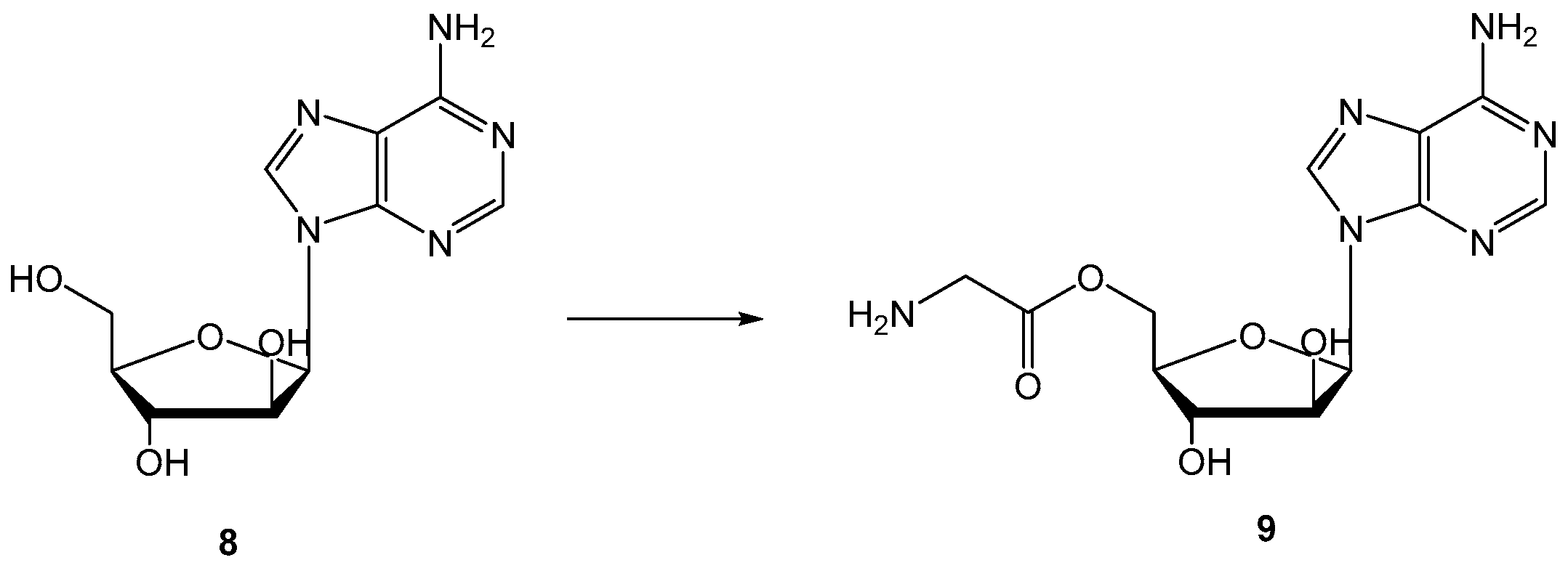{kind=link}
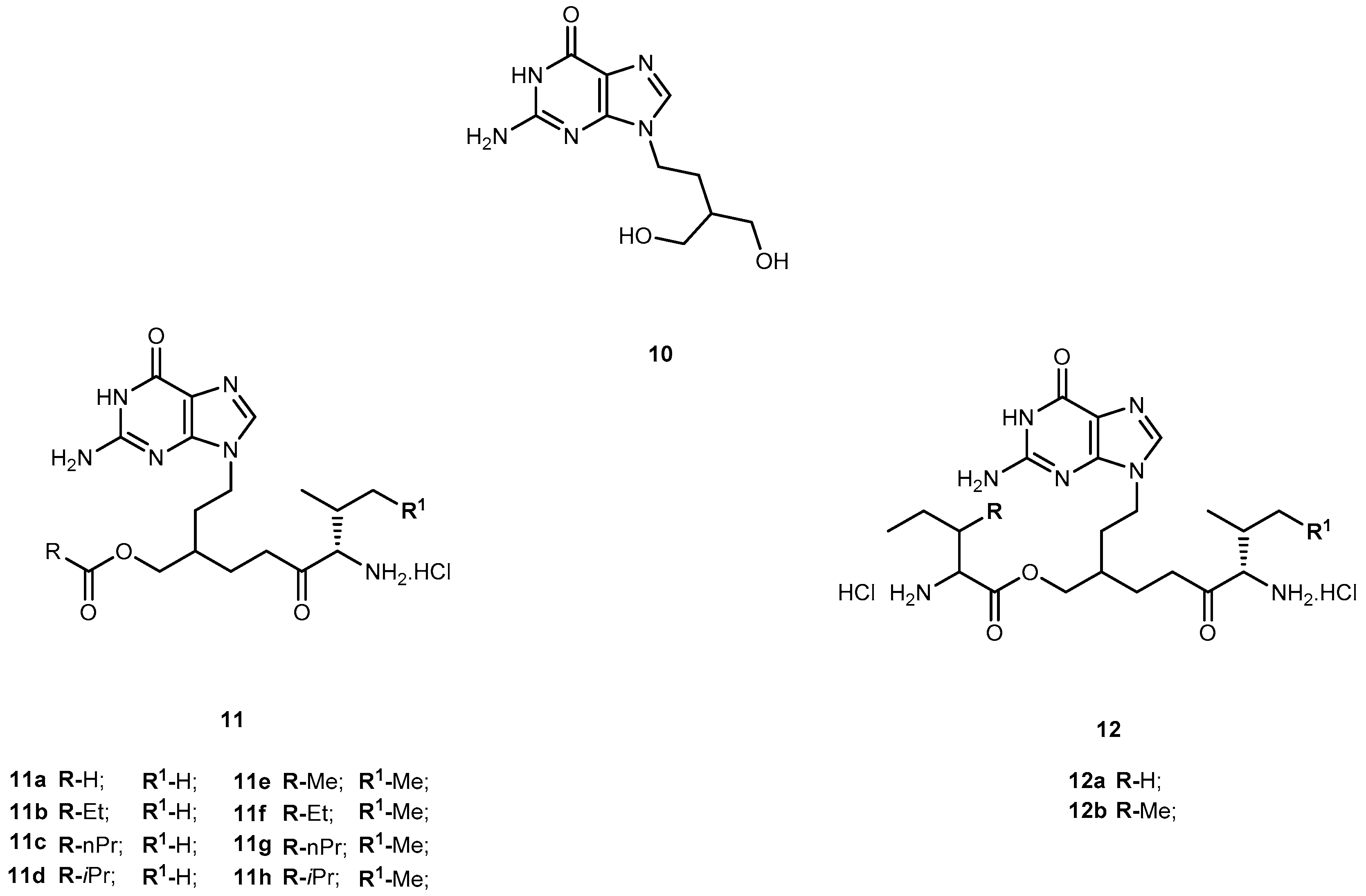{kind=link}
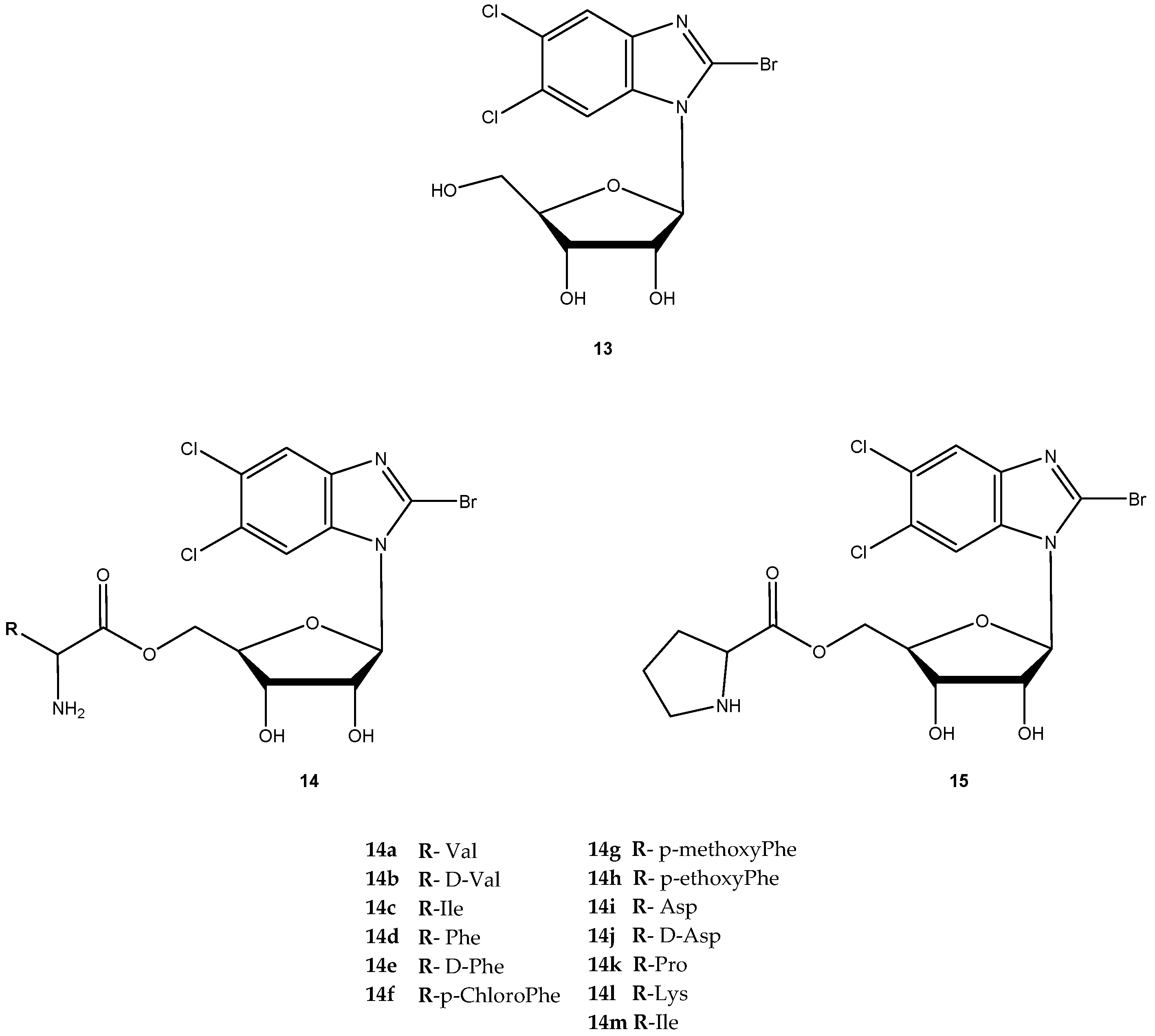{kind=link}
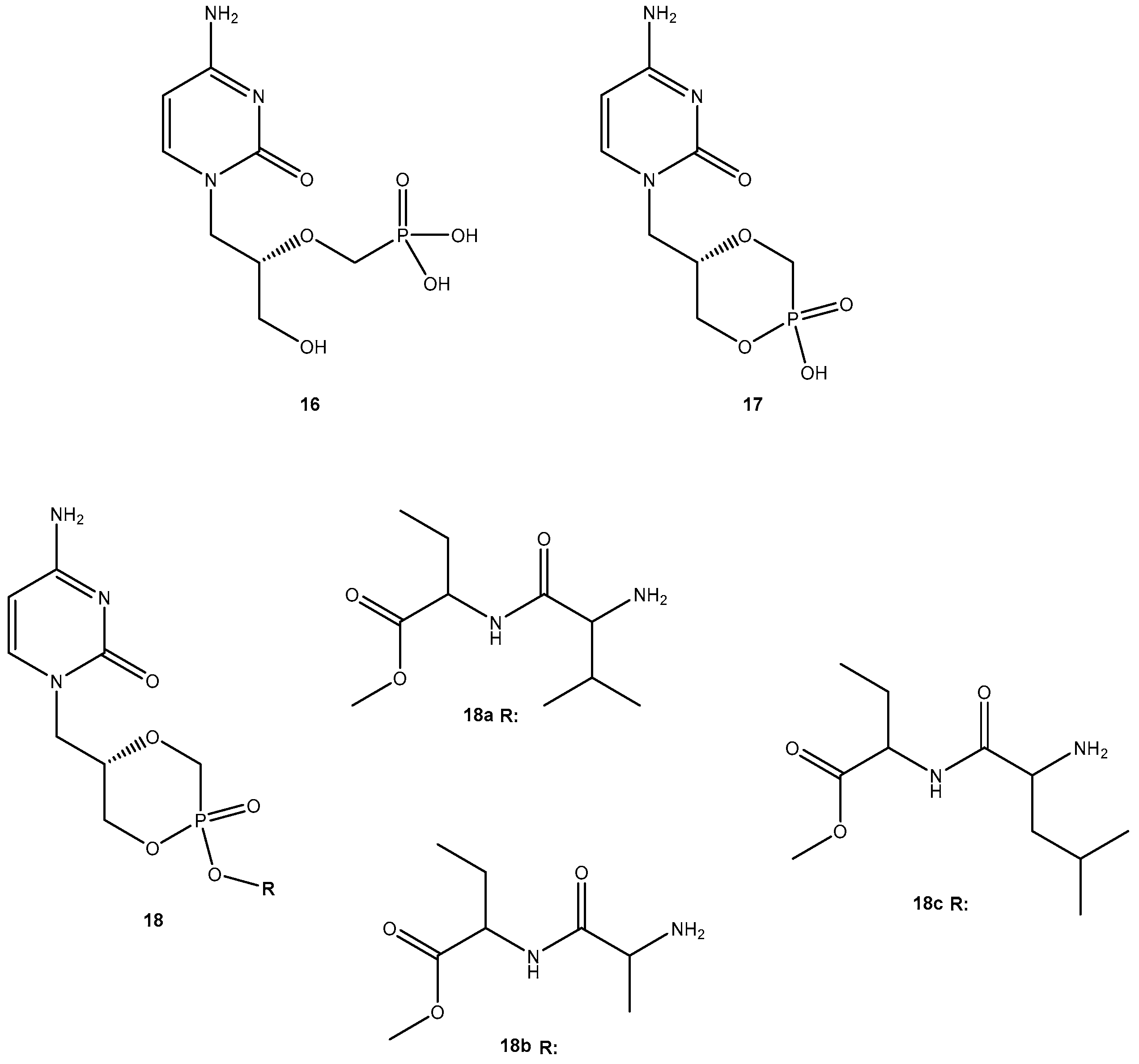{kind=link}
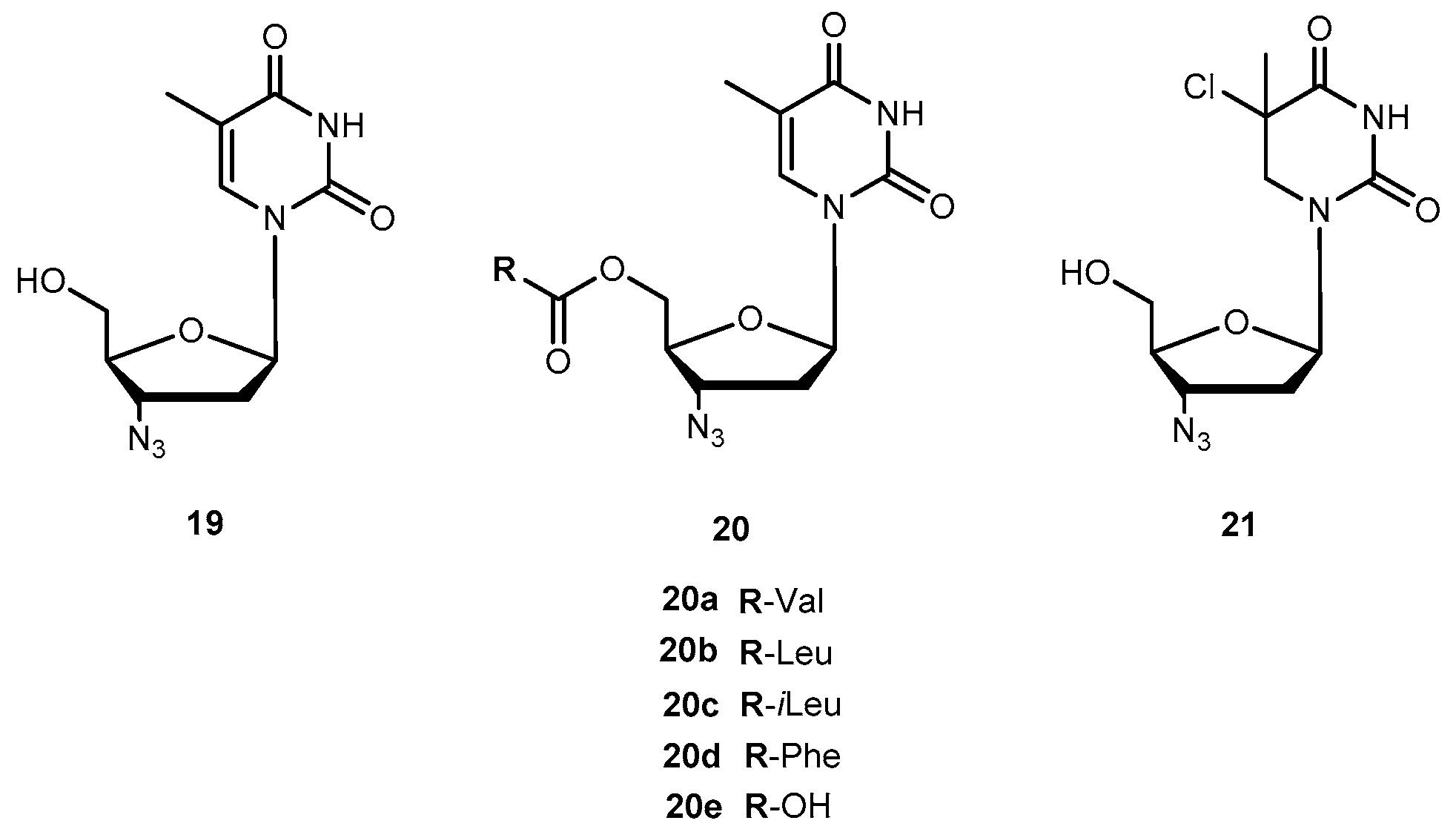{kind=link}
{kind=link}
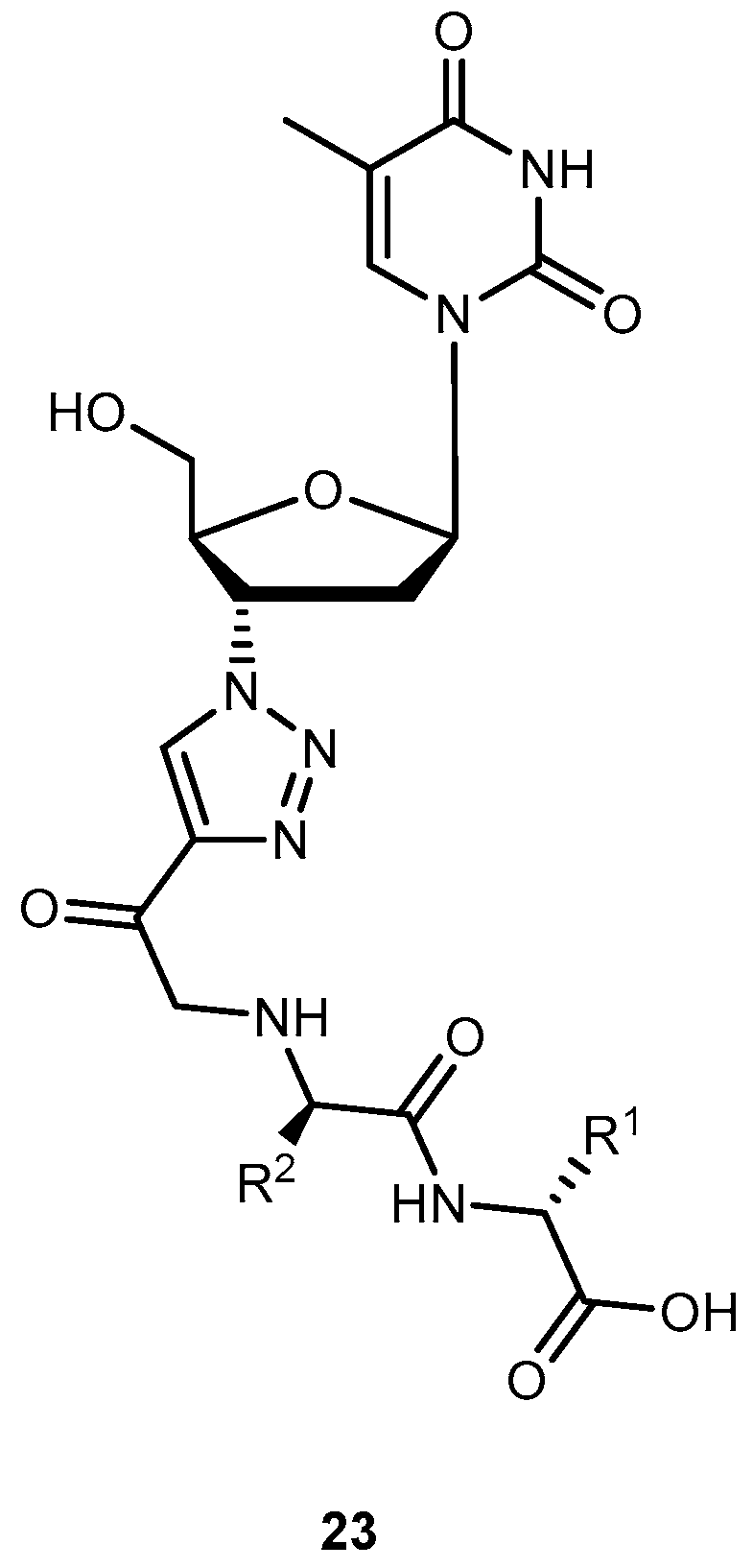{kind=link}
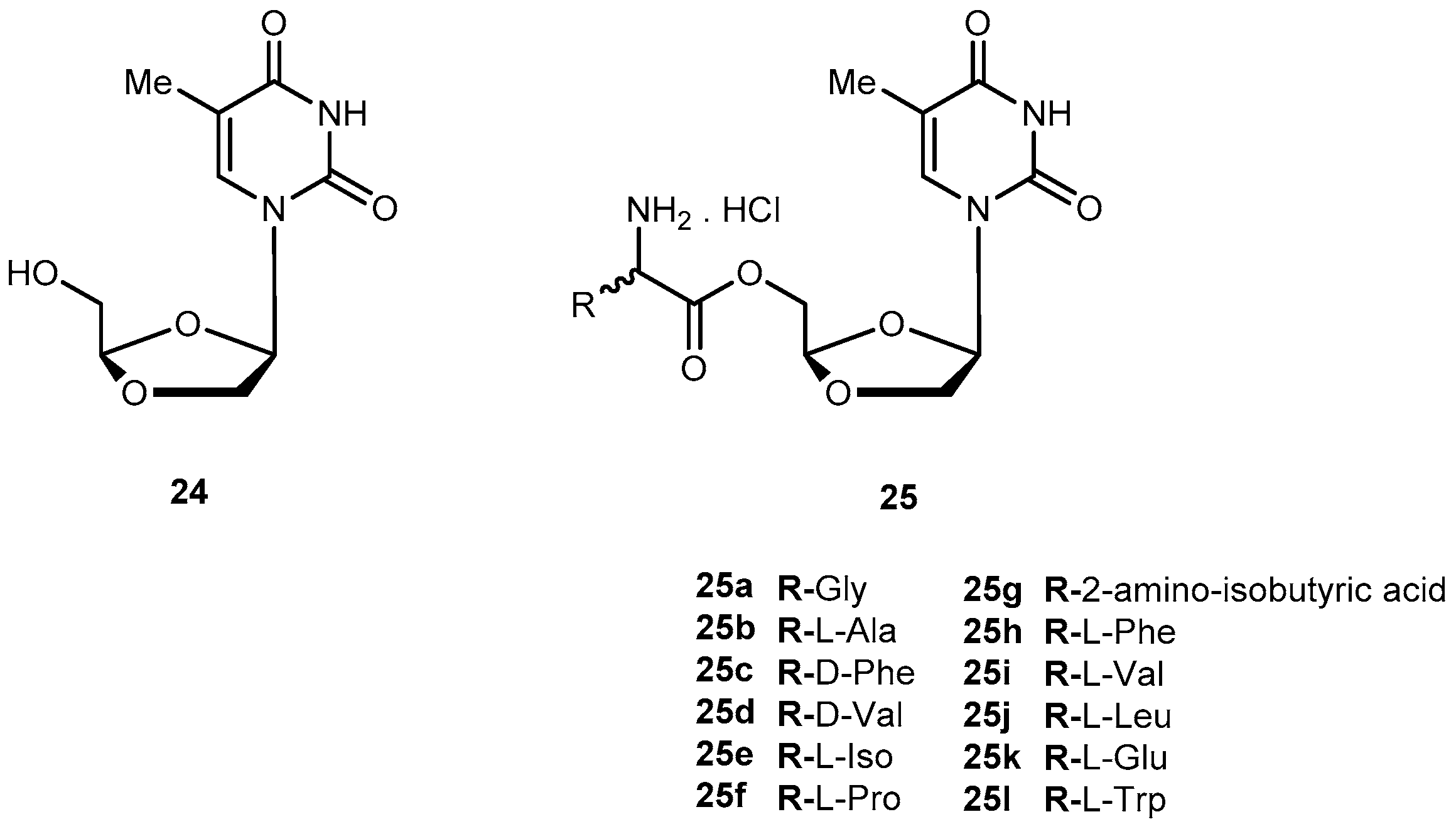{kind=link}
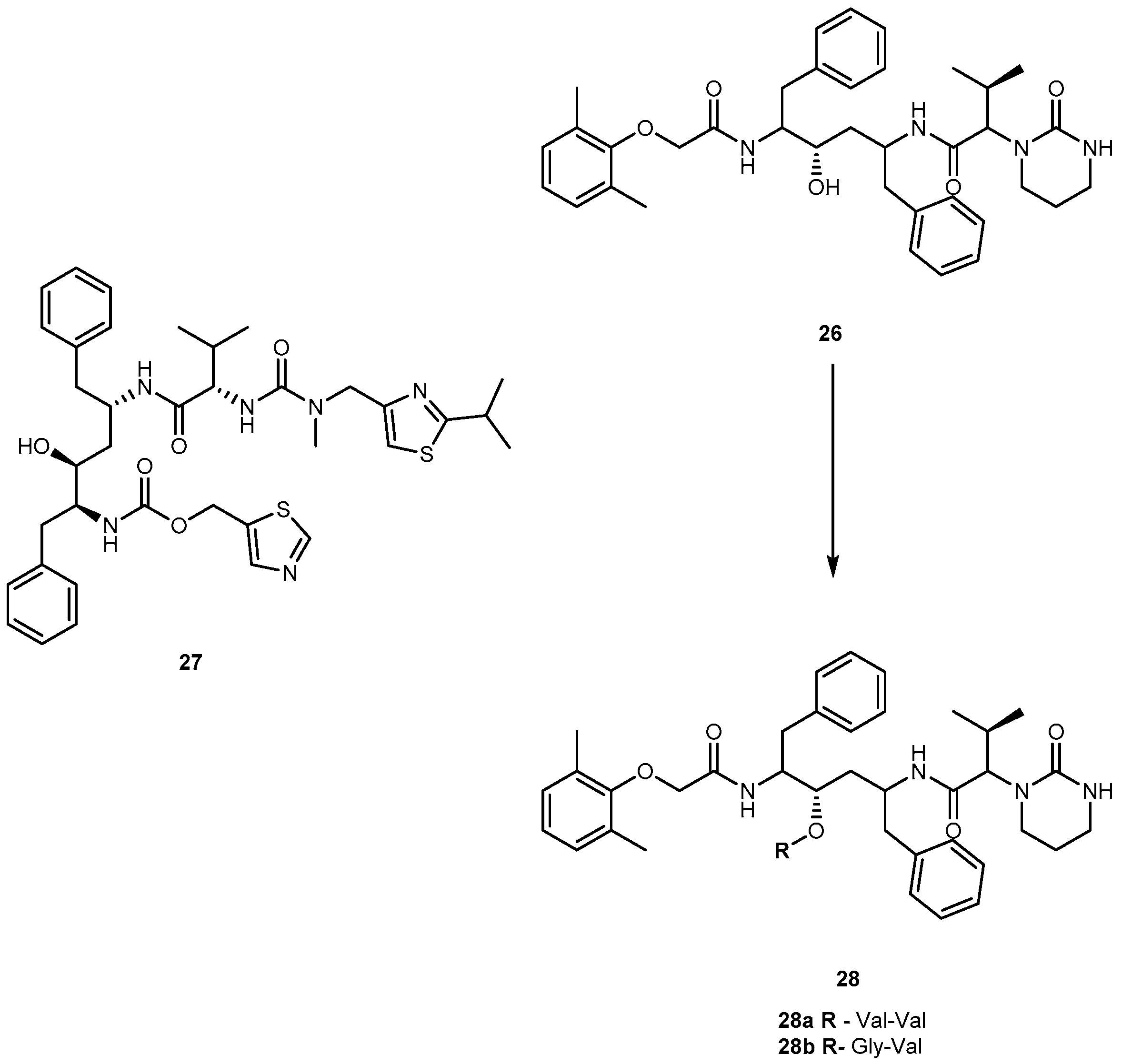{kind=link}
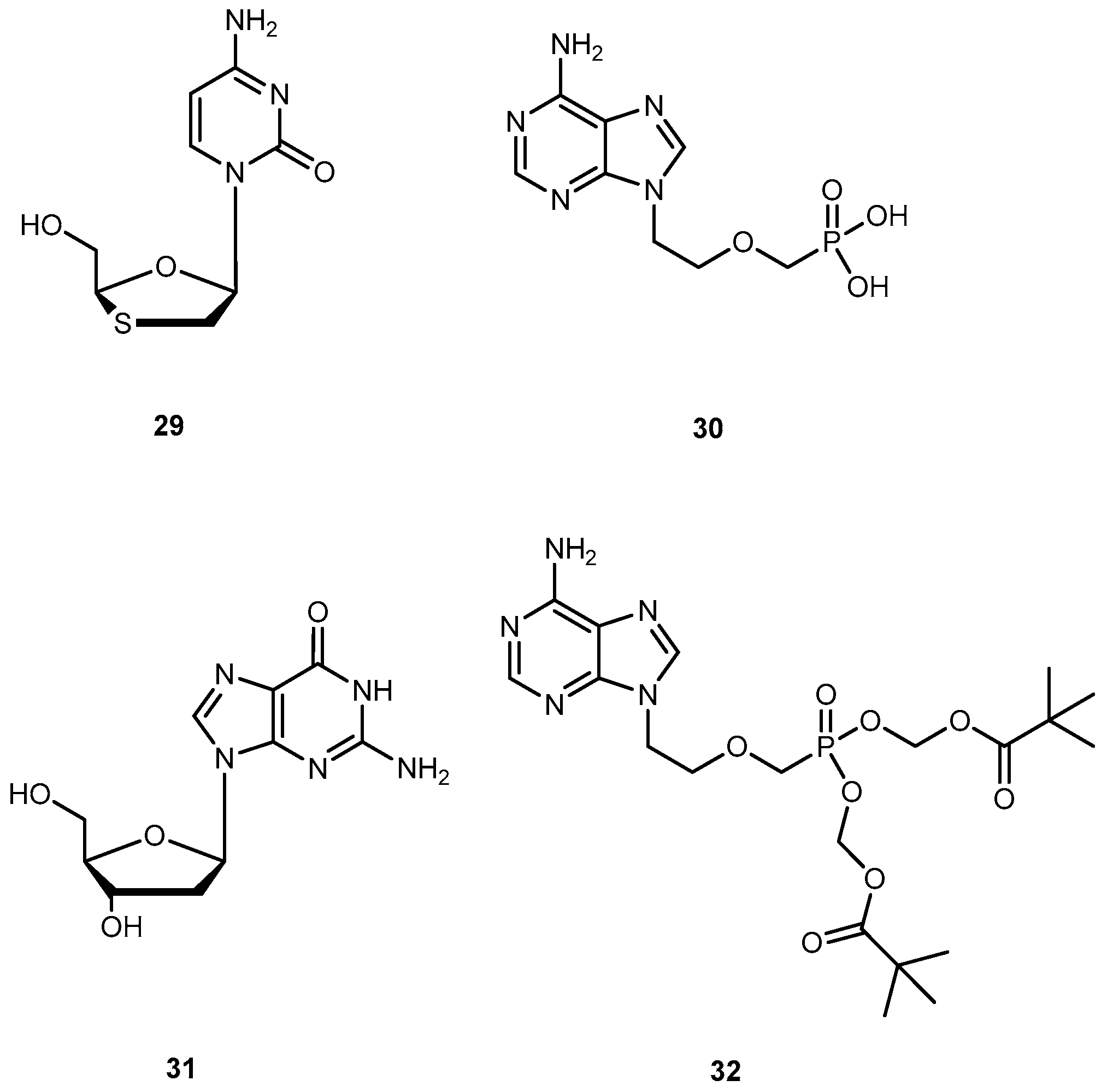{kind=link}
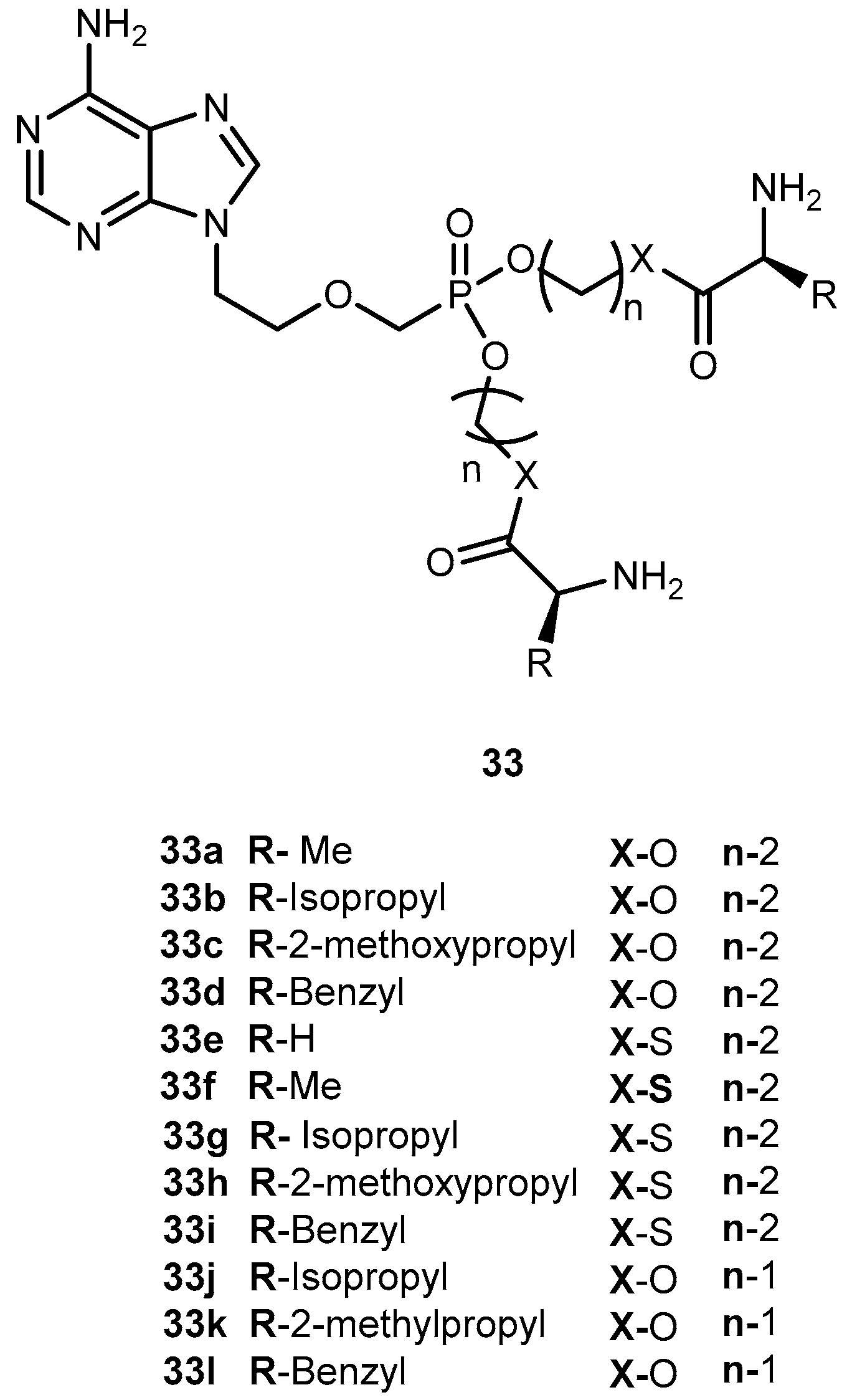{kind=link}
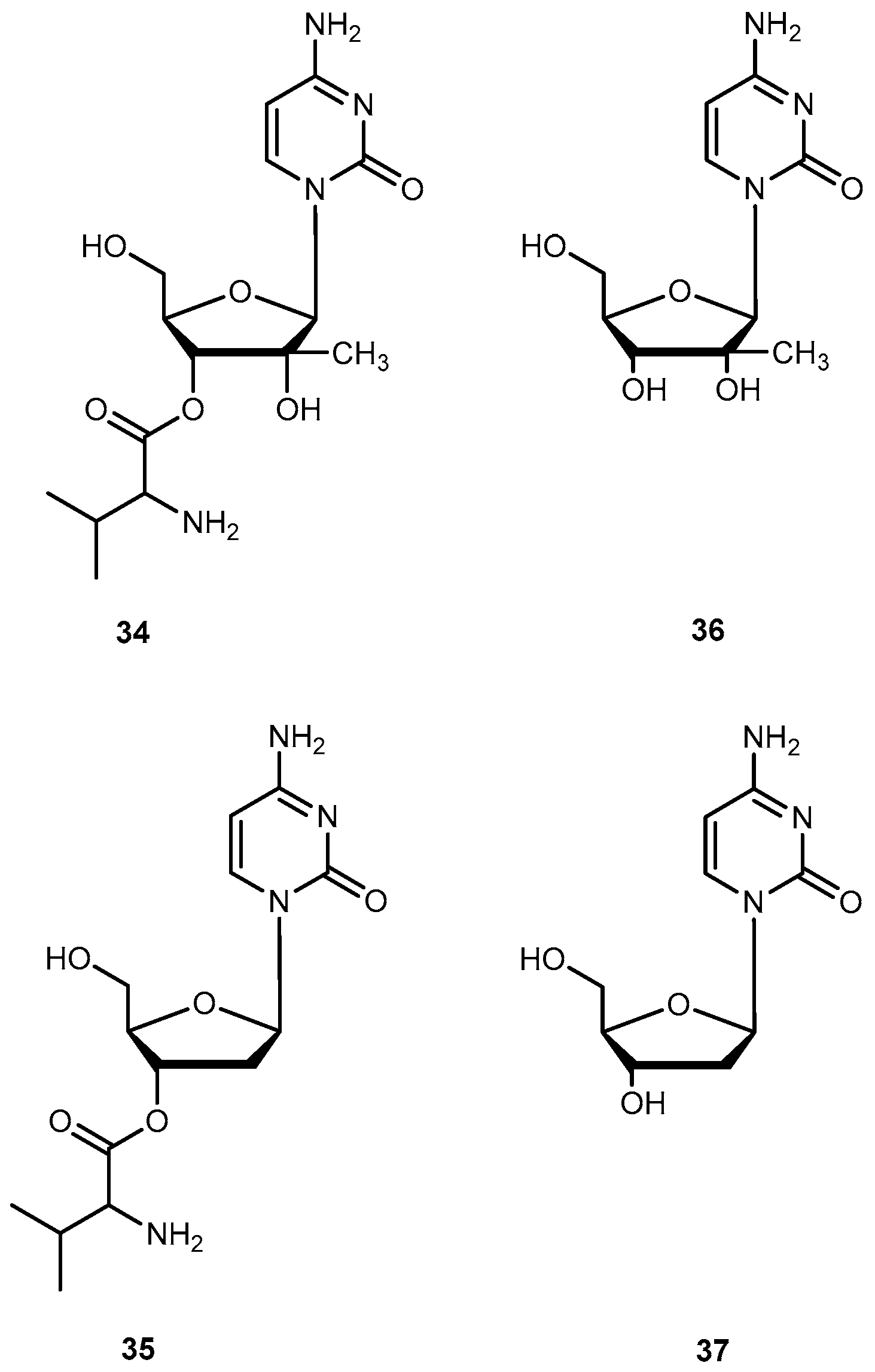{kind=link}
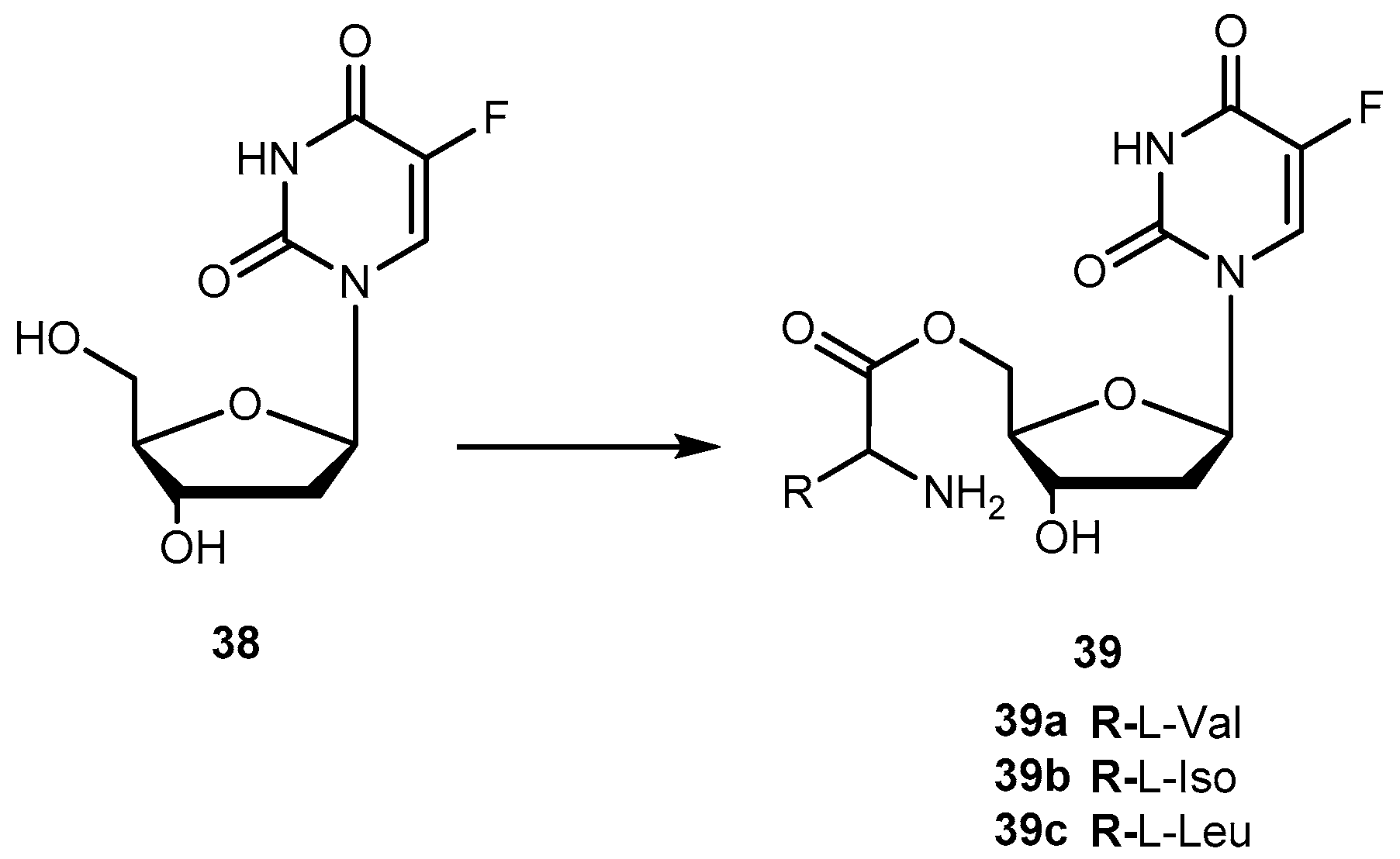{kind=link}
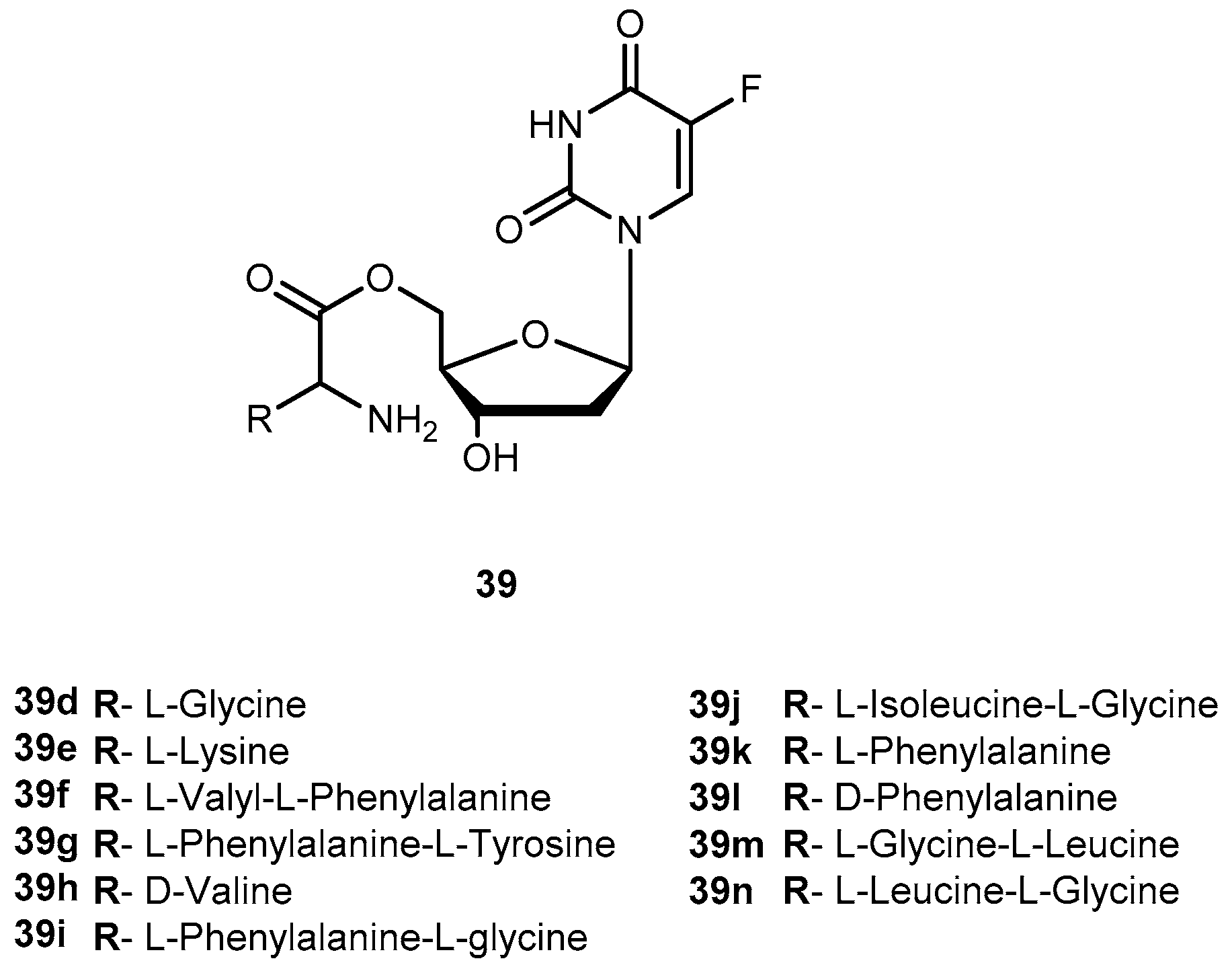{kind=link}
{kind=link}
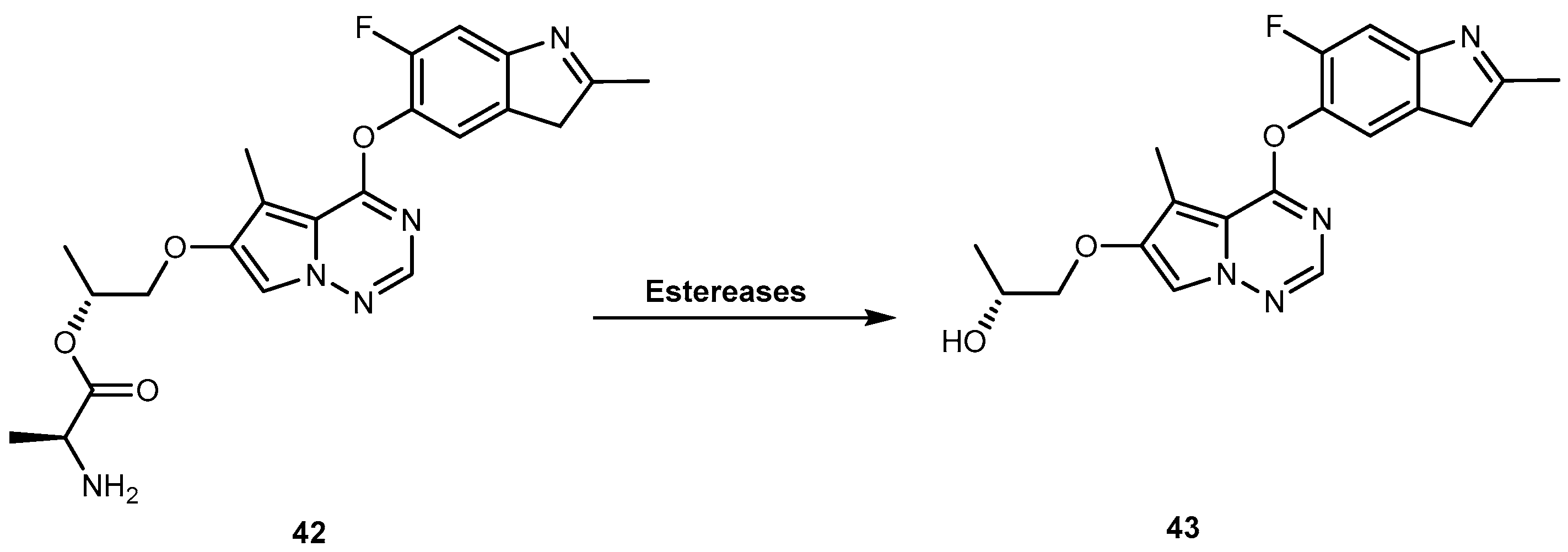{kind=link}
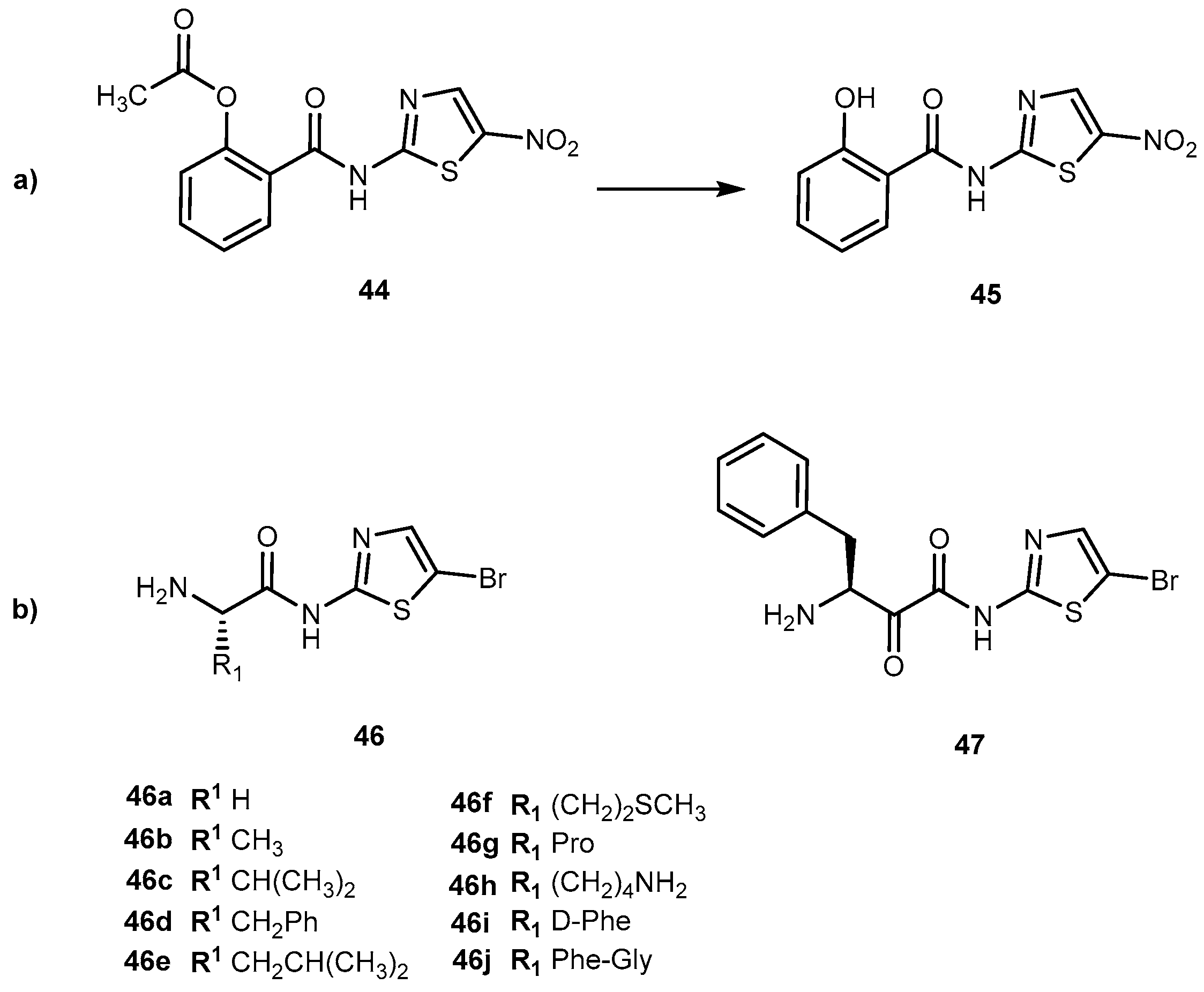{kind=link}
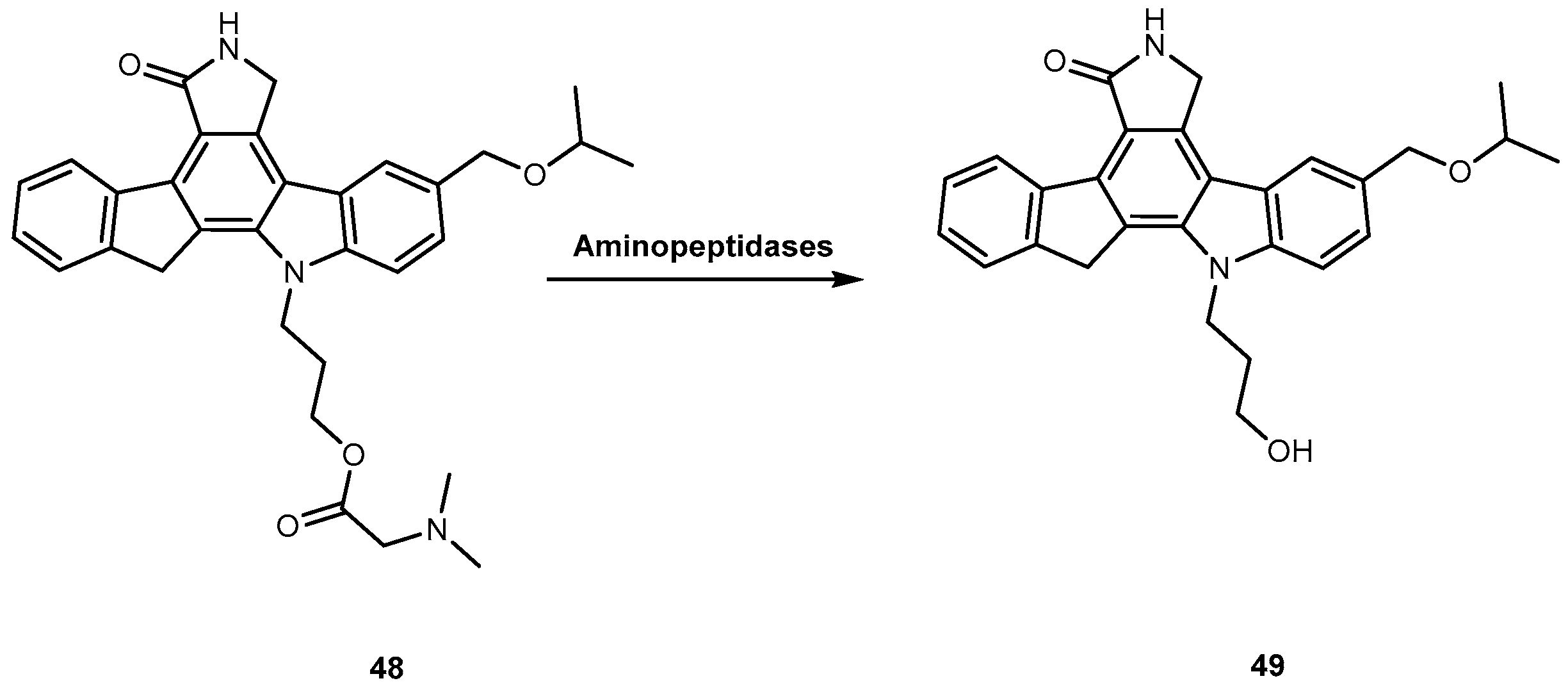{kind=link}
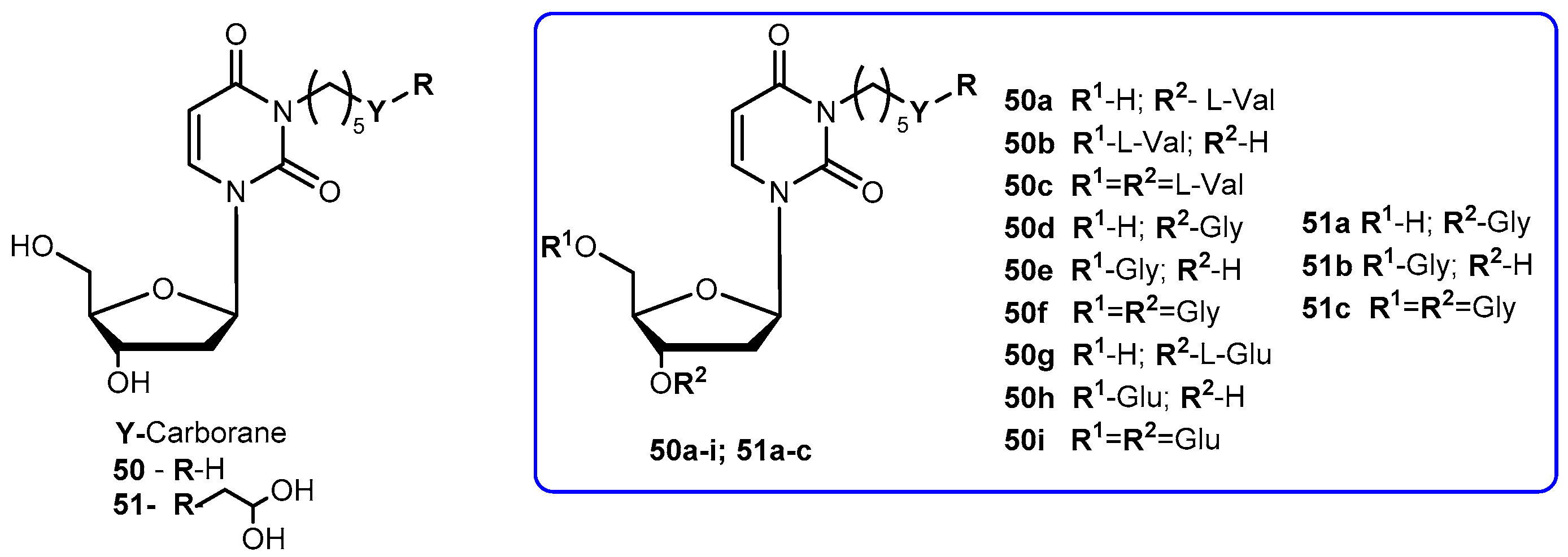{kind=link}
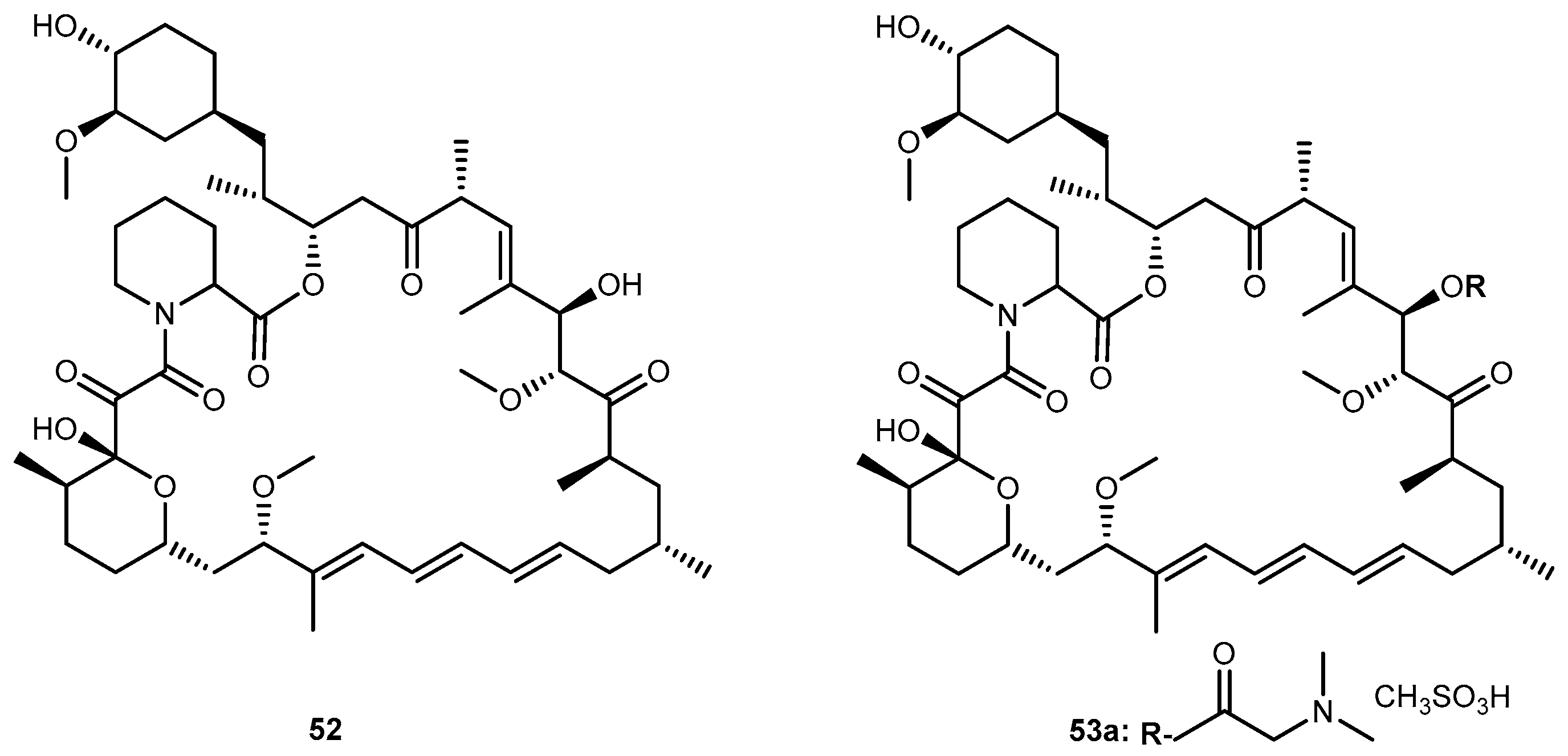{kind=link}
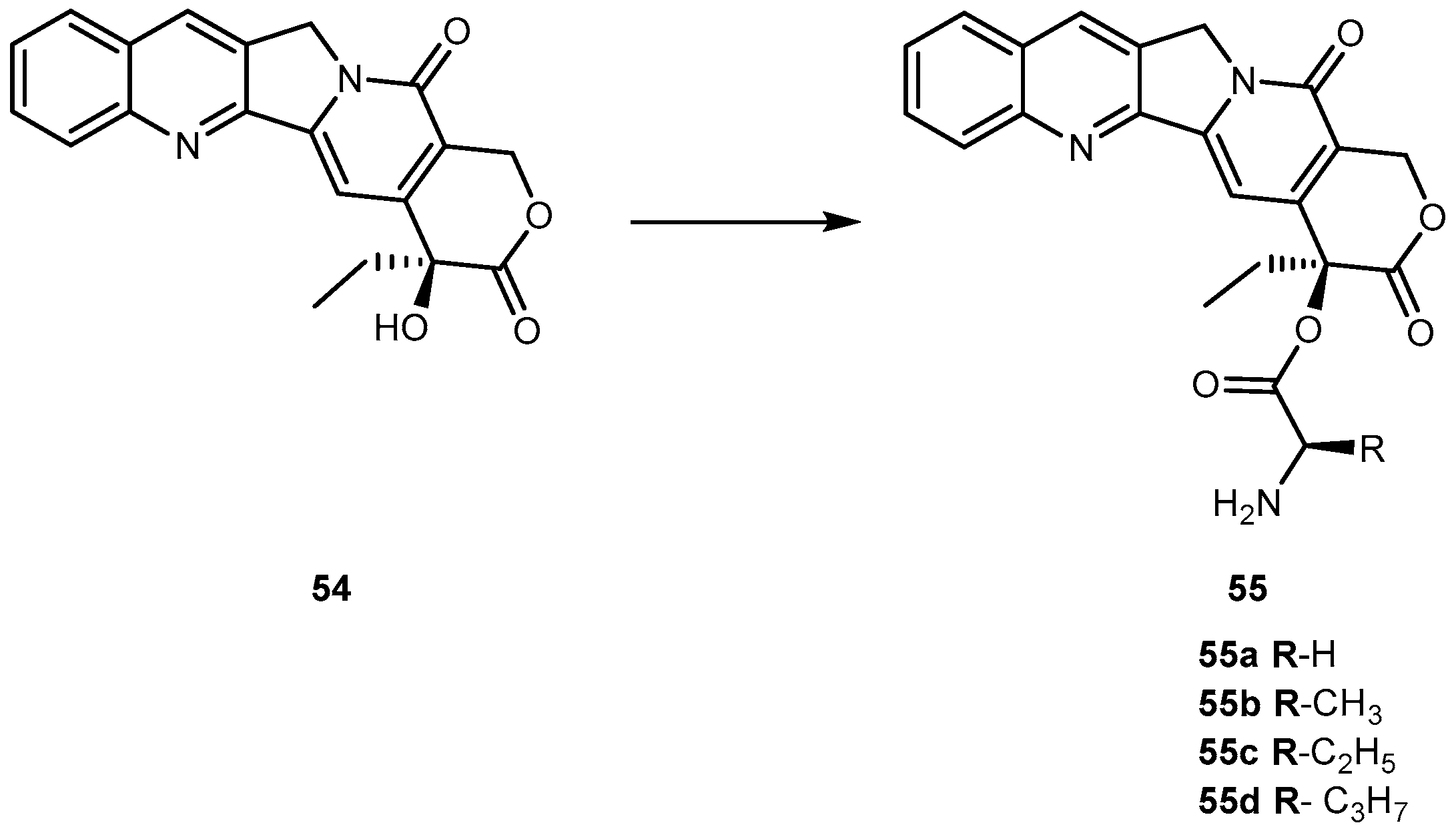{kind=link}
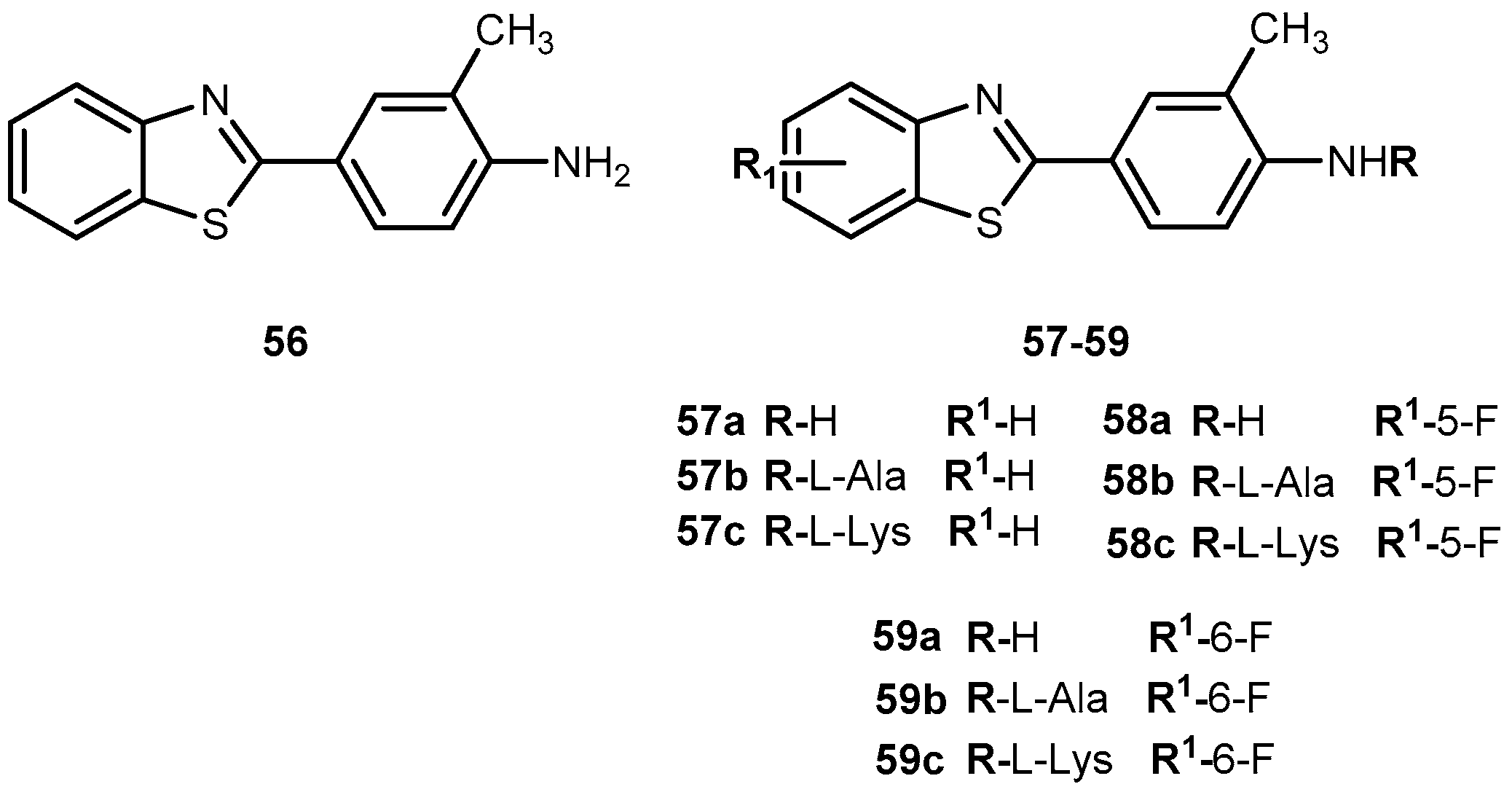{kind=link}
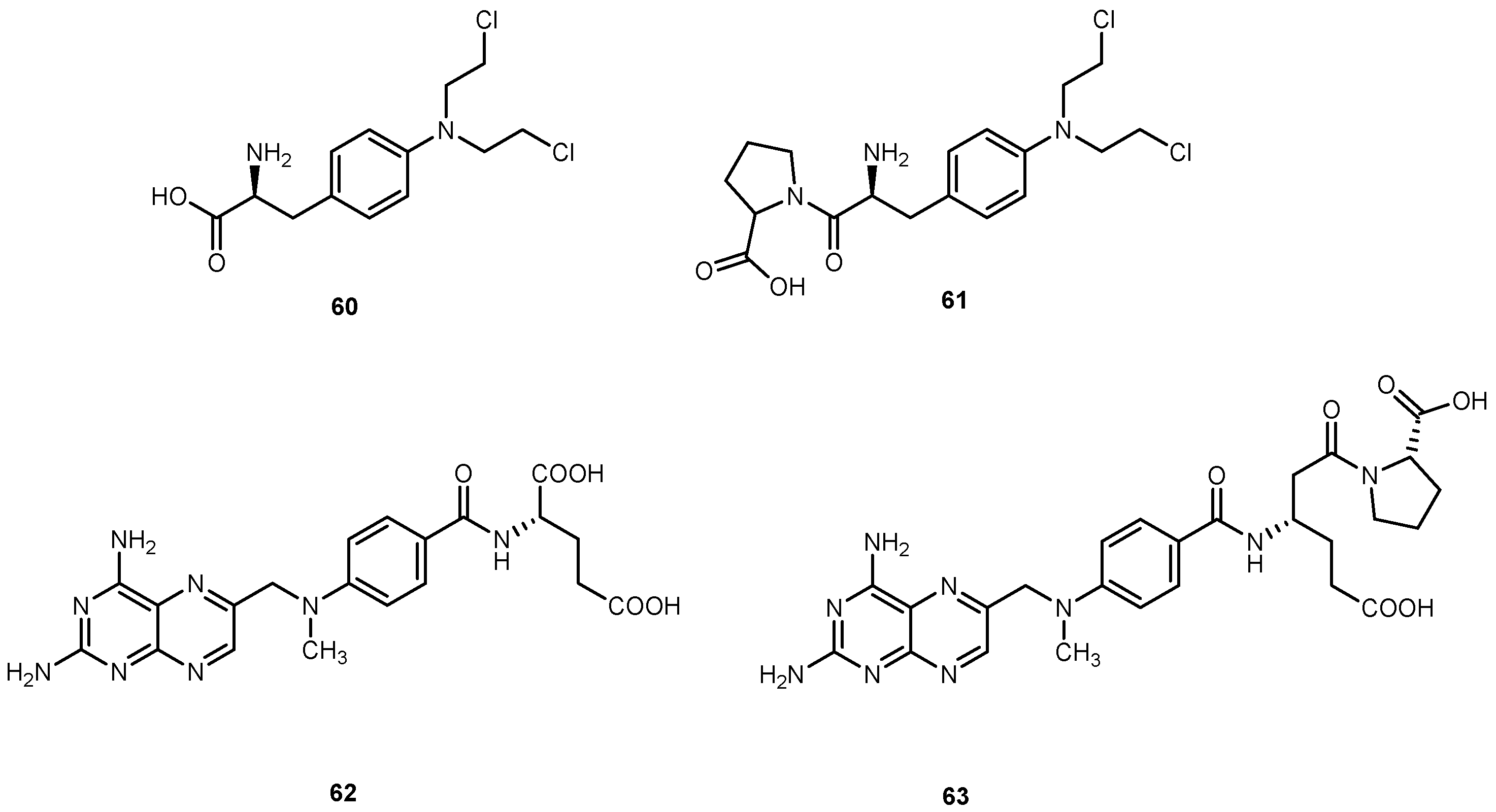{kind=link}
{kind=link}
{kind=link}
{kind=link}
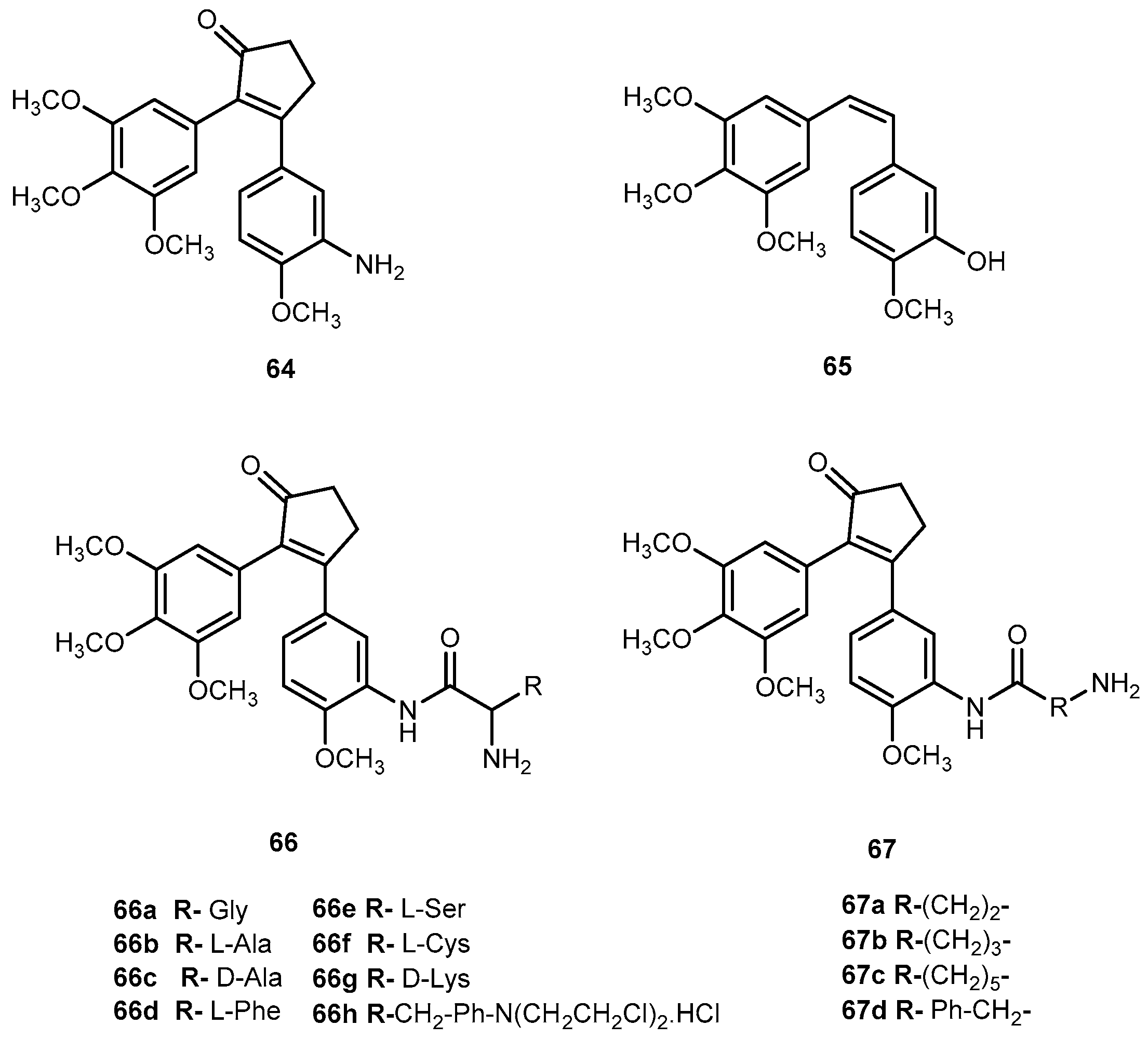
{kind=link}
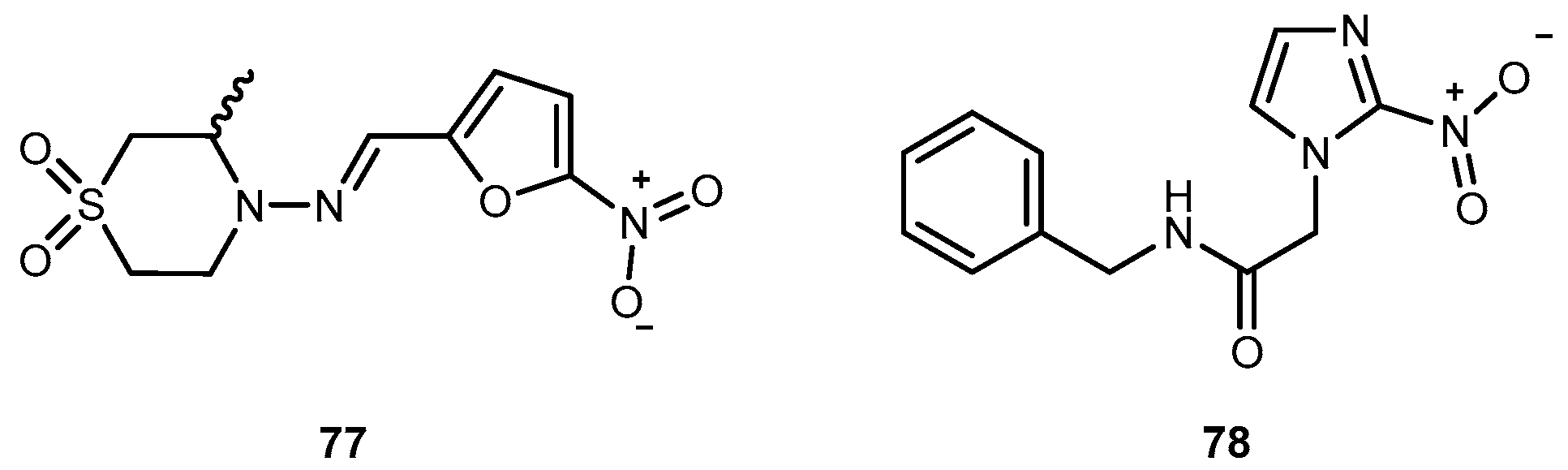{kind=link}
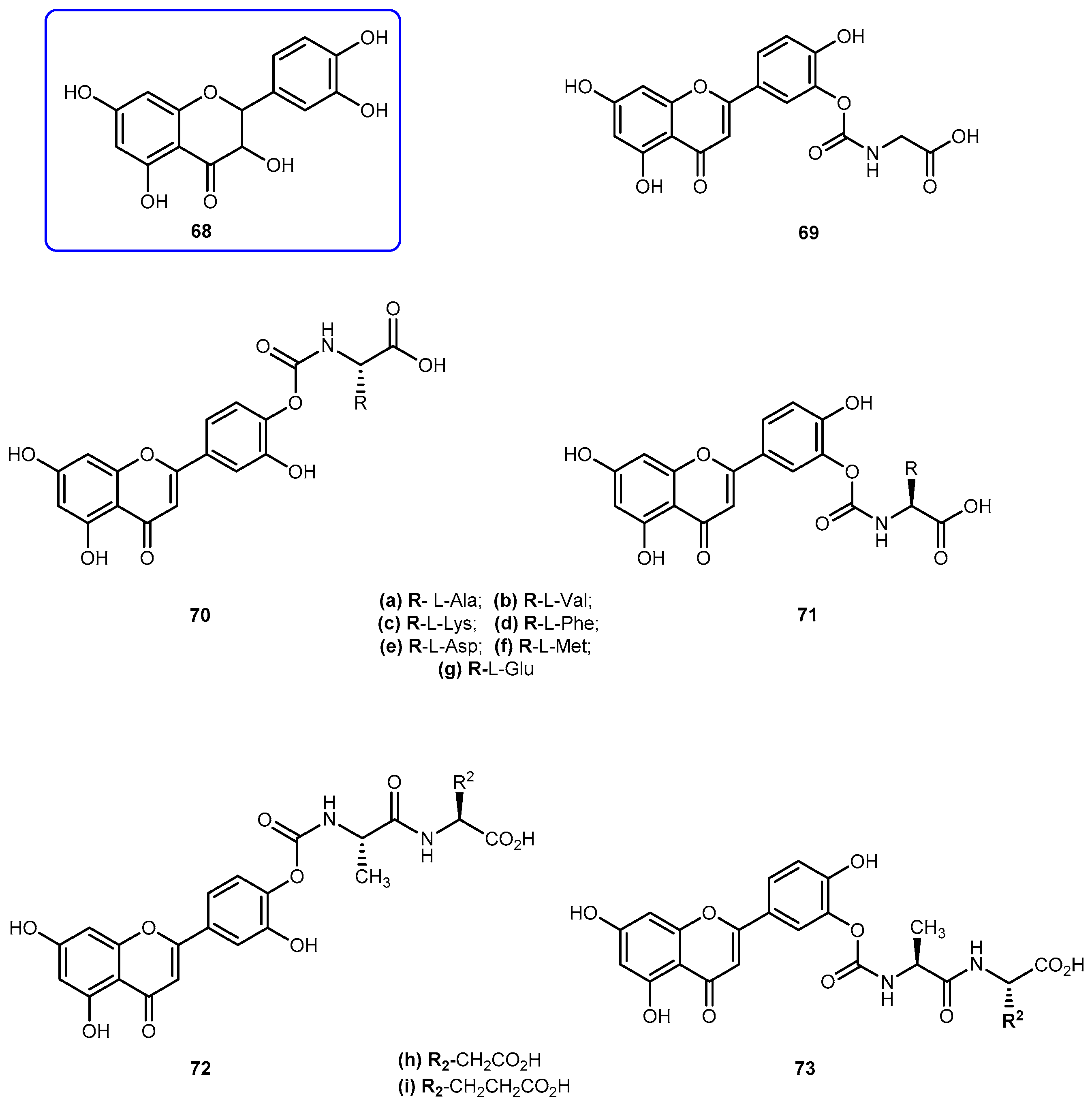
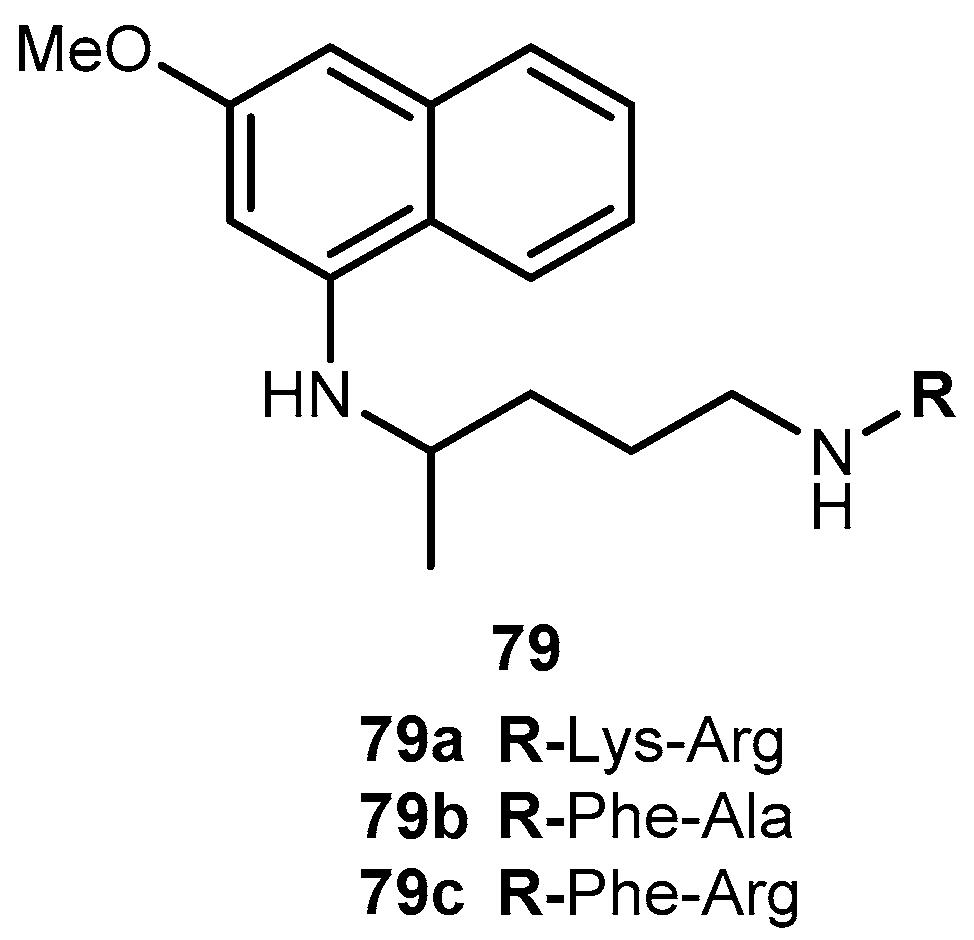{kind=link}
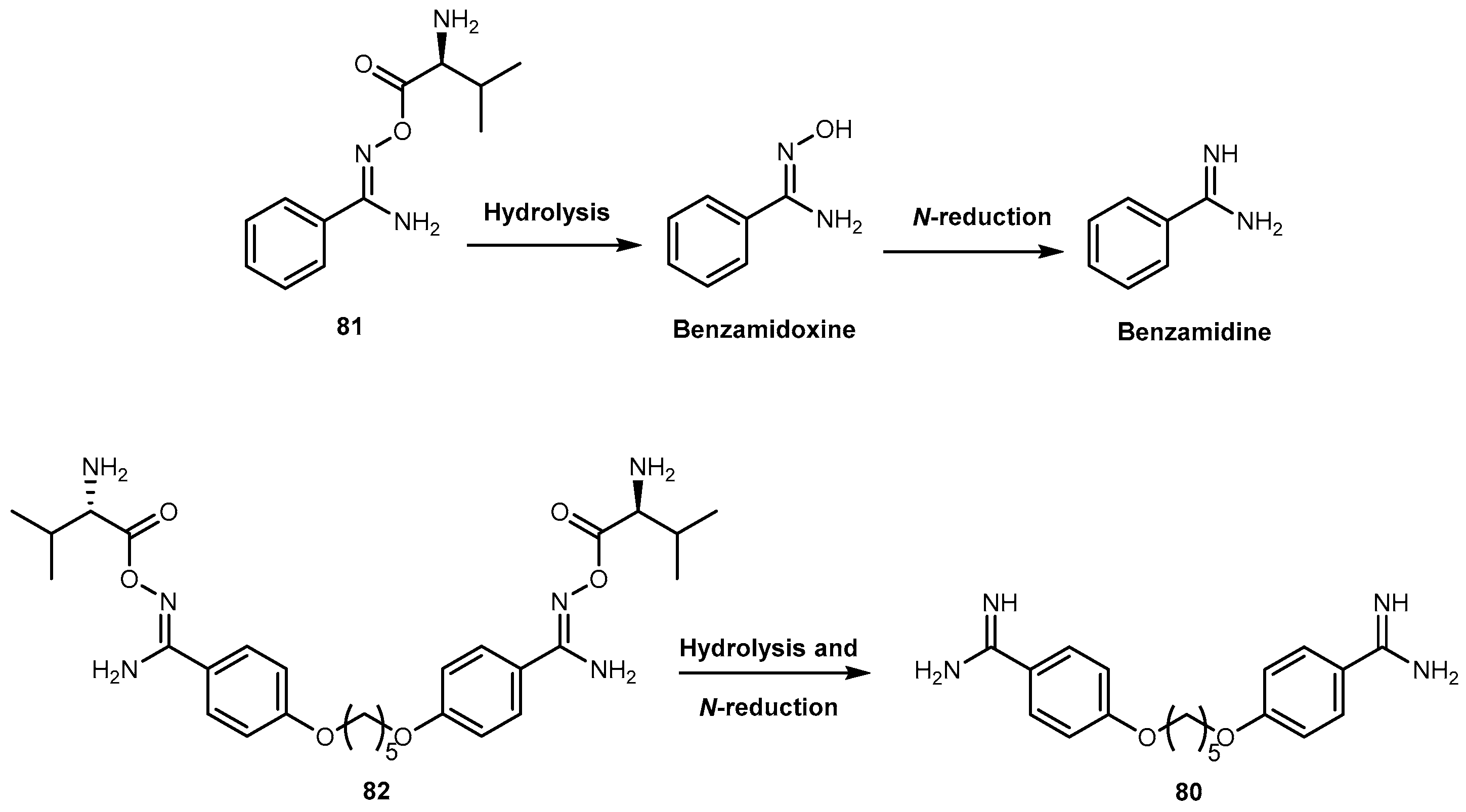{kind=link}
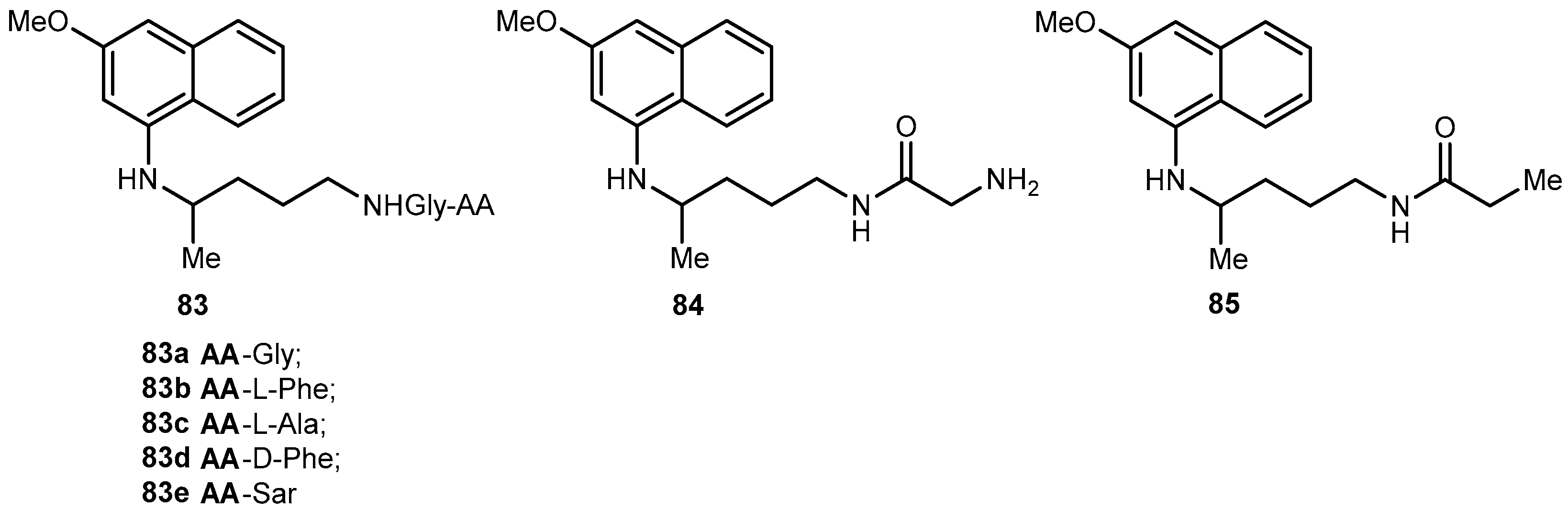{kind=link}
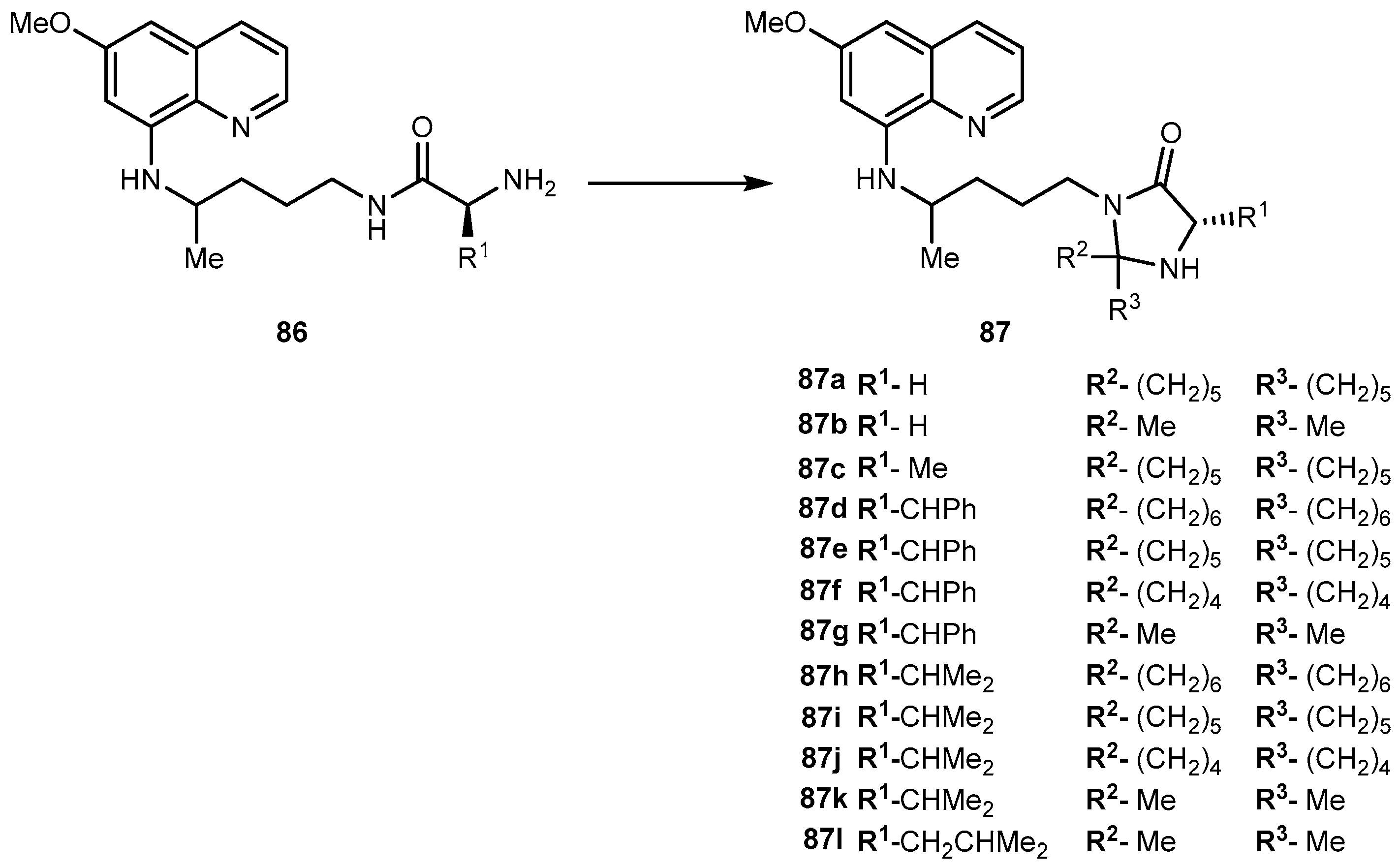{kind=link}
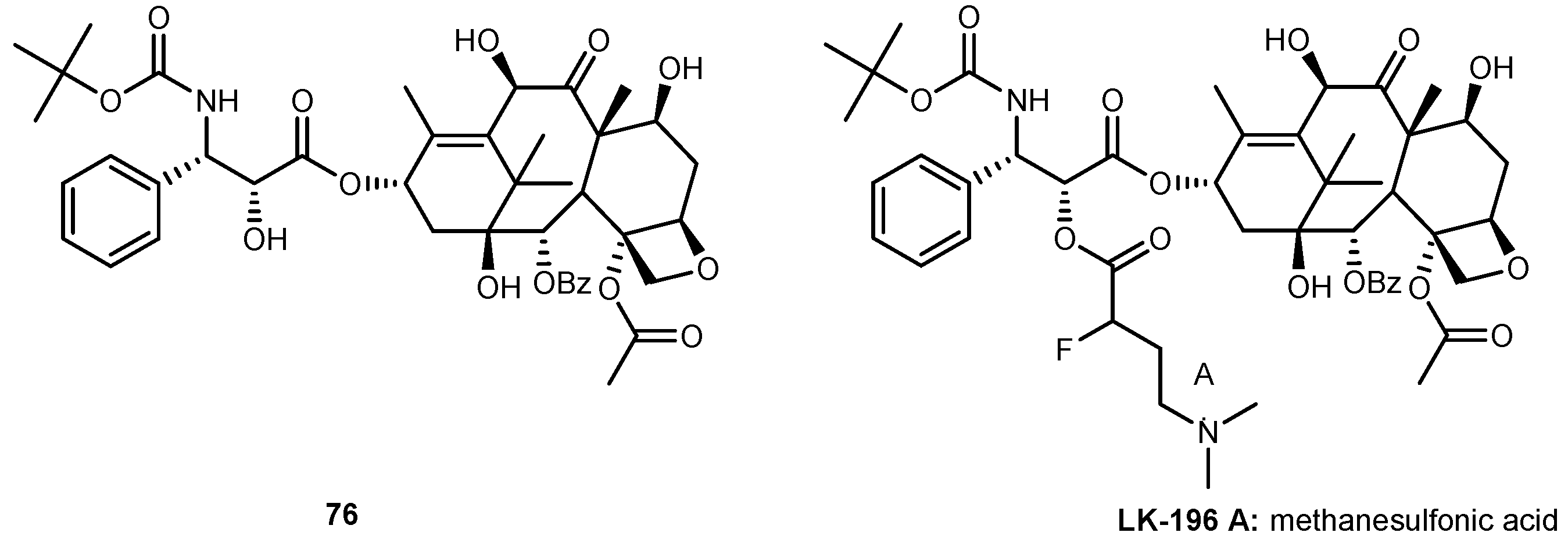
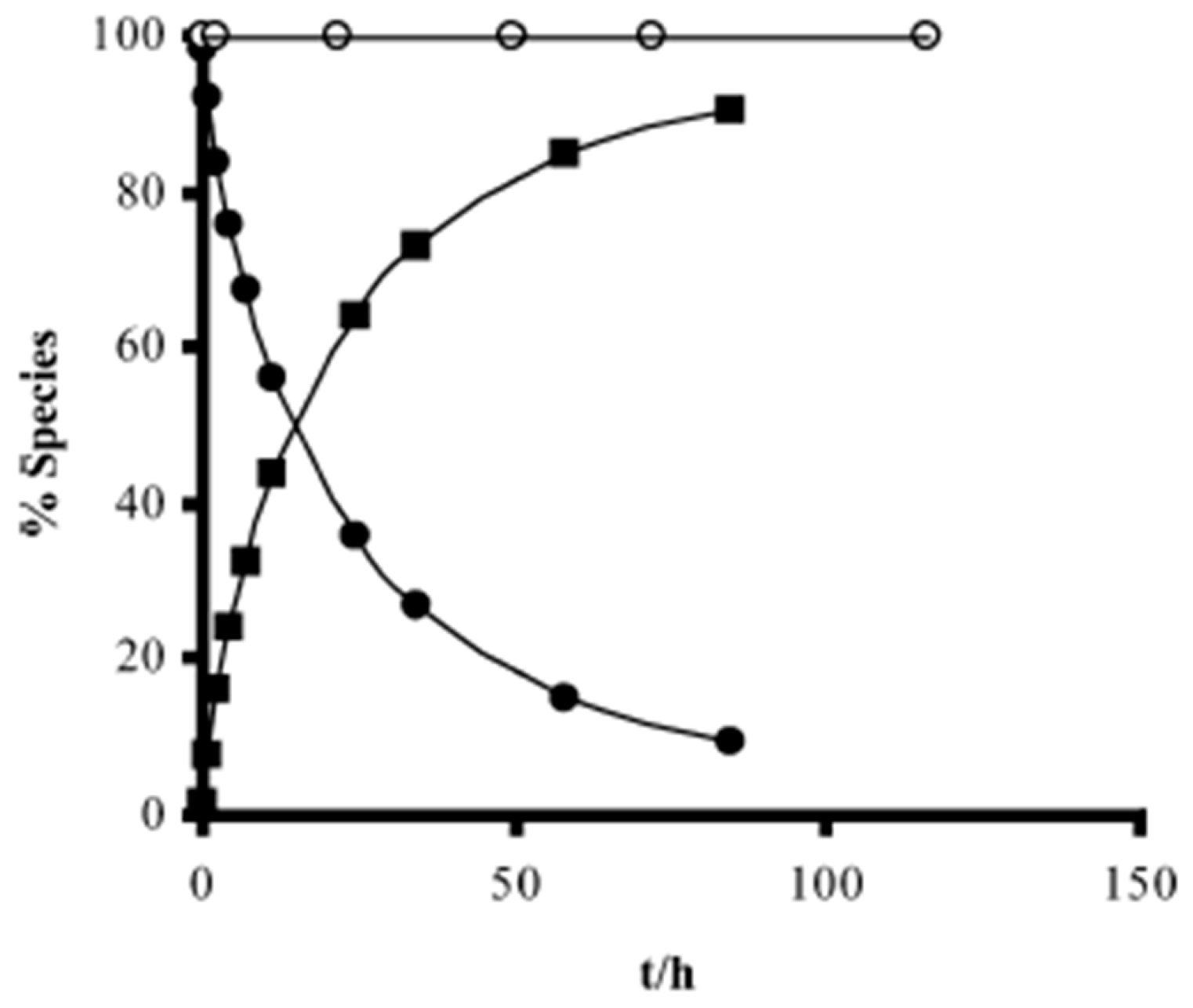{kind=link}
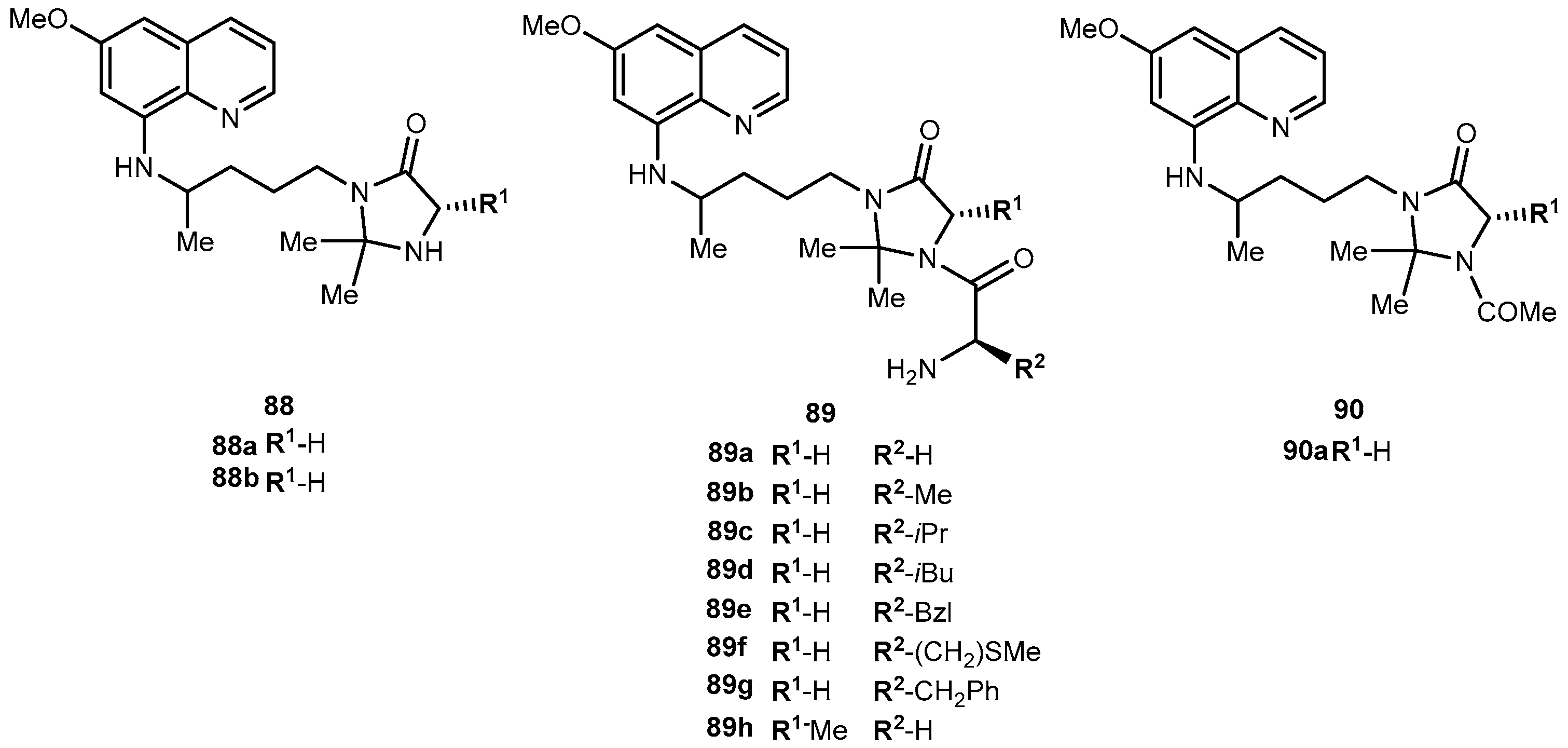{kind=link}
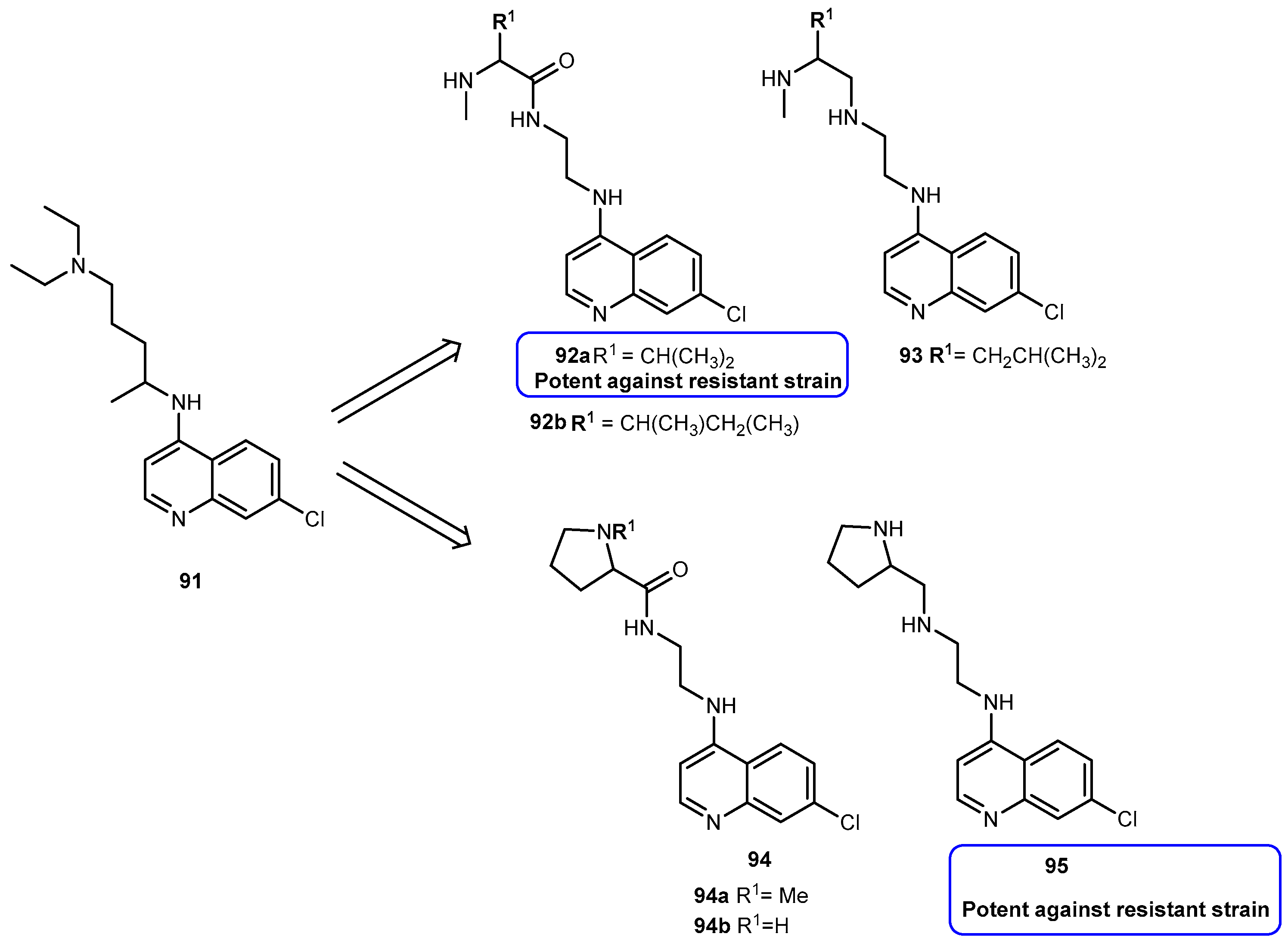{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
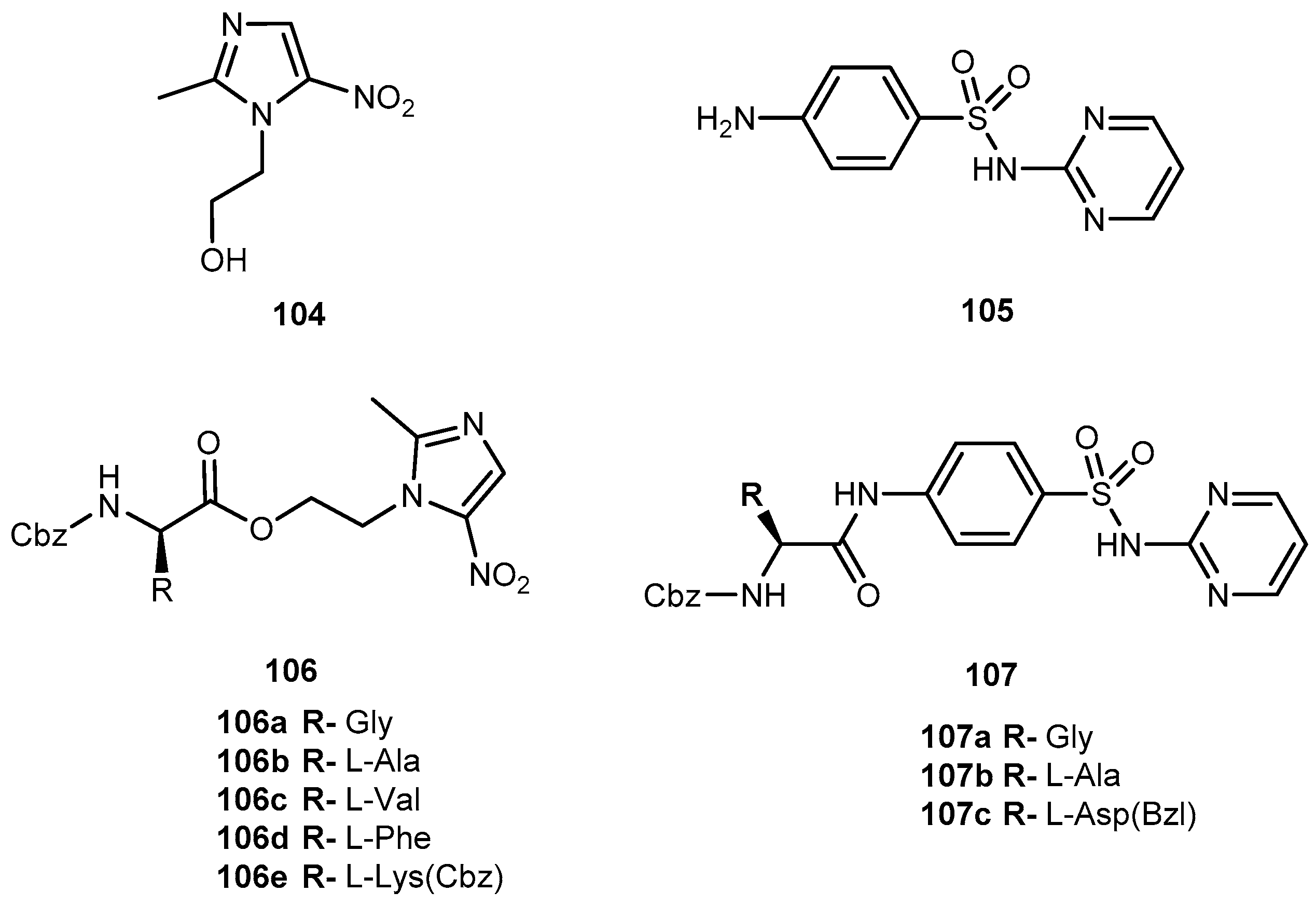{kind=link}
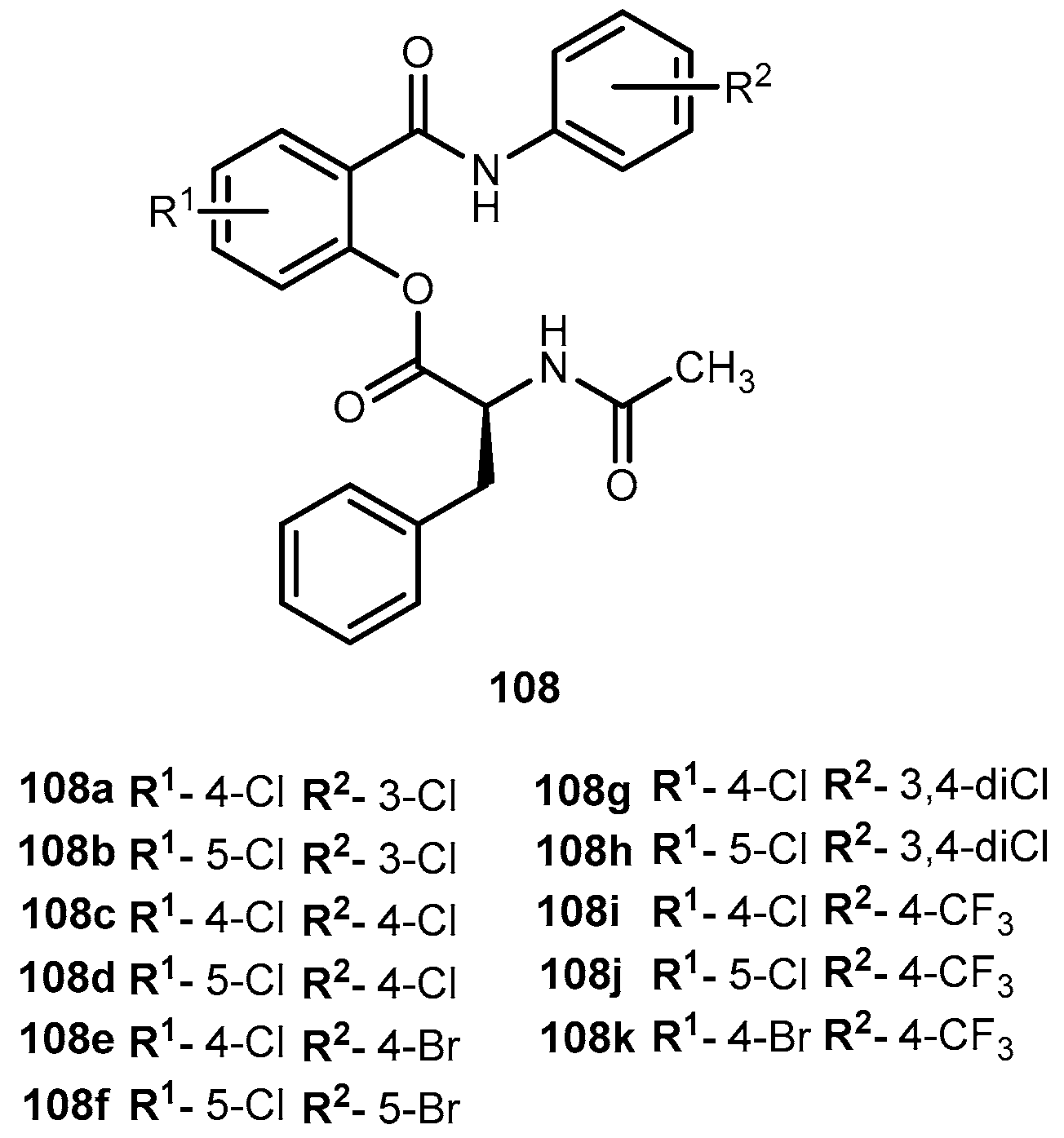{kind=link}
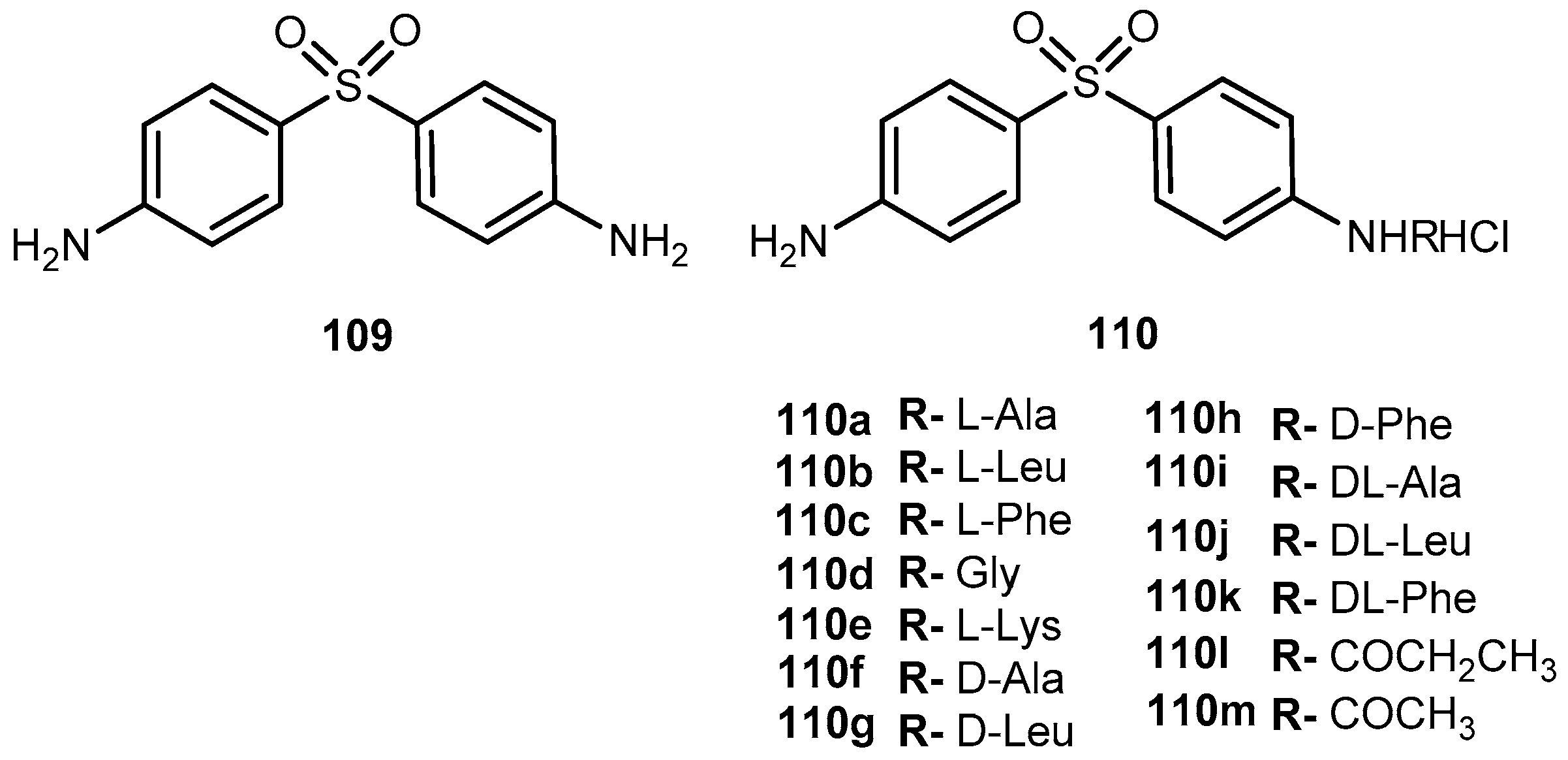{kind=link}
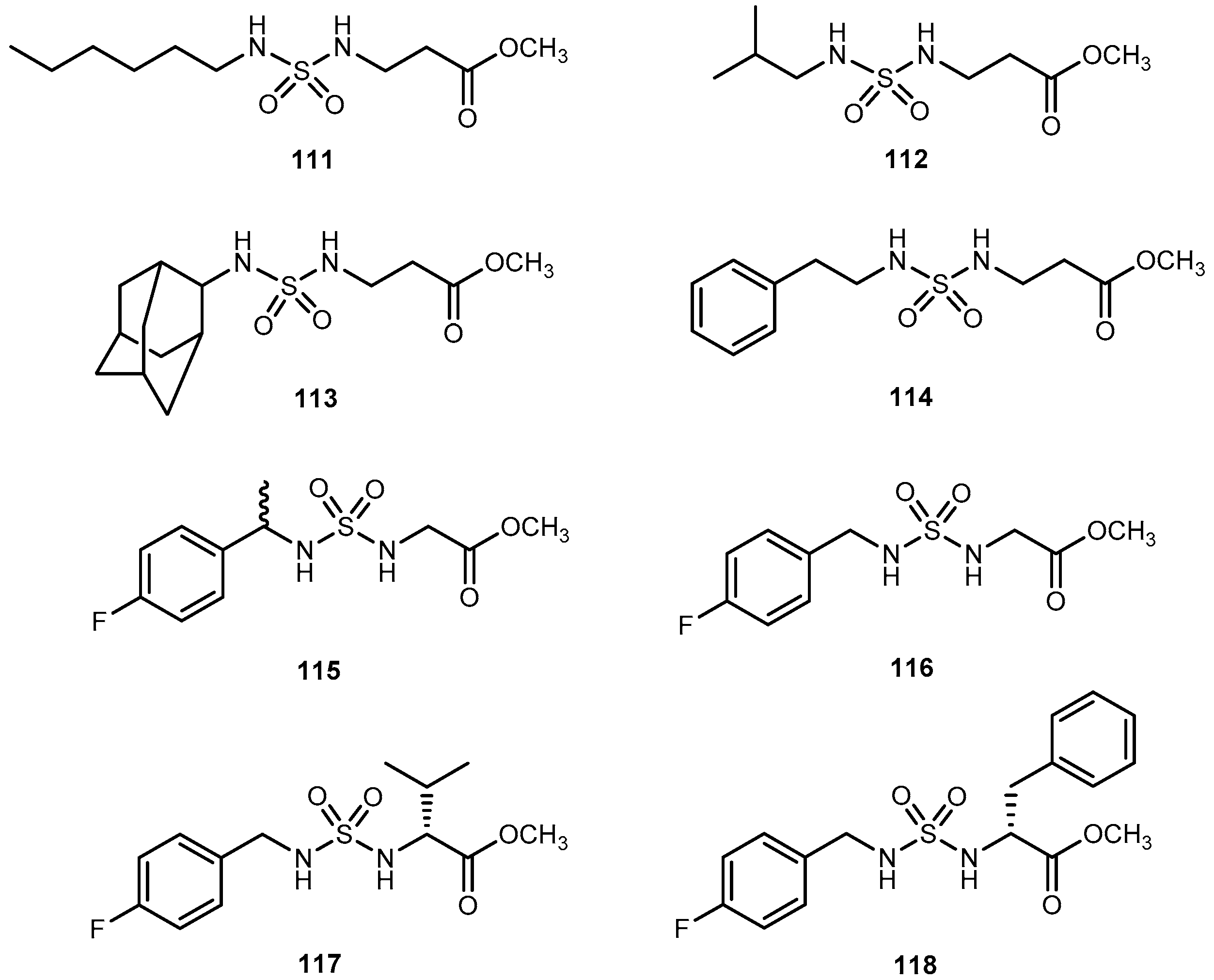{kind=link}
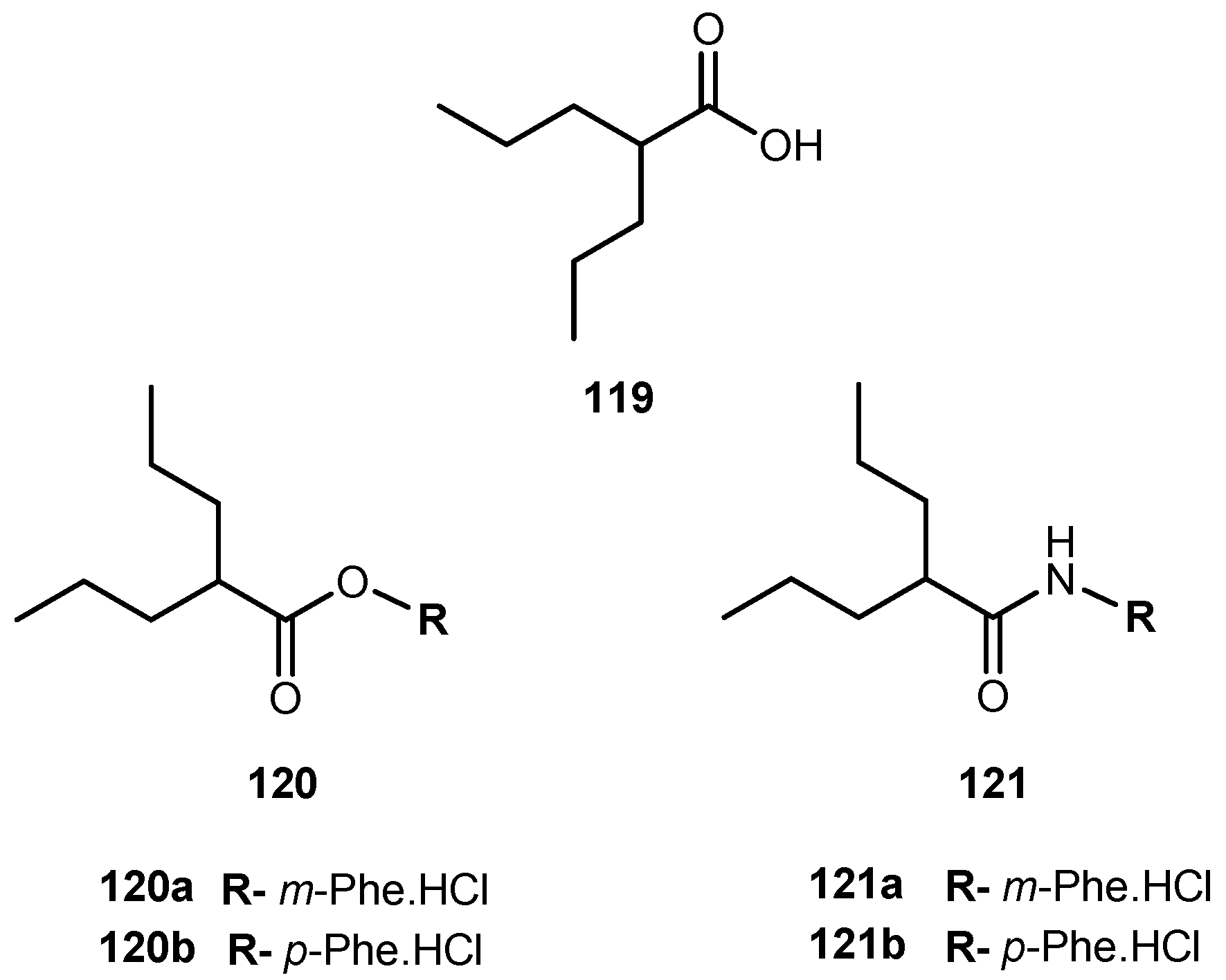{kind=link}
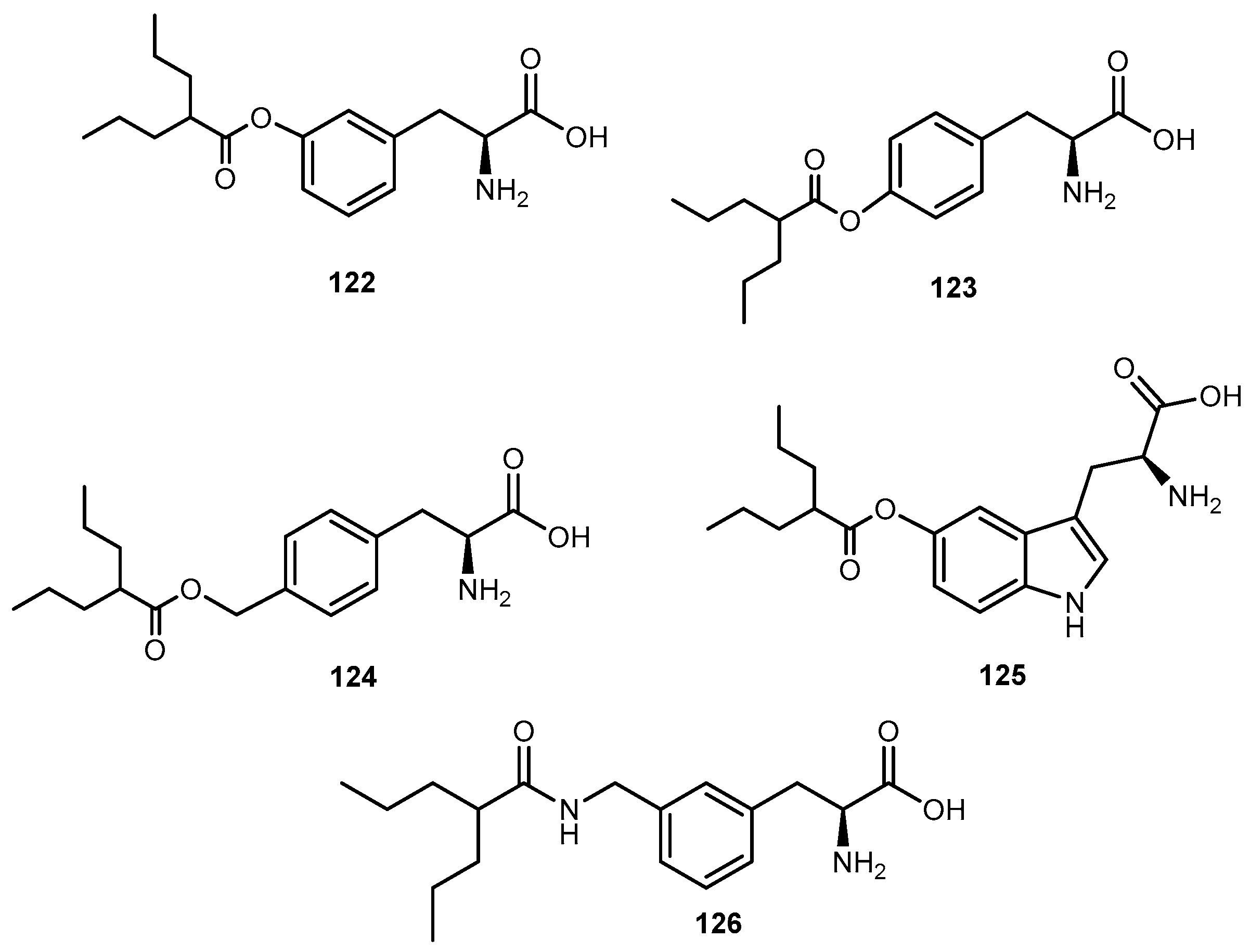{kind=link}
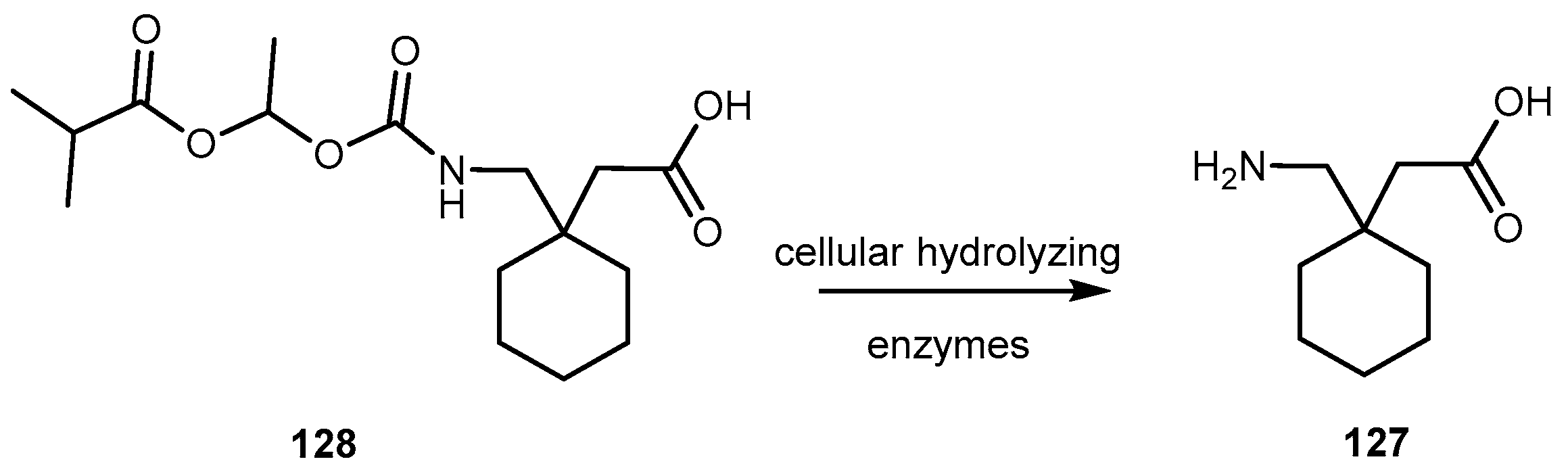{kind=link}
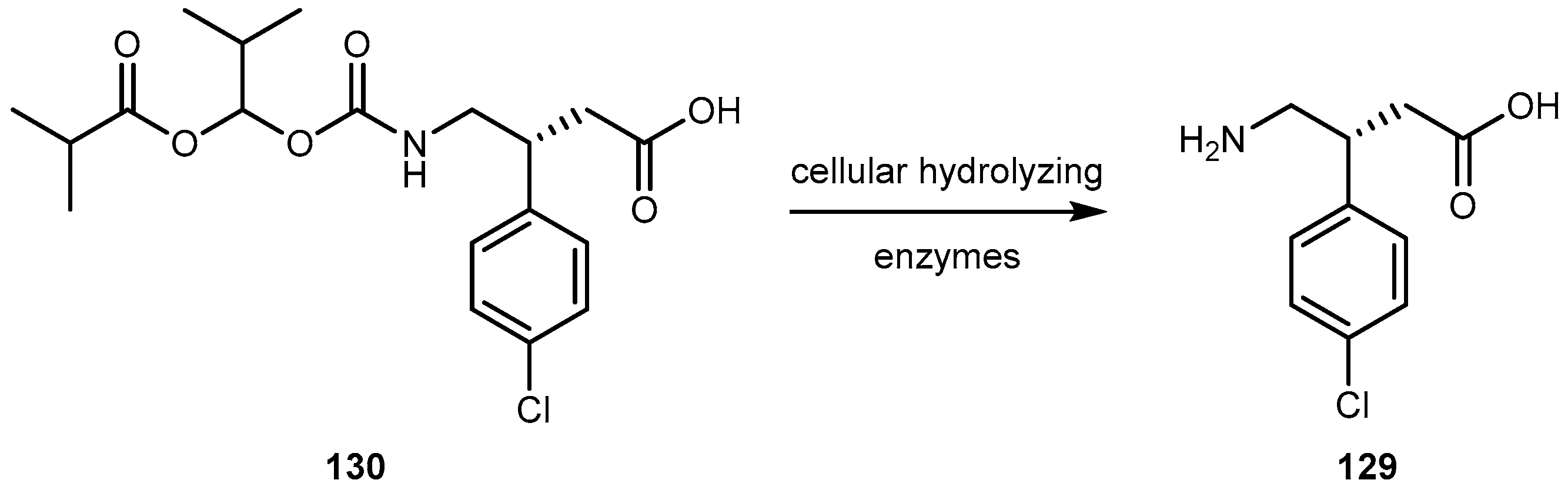{kind=link}
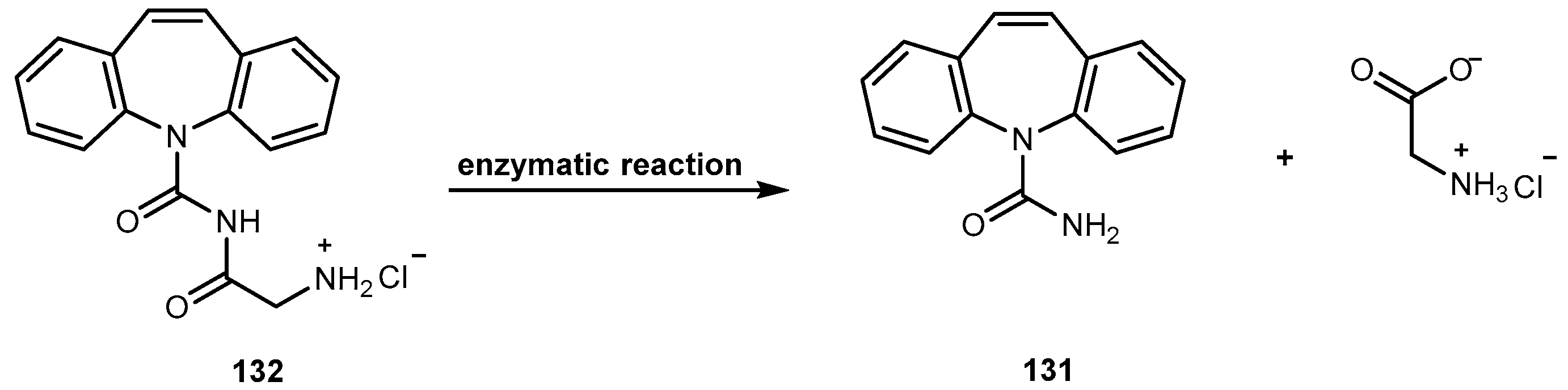{kind=link}
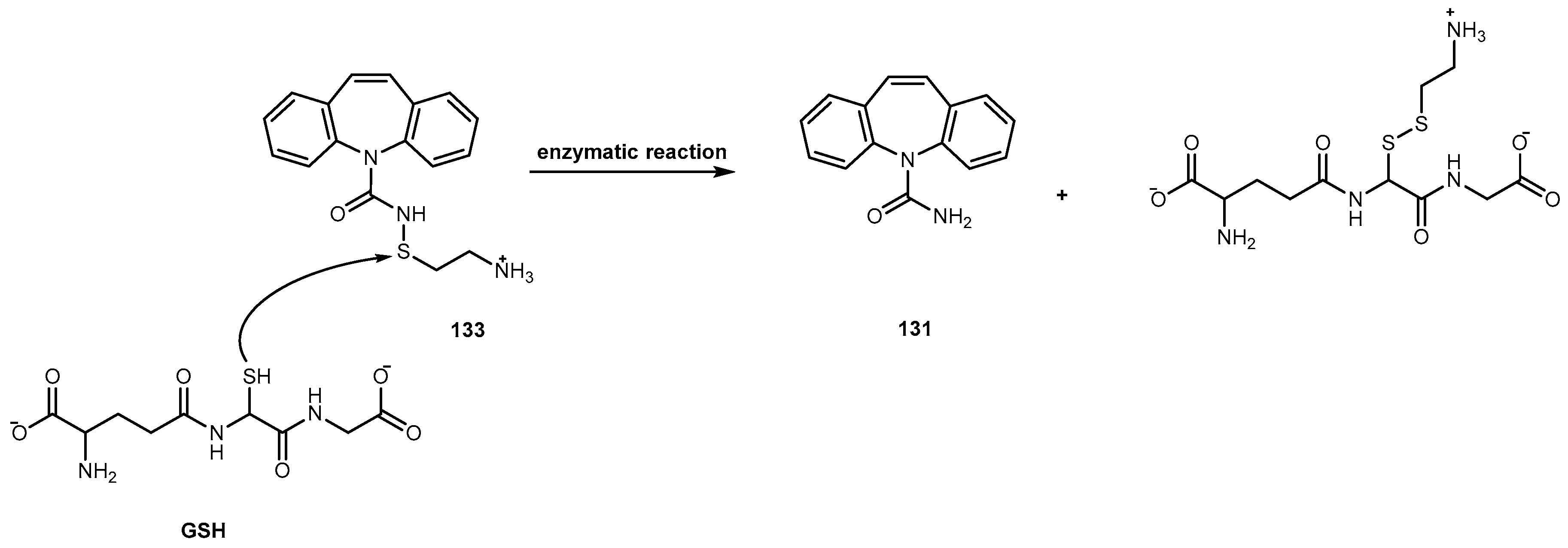{kind=link}
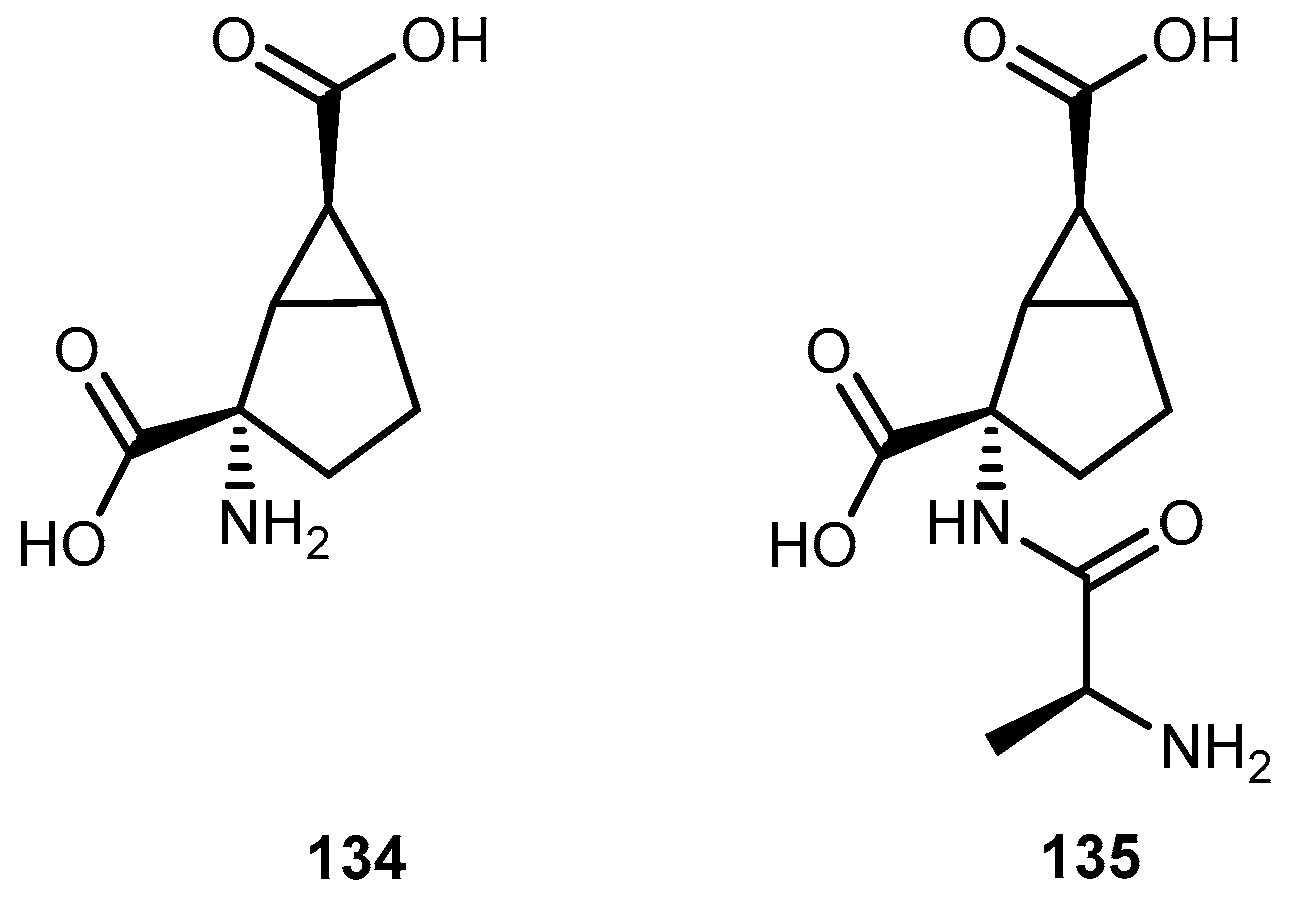{kind=link}
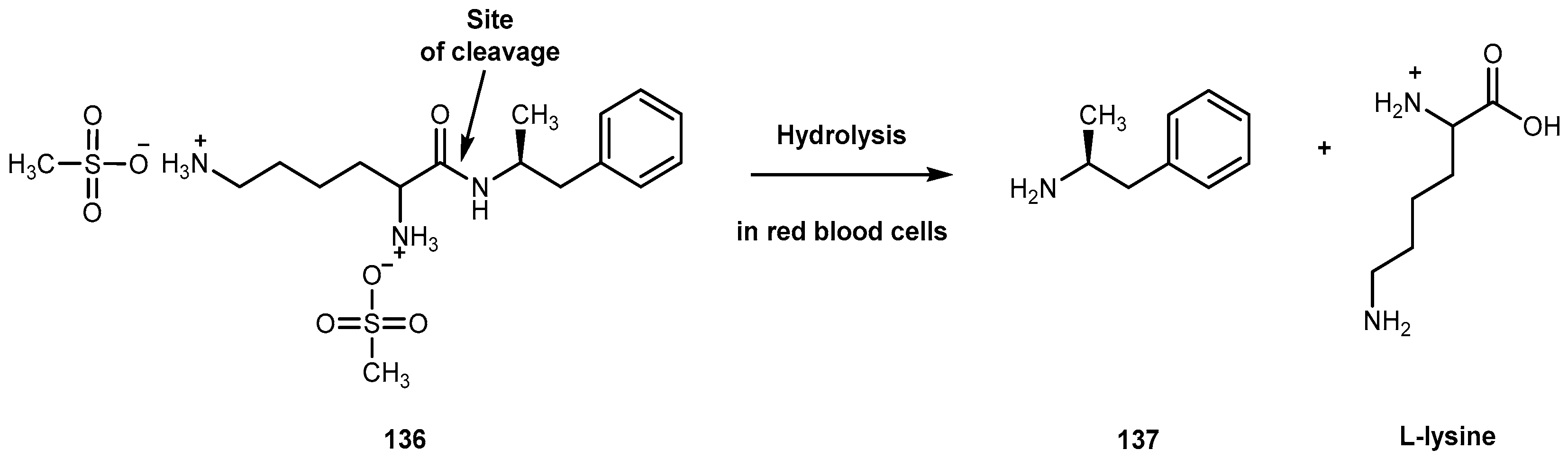{kind=link}
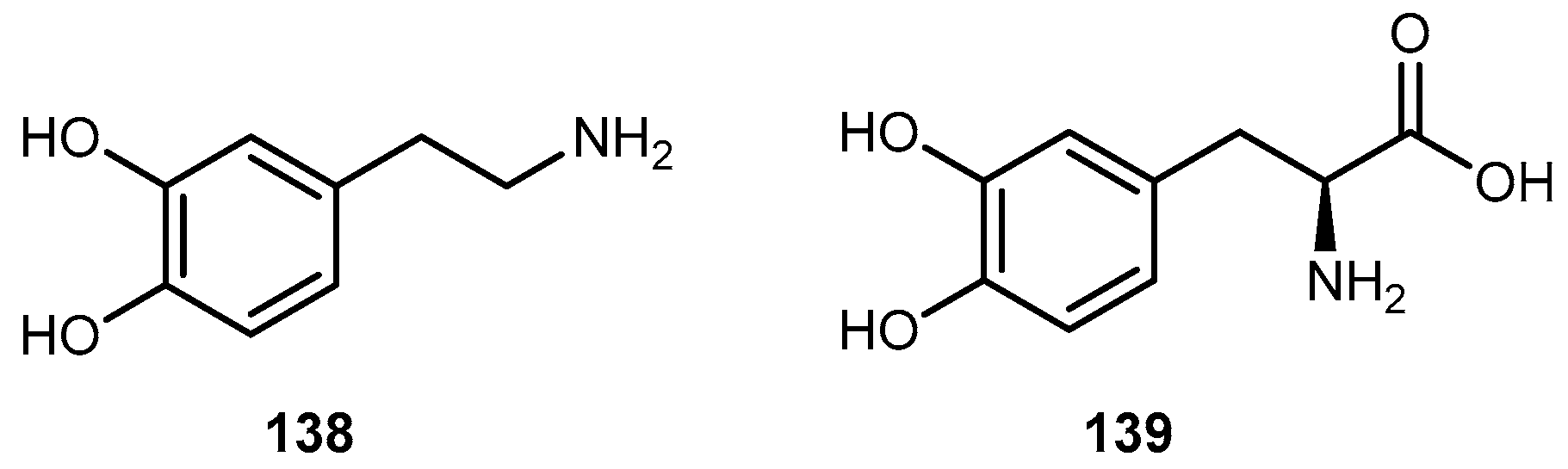{kind=link}
{kind=link}
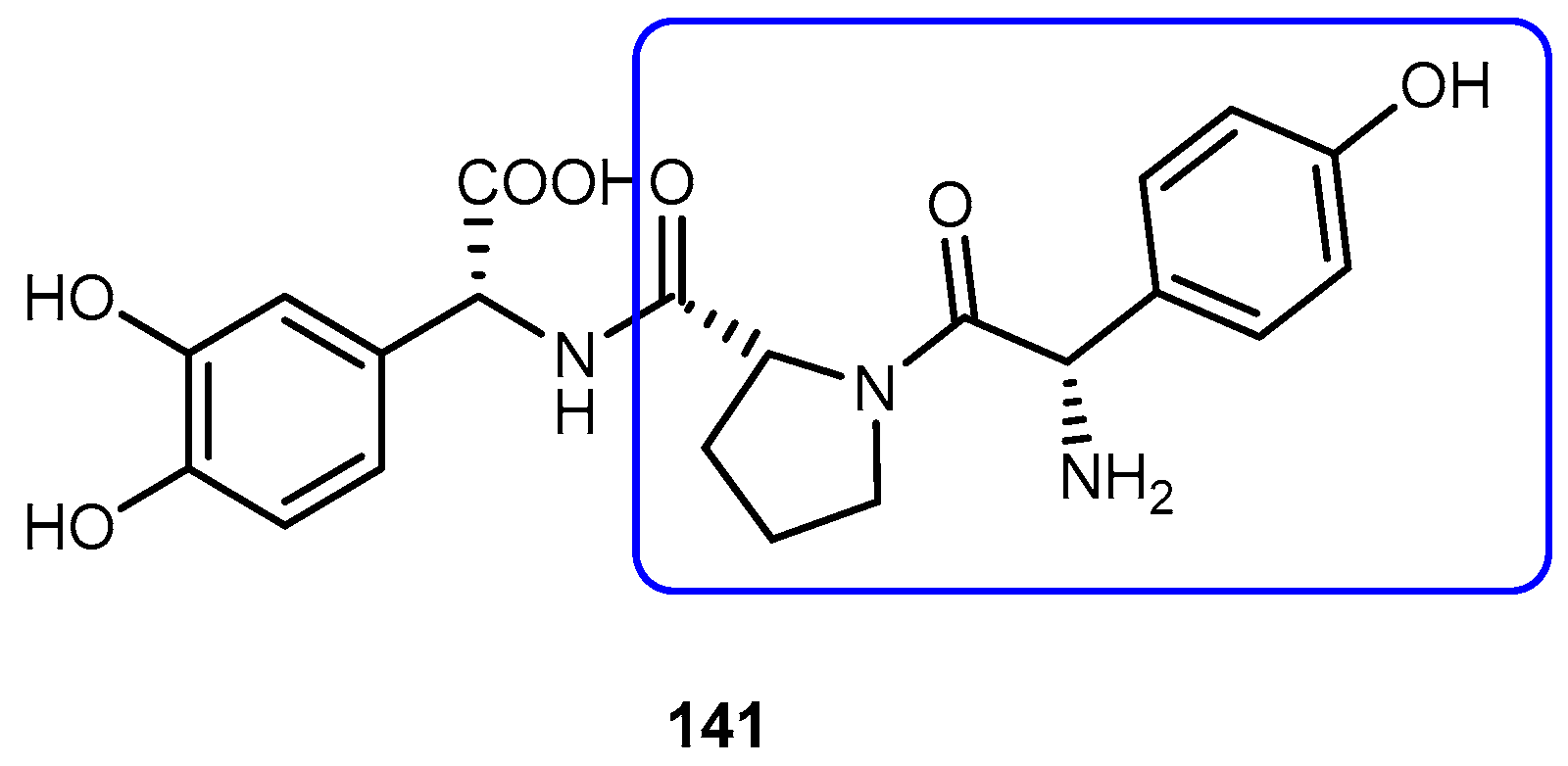{kind=link}
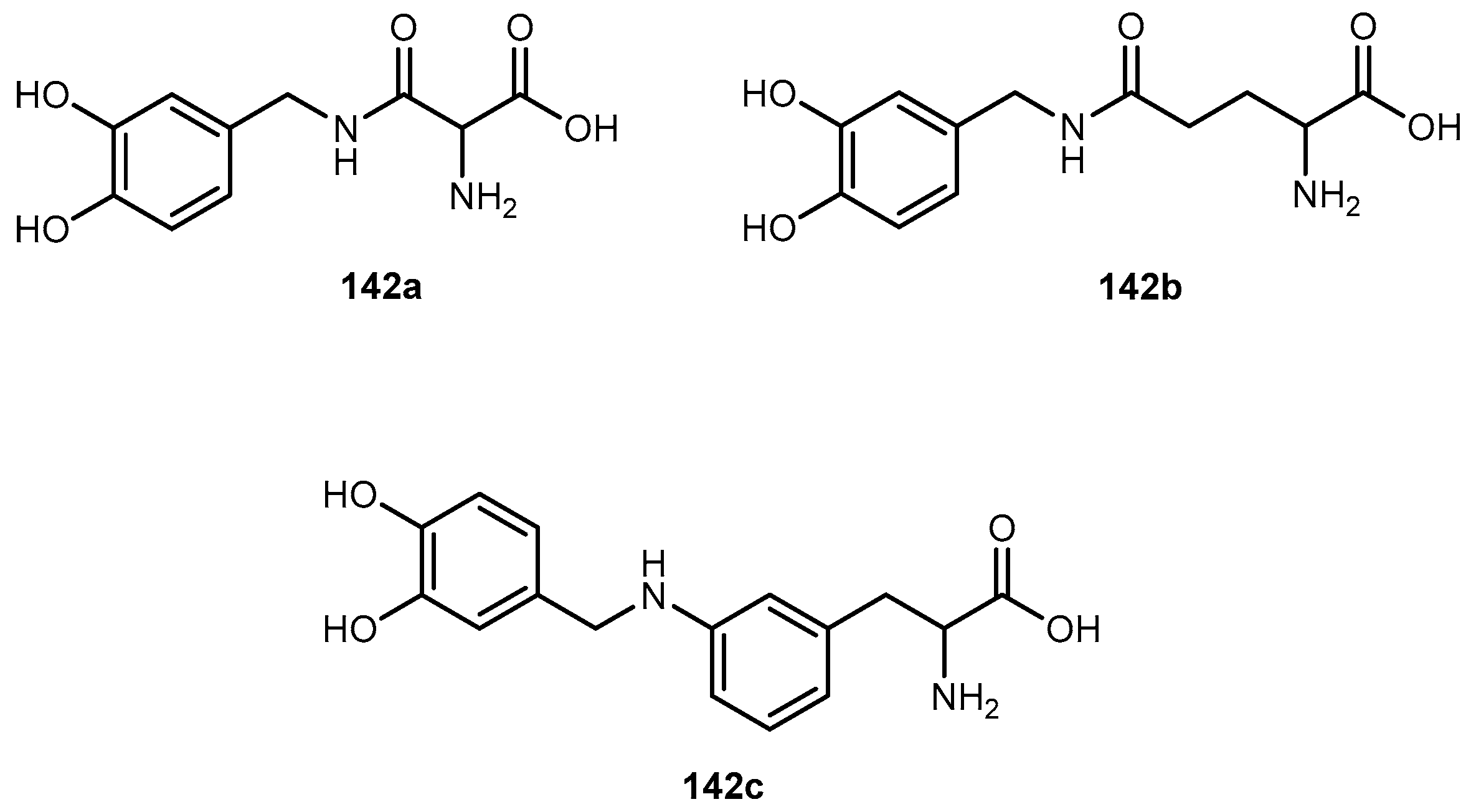{kind=link}
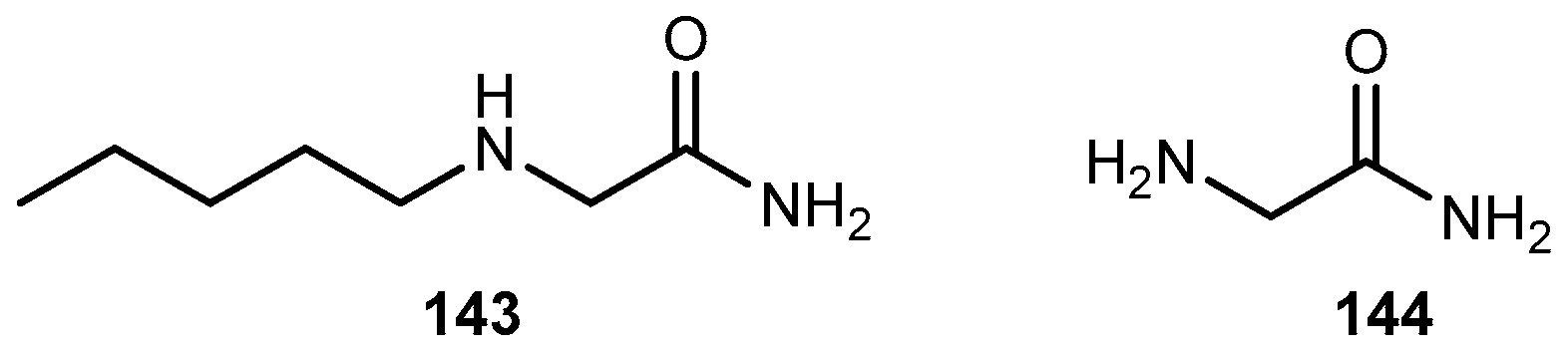{kind=link}
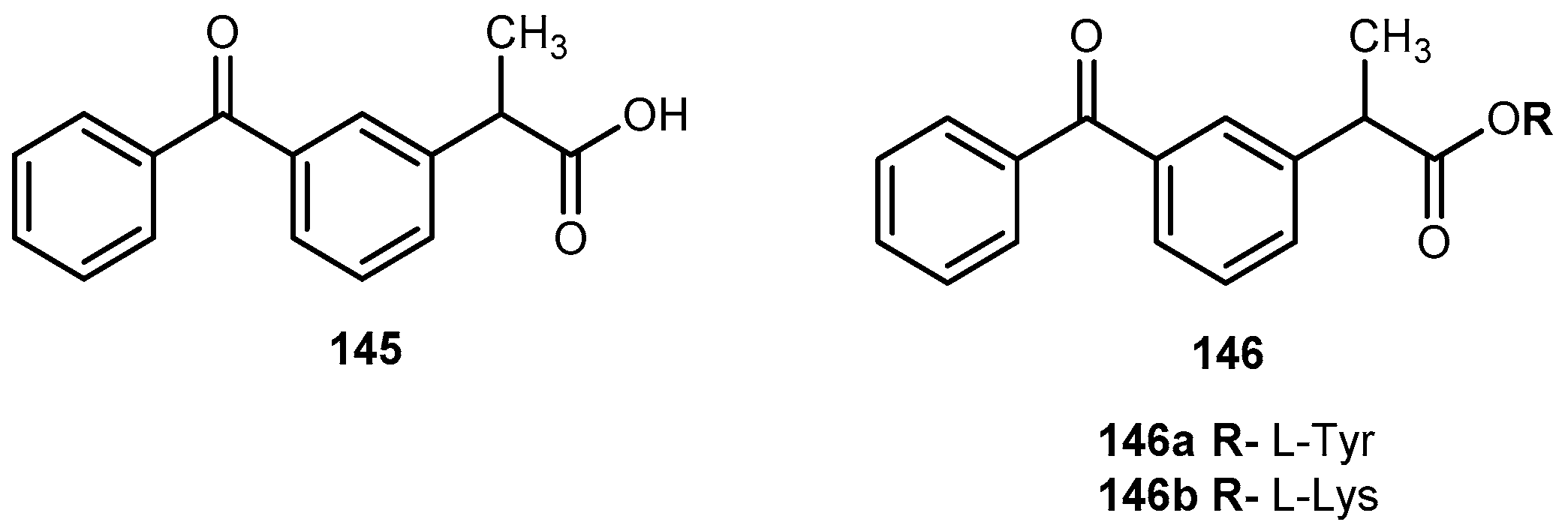{kind=link}
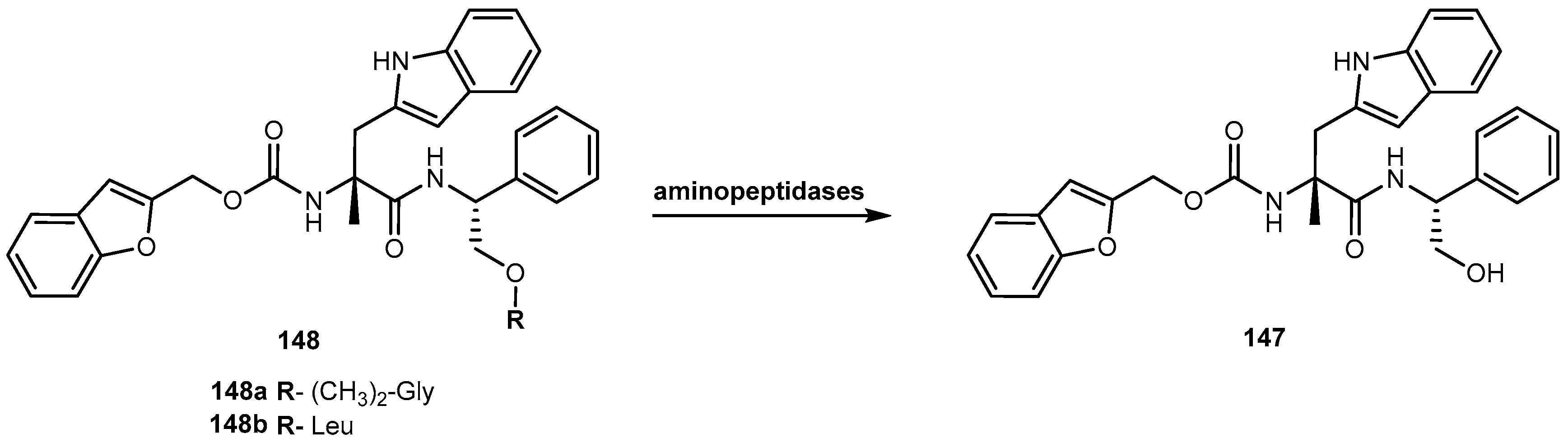{kind=link}
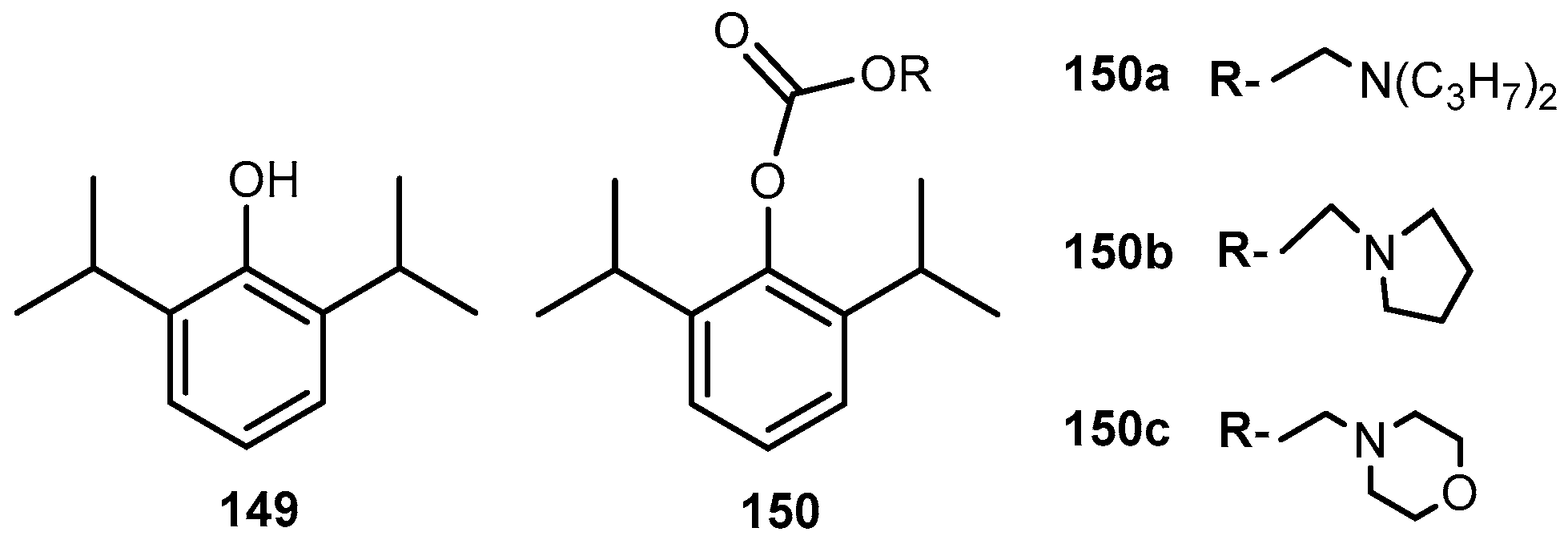{kind=link}
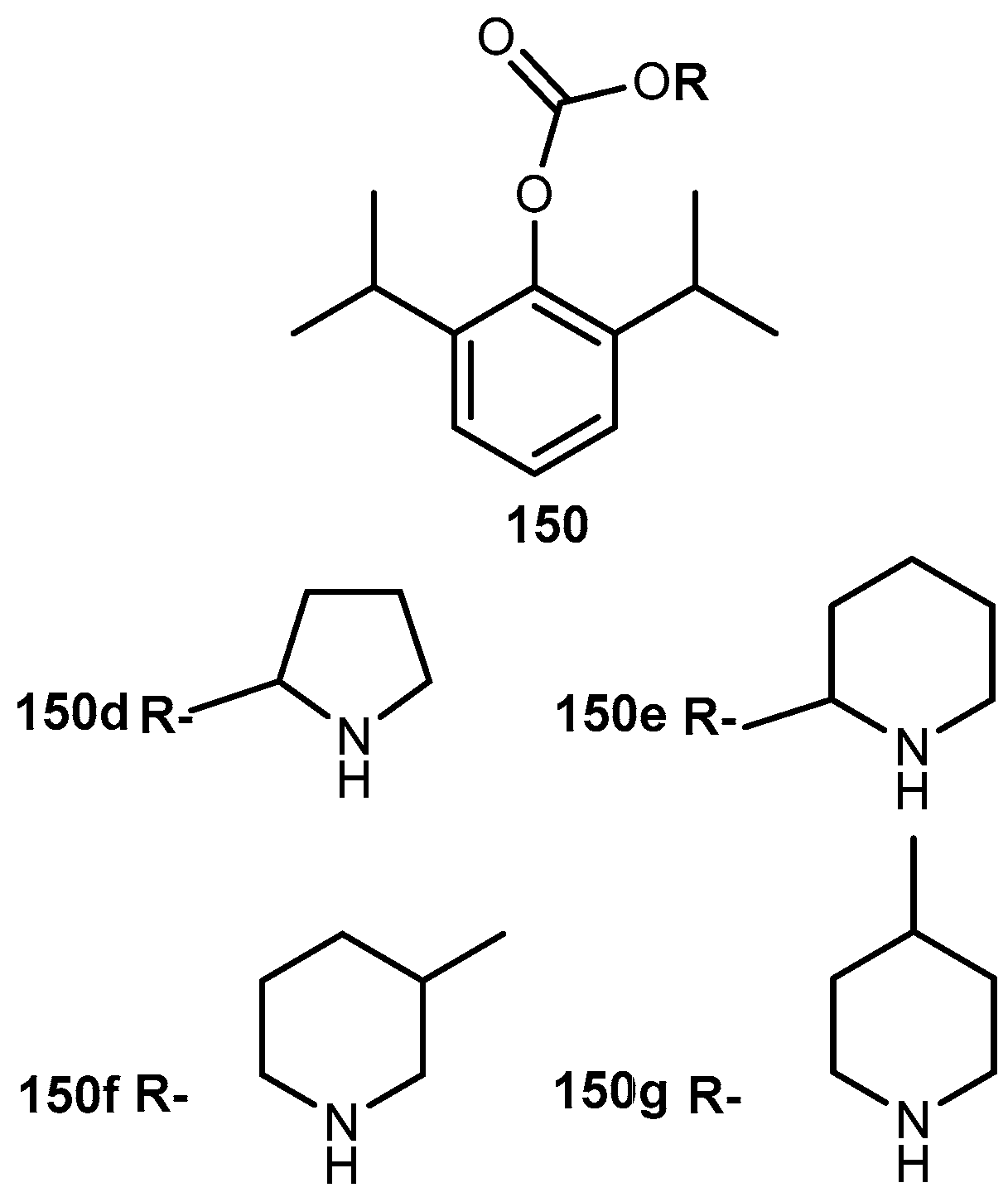{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
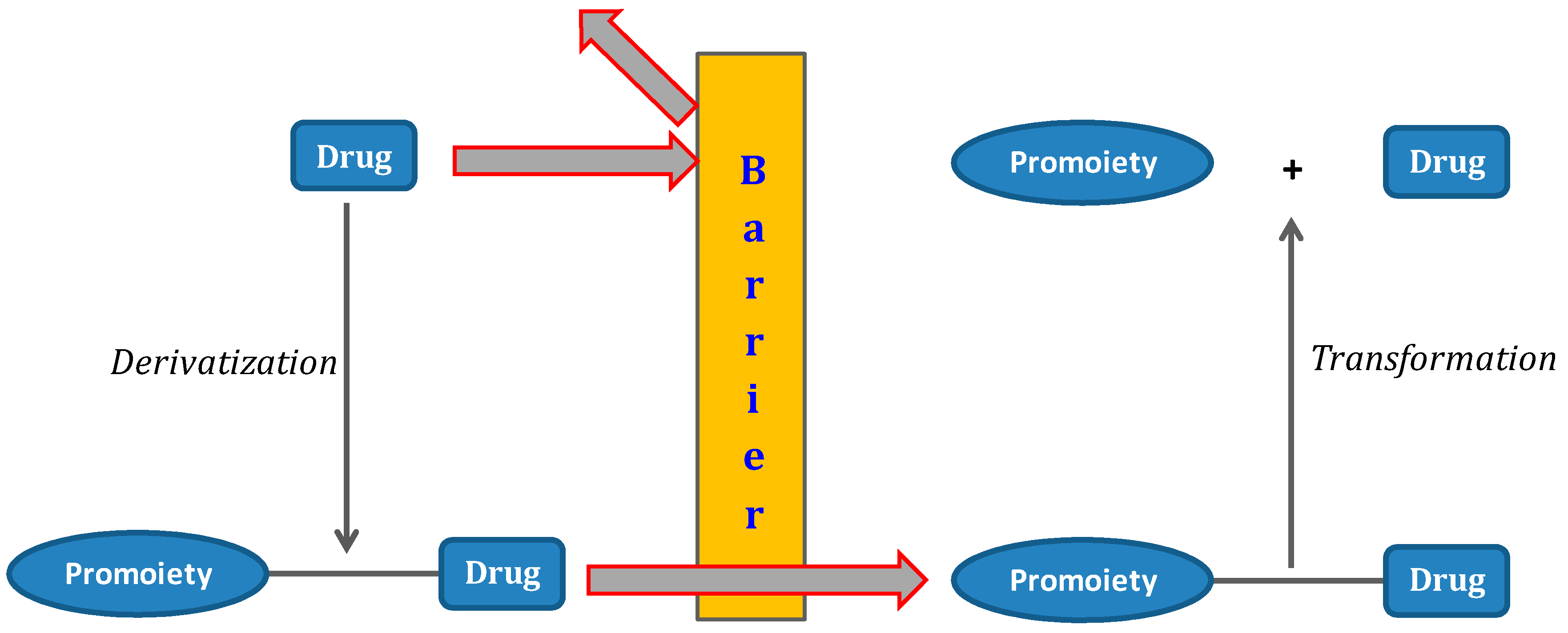
1. The Prodrug Concept: An Overview
2. Amino Acids and Routes of Transport
3. Drugs and Diseases
3.1. Anti-Viral Drugs
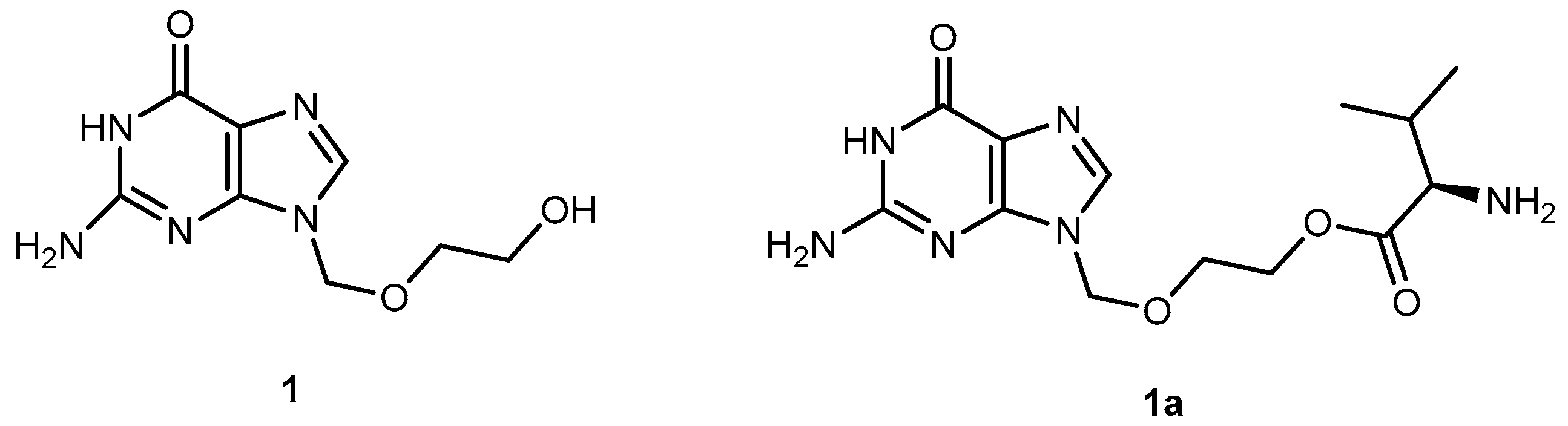
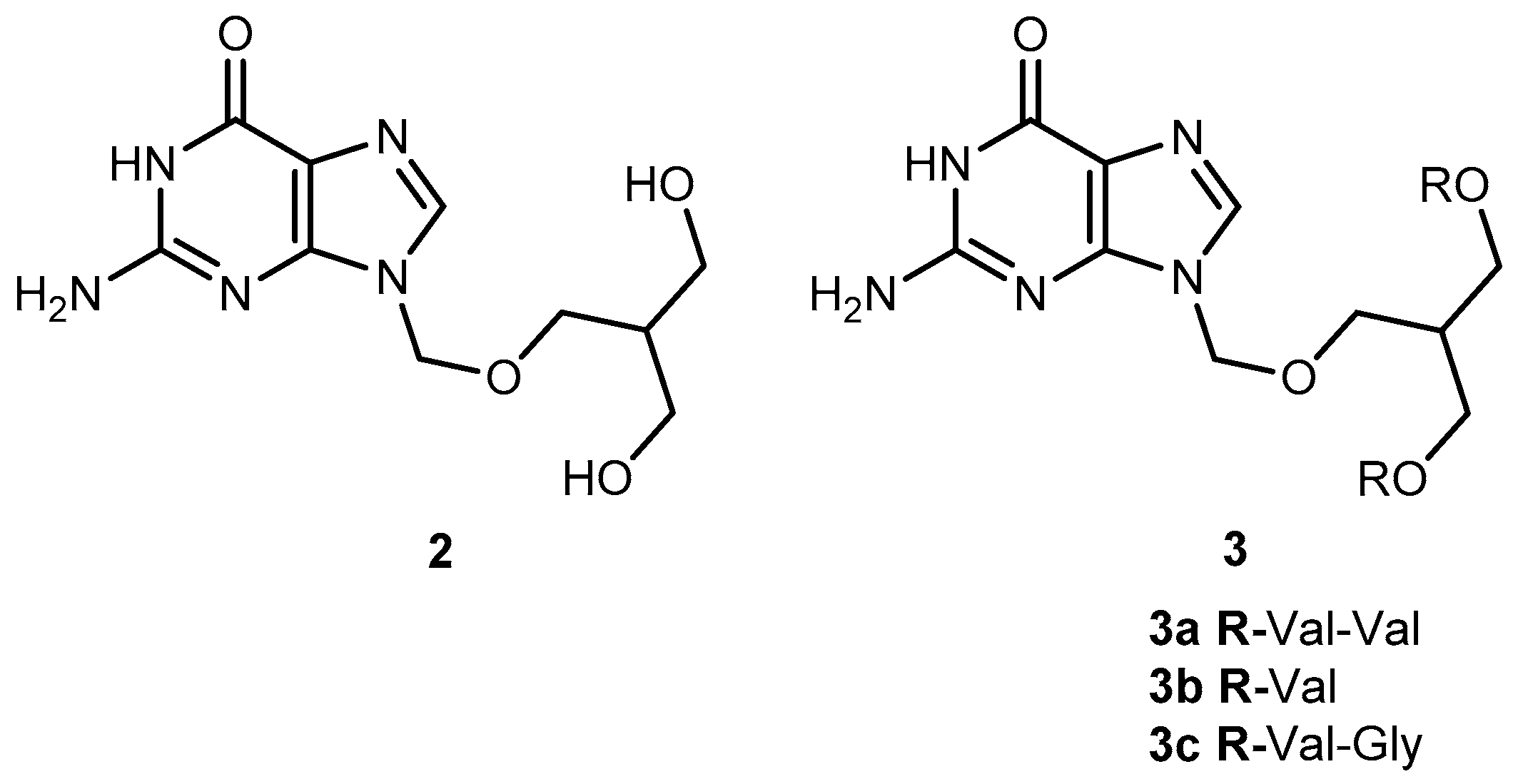
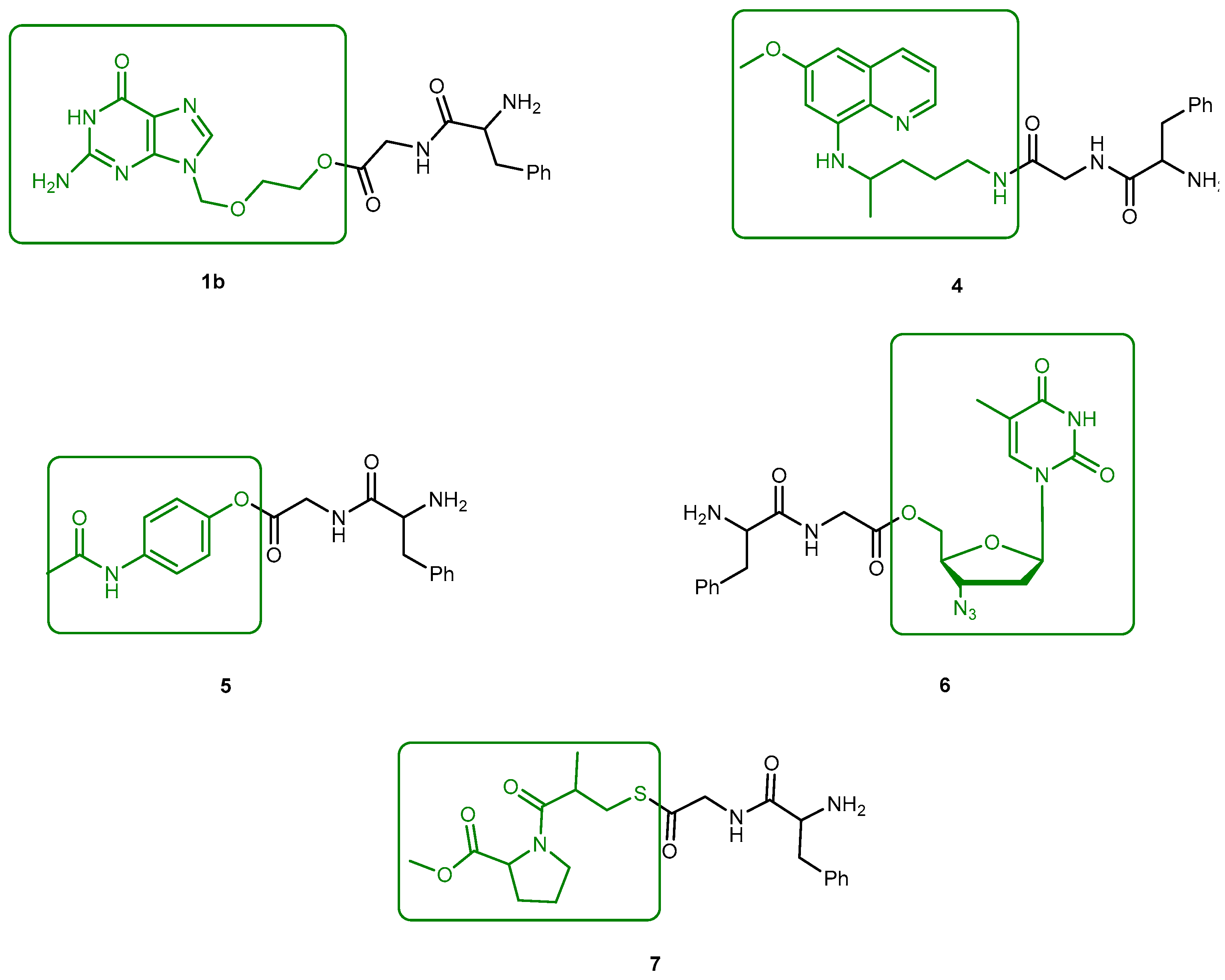
3.1.1. Herpes Viruses Infections (Herpes Simplex or Zoster and Cytomegalovirus)
3.1.2. Human Immunodeficiency Virus (HIV)
3.1.3. Hepatitis Virus (B and C)
3.2. Anticancer
3.2.1. Hepatocellular, Pancreatic Carcinoma and Colon-Rectal Cancer
3.2.2. Brain Cancer
3.2.3. Human Lung Carcinoma
3.2.4. Breast Cancer
3.2.5. Melanoma
3.2.6. Other Compounds
3.3. Antiparasitic Drugs
3.3.1. Trypanosomiasis
3.3.2. Malaria
3.3.3. Leishmaniasis
3.4. Bacterial Diseases
3.4.1. Gram(+) and Gram(−) Microorganisms
3.4.2. Tuberculosis (TB)
3.4.3. Leprosy
3.5. Brain and Central Nervous System (CNS) Diseases
3.5.1. Epilepsy and Bipolar Disease (Anticonvulsants)
3.5.2. Anxiety Disorders
3.5.3. Attention Deficit/Hyperactivity Disorder (ADHD)
3.5.4. Parkinson’s Disease (PD)
3.5.5. Neuropathic Pain
3.5.6. Depression
3.5.7. Anaesthetic
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Han, H.-K.; Amidon, G.L. Targeted prodrug design to optimize drug delivery. AAPS Pharm. Sci. 2000, 2, 48–58. [Google Scholar] [CrossRef]
- Longqin, H. The prodrug approach to better targeting. Curr. Drug Dis. 2004, 4, 28–32. [Google Scholar]
- Rautio, J.; Kumpulainen, H.; Heimbach, T.; Oliyai, R.; Oh, D.; Järvinen, T.; Savolainen, J. Prodrugs: Design and clinical applications. Nat. Rev. Drug Discov. 2008, 7, 255–270. [Google Scholar] [CrossRef] [PubMed]
- Vig, B.S.; Huttunen, K.M.; Laine, K.; Rautio, J. Amino acids as promoities in prodrug design and development. Adv. Drug Deliv. Rev. 2013, 65, 1370–1385. [Google Scholar] [CrossRef] [PubMed]
- Huttunen, K.M.; Raunio, H.; Rautio, J. Prodrugs—From serendipity to rational design. Pharmacol. Rev. 2011, 63, 750–771. [Google Scholar] [CrossRef] [PubMed]
- Mazák, K.; Noszál, B. Drug delivery: A process governed by species-specific lipophilicities. Eur. J. Pharm. Sci. 2014, 62, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Buckley, S.T.; Fischer, S.M.; Fricker, G.; Brandl, M. In vitro models to evaluate the permeability of poorly soluble drug entities: Challenges and perspectives. Eur. J. Pharm. Sci. 2012, 45, 235–250. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Nielsen, P.B.; Müllertz, A.; Norling, T.; Kristensen, H.G. The effect of alpha-tocopherol on the in vitro solubilisation of lipophilic drugs. Int. J. Pharm. 2001, 222, 217–224. [Google Scholar] [CrossRef]
- Saetern, A.M.; Flaten, G.E.; Brandl, M. A method to determine the incorporation capacity of camptothecin in liposomes. AAPS PharmSciTech 2004, 5, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Rathore, M.S.; Gupta, V.B. Functional characterization of amino acid transport system for transport of phenylalanine on mammalian cornea for better ocular drug delivery. J. Pharm. Sci. Res. 2010, 2, 329–337. [Google Scholar]
- Gupta, G.; Gupta, S.V.; Lee, K.D.; Amidon, G.L. Chemical and enzymatic stability of amino acid prodrugs containing methoxy, ethoxy and propylene glycol linkers. Mol. Pharm. 2009, 6, 1604–1611. [Google Scholar] [CrossRef] [PubMed]
- Del Amo, E.M.; Urtti, A.; Yliperttula, M. Pharmacokinetic role of L-type amino acid transporters LAT1 and LAT2. Eur. J. Pharm. Sci. 2008, 35, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Ylikangas, H.; Malmioja, K.; Peura, L.; Gynther, M.; Nwachukwu, E.O.; Leppänen, J.; Laine, K.; Rautio, J.; Lathela-Kakkonen, M.; Huttunen, K.M.; et al. Quantitative insight into design of compounds recognized by L-Type Amino Acids Transporter 1 (LAT1). ChemMedChem 2014, 9, 2699–2707. [Google Scholar] [CrossRef] [PubMed]
- Amidon, G.L.; Lennernäs, H.; Shah, V.P.; Crison, J.R. A theoretical basis for a biopharmaceutic drug classification: The correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm. Res. 1995, 12, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Mitra, A.K. Role of transporters in ocular drug delivery system. Pharm. Res. 2009, 26, 1192–1196. [Google Scholar] [CrossRef] [PubMed]
- Katragadda, S.; Gunda, S.; Hariharau, S.; Mitra, A.K. Ocular pharmacokinetics of acyclovir amino acid ester prodrugs in the anterior chamber. Evaluation of their utility in treating ocular HSV infections. Int. J. Pharm. 2008, 359, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Katragadda, S.; Xiadong, Z.; Ravi, T.S.; Mitra, A.K. Small neutral amino acid ester prodrugs of acyclovir targeting amino acid transporters on the cornea: Possible antiviral agents against ocular HSV-1 infections. Ophthal. Eye Dis. 2010, 2, 43–56. [Google Scholar]
- Bier, D.M.; Young, V.R. A kinetic approach to assessment of amino acid and protein replacement needs of individual sick patient. J. Parenter. Enteral. Nutr. 1987, 11, 95S–97S. [Google Scholar] [CrossRef] [PubMed]
- Hatanaka, T.; Haramura, M.; Fei, Y.J.; Miyauchi, S.; Bridges, C.C.; Ganapathy, P.S.; Smith, S.B.; Ganapathy, V.; Ganapathy, M.E. Transport of amino acid-based prodrugs by the Na+ and Cl− coupled amino acid transporter ATB0,+ and expression of the transporter in tissues amenable for drug delivery. J. Pharmacol. Exp. Ther. 2004, 308, 1138–1147. [Google Scholar] [CrossRef] [PubMed]
- Nashed, Y.E.; Mitra, A.K. Synthesis and characterization of novel dipeptide ester prodrugs of acyclovir. Spectrochim. Acta Part A 2003, 59, 2033–2039. [Google Scholar] [CrossRef]
- Colla, L.; De Clercq, E.; Busson, R.; Vanderhaeghe, H. Synthesis and antiviral activity of water-soluble esters of acyclovir [9-[(2-hydroxyethoxy)methyl]guanine]. J. Med. Chem. 1983, 26, 602–604. [Google Scholar] [CrossRef] [PubMed]
- Maudgal, P.C.; De Clerqc, E.; Descamps, J.; Missotten, L. Topical treatment of experimental herpes simplex keratouveities with 2’-O-glycylacyclovir. A water-soluble ester of acyclovir. Arch. Ophtalmol. 1984, 102, 140–142. [Google Scholar] [CrossRef]
- Shao, Z.; Park, G.-B.; Krishnamoorthy, R.; Mitra, A.K. The physicochemical properties, plasma enzymatic hydrolysis, and nasal absorption of acyclovir and its 2-ester prodrugs. Pharm. Res. 1994, 11, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Mitra, A.K. Regioselective synthesis of acyclovir and its prodrugs. Synth. Commun. 2001, 31, 1399–1419. [Google Scholar] [CrossRef]
- Beutner, K.R.; Friedman, D.J.; Forszpaniak, C.; Andersen, P.L.; Wood, M.J. Valaciclovir compared with acyclovir for improved therapy for herpes zoster in immunocompetent adults. Antimicrob. Agents Chemother. 1995, 39, 1546–1553. [Google Scholar] [CrossRef] [PubMed]
- Soul-Lawton, J.; Seaber, E.; On, N.; Wootton, R.; Rolan, P.; Posner, J. Absolute bioavailability and metabolic disposition of valaciclovir, the L-valyl ester of acyclovir, following oral administration to humans. Antimicrob. Agents Chemother. 1995, 39, 2759–2764. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.H.; Schultz, M.; Weller, S.; Smiley, M.L.; Blum, M.R. Pharmacokinetics and safety of multiple dose valaciclovir in geriatric volunteers with and without concomitant diuretic therapy. Antimicrob. Agents Chemother. 1996, 40, 80–85. [Google Scholar] [PubMed]
- Anand, B.; Nashed, Y.; Mitra, A. Novel dipeptide prodrugs of acyclovir for ocular herpes infections: Bioreversion, antiviral activity and transport across rabbit cornea. Curr. Eye Res. 2003, 26, 151–163. [Google Scholar] [CrossRef] [PubMed]
- Ganapathy, M.E.; Huang, W.; Wang, H.; Ganapathy, V.; Leibach, F.H. Valacyclovir: A substrate for the intestinal and renal peptide transporters PEPT1 and PEPT2. Biochem. Biophys. Res. Commun. 1998, 246, 470–475. [Google Scholar] [CrossRef] [PubMed]
- Han, H.K.; Oh, D.M.; Amidon, G.L. Cellular uptake mechanism of amino acid ester prodrugs in Caco-2/hPEPT1 cells overexpressing a human peptide transporter. Pharm. Res. 1998, 15, 1382–1386. [Google Scholar] [CrossRef] [PubMed]
- Vrueh, R.L.; Smith, P.L.; Lee, C.P. Transport of L-valine-acyclovir via the oligopeptide transporter in the human intestinal cell line Caco-2. J. Pharmacol. Exp. Ther. 1998, 286, 1166–1170. [Google Scholar] [PubMed]
- Han, H.; Oh, D.M.; Amidon, G.L. 5′-amino acid esters of antiviral nucleosides, acyclovir, and AZT are absorbed by the intestinal PEPT1 peptide transporter. Pharm. Res. 1998, 15, 1154–1159. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.; Trivedi, S.; Luo, S.; Zhu, X.; Pal, D.; Kern, E.R.; Mitra, A.K. Synthesis, physicochemical properties and antiviral activities of ester prodrugs of ganciclovir. Int. J. Pharm. 2005, 305, 75–89. [Google Scholar] [CrossRef] [PubMed]
- Santos, C.R.; Capela, R.; Pereira, C.S.; Valente, E.; Gouveia, L.; Pannecouque, C.; De Clercq, E.; Moreira, R.; Gomes, P. Structure-activity relationships for dipeptide prodrugs of acyclovir: Implications for prodrug design. Eur. J. Med. Chem. 2009, 44, 2339–2346. [Google Scholar] [CrossRef] [PubMed]
- Larsen, S.W.; Ankersen, M.; Larsen, C. Kinetics of degradation and oil solubility of ester prodrugs of a model dipeptide (Gly-Phe). Eur. J. Pharm. Sci. 2004, 22, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Portela, M.J.; Moreira, R.; Valente, E.; Constantino, L.; Iley, J.; Pinto, J.; Rosa, R.; Cravo, P.; Rosário, V.E. Dipeptide derivatives of primaquine as transmission-blocking antimalarials: Effect of aliphatic side-chain acylation on the gametocytocidal activity and on the formation of carboxyprimaquine in rat liver homogenates. Pharm. Res. 1999, 16, 942–954. [Google Scholar] [CrossRef]
- Thomsen, A.E.; Christensen, M.S.; Bagger, M.A.; Steffansen, B. Acyclovir prodrug for the intestinal di/tri-peptide transporter PEPT1: Comparison of the in vivo bioavailability in rats and transport in Caco-2 cells. Eur. J. Pharm. Sci. 2004, 23, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Steingrimsdottir, H.; Gruber, A.; Palm, C.; Grimfors, G.; Kalin, M.; Eksborg, S. Bioavailability of aciclovir after oral administration of aciclovir and its prodrugs valaciclovir to patients with leucopenia after chemotherapy. Antimicrob. Agents Chemother. 2000, 44, 207–209. [Google Scholar] [CrossRef] [PubMed]
- Höglund, M.; Ljungman, P.; Weller, S. Comparable aciclovir exposures by oral valaciclovir and intravenous acyclovir in immunocompromised cancer patients. J. Antimicrob. Chemother. 2001, 47, 855–861. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Kim, J.-S.; Kish, P.E.; Zhang, J.; Mitchell, S.; Gentry, B.G.; Breitenbach, J.M.; Drach, J.C.; Hilfinger, J. Design and synthesis of vidarabine prodrugs as antiviral agents. Bioorg. Med. Chem. Lett. 2009, 19, 792–796. [Google Scholar] [CrossRef] [PubMed]
- Boyd, M.R.; Bacon, T.H.; Sutton, D.; Cole, M. Antiherpesvirus activity of 9-(4-hydroxy-3-hydroxy-methylbut-1-yl)guanine (BRL39123) in cell culture. Antimicrob. Agents Chemother. 1987, 31, 1238–1242. [Google Scholar] [CrossRef] [PubMed]
- Harnden, M.R.; Jarvest, R.L.; Boyd, M.R.; Sutton, D.; Hodge, R.A.V. Prodrugs of selective antiherpesvirus agent 9-[4-hydroxy-3-(hydroxymethoxyl)but-1-yl]guanidine (BRL-39123) with improved gastroinstestinal absorption properties. J. Med. Chem. 1989, 30, 1738–1743. [Google Scholar] [CrossRef]
- Beauchamp, L.M.; Orr, G.F.; de Miranda, P.; Burnette, T.; Krenitsky, T.A. Amino acid ester prodrugs of acyclovir. Antiviral Chem. Chemother. 1992, 3, 157–169. [Google Scholar] [CrossRef]
- Kim, D.-K.; Lee, N.; Im, G.J.; Kim, Y.-G.; Chang, K.; Kim, H.-T.; Cho, Y.-G.; Choi, W.-S.; Jung, I.; Kim, K.H. Synthesis and evaluation of amino acid ester prodrugs of penciclovir. Bioorg. Med. Chem. Lett. 1996, 6, 1849–1854. [Google Scholar] [CrossRef]
- Zou, R.; Drach, J.C.; Townsend, L.B. Design, synthesis and viral evaluation of 2-chloro-5,6-dihalo-1-(β-d-ribofuranosyl)-benzimidazoles as potential agents for human cytomegalovirus infection. J. Med. Chem. 1997, 40, 811–818. [Google Scholar] [CrossRef] [PubMed]
- Underwood, M.R.; Harvey, R.J.; Stanat, S.C.; Hemphil, M.L.; Miller, T.; Drach, J.C.; Townsend, L.B.; Biron, K.K. Inhibition of human cytomegalovirus DNA maturation by a benzimidazole ribonucleoside is mediated through the UL89 gene product. J. Virol. 1998, 72, 717–725. [Google Scholar] [PubMed]
- Song, X.; Vig, B.S.; Lorenzi, P.L.; Drach, J.C.; Townsend, L.B.; Amidon, G.L. Amino acid ester prodrugs of the antiviral agent 2-bromo-5,6-dichloro-1-(β-d-ribofuranosyl)benzimidazole as potential substrates of hPEPT1 transporter. J. Med. Chem. 2005, 48, 1274–1277. [Google Scholar] [CrossRef] [PubMed]
- Lorenzi, P.L.; Landowski, C.P.; Song, X.; Borysko, K.Z.; Breitenbach, J.M.; Kim, J.S.; Hilfinger, J.M.; Townsend, L.B.; Drach, J.C.; Amidon, G.L. Amido acid ester prodrugs of 2-bromo-5,6-dichloro-1-(β-d-ribofuranosyl)benzimidazole enhance metabolic stability in vitro and in vivo. J. Pharm. Exp. Ther. 2005, 314, 883–890. [Google Scholar] [CrossRef] [PubMed]
- McKenna, C.E.; Kashemirov, B.A.; Eriksson, U.; Amidon, G.L.; Kish, P.E.; Mitchell, S.; Kim, J.-S.; Hilfinger, J.M. Cidofovir peptide conjugates as prodrugs. J. Organomet. Chem. 2005, 690, 2673–2678. [Google Scholar] [CrossRef]
- De Clerqc, E. Antiviral drugs: Current state of the art. J. Clin. Virol. 2001, 22, 73–89. [Google Scholar] [CrossRef]
- Hasegawa, T.; Kawaguchi, T. Future of prodrugs in antiviral therapy. Clin. Pharmacokinet. 1994, 27, 331–336. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization (WHO). Malaria and HIV Interactions and Their Implications for Public Health Policy; Report of a Technical Consultation; WHO: Geneva, Switzerland, 2005. [Google Scholar]
- Fischl, M.A.; Richman, D.D.; Grieco, M.H.; Gottlieb, M.S.; Volberding, P.A.; Laskin, O.L.; Leedom, J.M.; Groopman, J.E.; Mildvan, D.; Schooley, R.T.; et al. The efficacy of azidothymidine (AZT) in treatment of patients with AIDS and AIDS-related complex. A double-blind, placebo-controlled trial. N. Engl. J. Med. 1987, 317, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Mohan, P. Problems and perspectives in the design of anti-HIV-1 agents. Drug Dev. Rev. 1993, 29, 1–17. [Google Scholar] [CrossRef]
- Klecker, R.W., Jr.; Collins, J.M.; Yarchoan, R.; Thomas, R.; Jenkins, J.F.; Broder, S.; Meyers, C.E. Plasma and cerebrospinal fluid pharmacokinetics of 3′-azido-3′-deoxythymidine: A novel pyrimidine analog with potential application for the treatment of patients with AIDS and related diseases. Clin. Pharmacol. Ther. 1987, 41, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Styrt, B.A.; Piazza-Hepp, T.D.; Chikami, G.K. Clinical toxicity of antiretroviral nucleoside analogs. Antivir. Res. 1996, 31, 121–135. [Google Scholar] [CrossRef]
- Turk, G.; Moroni, G.; Pampuro, S.; Briñon, M.C.; Salomón, H. Antiretroviral activity and cytotoxicity of novel zidovudine (AZT) derivatives and their relation to their chemical structure. Int. J. Antimicrob. Agents 2002, 20, 282–288. [Google Scholar] [CrossRef]
- Santos, C.; Capela, R.; Pereira, C.S.; Valente, E.; Gouveia, L.; Pannecouque, C.; De Clercq, E.; Moreira, R.; Gomes, P. Dipeptide derivatives of AZT: Synthesis, chemical stability, activation in human plasma, hPEPT1 affinity, and antiviral activity. ChemMedChem 2008, 3, 970–978. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, L.; Luo, T.; Zhou, J.; Sun, L.; Xu, Y. Synthesis and evaluation of a dipeptide-drug conjugate library as substrates for PEPT1. ACS Comb. Sci. 2012, 14, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Lai, L.; Xu, Z.; Zhou, J.; Lee, K.D.; Amidon, G.L. Molecular basis of prodrug activation by human valacyclovirase, an alpha-amino acid ester hydrolase. J. Biol. Chem. 2008, 283, 9318–9327. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Sharon, A.; Grier, J.P.; Rapp, K.L.; Schinazi, R.F.; Chu, C.K. 5’-O-aliphatic and amino acid ester prodrugs of (−)-β-d-(2R,4R)-dioxolane-thymide (DOT): Synthesis, anti-HIV activity, cytotoxicity and stability studies. Bioorg. Med. Chem. 2009, 17, 1404–1409. [Google Scholar] [CrossRef] [PubMed]
- Kumar, G.N.; Jayanti, V.K.; Johnson, M.K.; Uchic, J.; Thomas, S.; Lee, R.D.; Grabowski, B.A.; Sham, H.L.; Kempf, D.-J.; Denissen, J.F.; et al. Metabolism and disposition of the HIV-1 protease inhibitor lopinavir (ABT-378) given in combination with ritonavir in rats, dogs, and humans. Pharm. Res. 2004, 21, 1622–1630. [Google Scholar] [CrossRef] [PubMed]
- Argawal, S.; Boddu, S.H.; Jain, R.; Samanta, S.; Pal, D.; Mitra, A.K. Peptide prodrugs: Improved oral absorption of lopinavir, a HIV protease inhibitor. Int. J. Pharm. 2008, 359, 7–14. [Google Scholar]
- Lavanchy, D. Hepatitis B virus epidemiology, disease burden, treatment, and current and emerging prevention and control measures. J. Virol. Hep. 2004, 11, 97–107. [Google Scholar] [CrossRef]
- Chen, Z.; Zheng, M. Patents and development of HBV and HCV clinical treatment: From 2001 to April 2005. Exp. Opin. Ther. Patents 2005, 15, 1027–1039. [Google Scholar] [CrossRef]
- Hwang, J.-T.; Choi, J.-R. Novel phosphate nucleoside as antiviral agents. Drugs Fut. 2004, 29, 163–177. [Google Scholar] [CrossRef]
- Fu, X.; Jiang, S.; Li, C.; Xin, J.; Yang, Y.; Ji, R. Design and synthesis of novel bis(L-amino acid) ester prodrug of 9-[2-phosphonomethoxy)ethyl)adenine (PMEA) with improved anti HBV-activity. Bioorg. Med. Chem. Lett. 2007, 17, 465–470. [Google Scholar] [CrossRef] [PubMed]
- Hodge, R.A. Telbivudine/torcitabine idenix/Novartis. Curr. Opin. Investig. Drug 2004, 5, 232–241. [Google Scholar]
- Pierra, C.; Amador, A.; Benzaria, S.; Cretton-Scott, E.; D’Amours, M.; Mao, J.; Mathieu, S.; Moussa, A.; Bridges, E.G.; Standring, D.N.; et al. Synthesis and pharmacokinetics of valopicitabine (NM 283), an efficient prodrug of the potent anti-HCV agent 2’-C-methylcytidine. J. Med. Chem. 2006, 49, 6614–6620. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Maag, H.; Alfredson, T. Prodrugs of nucleoside analogues for improved oral absorption and tissue targeting. J. Pharm. Sci. 2008, 97, 1109–1134. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.J.; Knox, S.; Fielman, B.; Chao, G.; Brown, N. Safety and pharmacokinetics of NM107 following intravenous infusion of escalating doses in healthy volunteers: Determination of absolute oral bioavailability of valopicitabine (NM283). J. Hepatol. 2006, 44, S231–S232. [Google Scholar] [CrossRef]
- Sorbera, L.A.; Castaner, J.; Leeson, P.A. Valopicitabine: Anti-hepatitis C virus drug, RNA-directed RBA polymerase (NS5B) inhibitor. Drugs Fut. 2006, 3, 320–324. [Google Scholar] [CrossRef]
- Sinhababu, A.K.; Thakker, D.R. Prodrugs of anticancer agents. Adv. Drug Deliv. Res. 1996, 19, 241–273. [Google Scholar] [CrossRef]
- Landowski, C.P.; Lorenzi, P.L.; Song, X.; Amidon, G.L. Nucleoside ester prodrugs substrate specificity of liver carboxylesterase. J. Pharmacol. Exp. Ther. 2006, 316, 241–273. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, D.E.; Covitz, K.-M.; Sadée, W.; Mrsny, R.J. An oligopeptide transporter is expressed at high levels in the pancreatic carcinoma cell lines AsPc-1 and Capan-2. Cancer Res. 1998, 58, 519–525. [Google Scholar] [PubMed]
- Nakanishi, T.; Tamai, I.; Takaki, A.; Tsuji, A. Cancer cell-targeted drug delivery utilizing oligopeptide transport activity. Int. J. Cancer 2000, 88, 274–280. [Google Scholar] [CrossRef]
- Vig, B.S.; Lorenzi, P.L.; Mittal, S.; Landowski, C.P.; Shin, H.-C.; Mosberg, H.I.; Hilfinger, J.M.; Amidon, G.L. Amino acid ester prodrugs of floxuridine: Synthesis and effects of structure, stereochemistry and site of esterification on the rate of hydrolysis. Pharm. Res. 2003, 20, 1381–1388. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Lorenzi, P.L.; Landowski, C.P.; Vig, B.S.; Hilfinger, J.M.; Amidon, G.L. Amino acid ester prodrug of the anticancer agent gemcitabine: Synthesis, bioconversion, metabolic bioevasion and hPEPT1-mediated transport. Mol. Pharm. 2005, 2, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Landowski, C.P.; Vig, B.S.; Song, X.; Amidon, G.L. Targeted delivery to PEPT1-overexpresssing cells: Acidic, basic, and secondary floxuridine amino acid ester prodrugs. Mol. Cancer Ther. 2005, 4, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Landowski, C.P.; Song, X.; Lorenzi, P.L.; Hilfinger, J.M.; Amidon, G.L. Floxuridine amino acid ester prodrugs: Enhancing Caco-2 permeability and resistance to glycosidic bond metabolism. Pharm. Res. 2005, 22, 1510–1518. [Google Scholar] [CrossRef] [PubMed]
- Tsume, Y.; Hilfinger, J.M.; Amidon, G.L. Enhanced cancer cell growth inhibition by dipeptide prodrugs of Floxuridine: Increased transporter affinity and metabolic stability. Mol. Pharm. 2008, 5, 717–727. [Google Scholar] [CrossRef] [PubMed]
- Tsume, Y.; Hilfinger, J.M.; Amidon, G.L. Potential of amino acid/dipeptide monoester prodrug of floxuridine in facilitating enhanced delivery of active drug to interior sites of tumors: A two-tier monololayer in vitro stability. Pharm. Res. 2011, 28, 2575–2588. [Google Scholar] [CrossRef] [PubMed]
- Tsume, Y.; Amidon, G.L. The feasibility of enzyme targeted activation for amino acid/dipeptide monoester prodrugs of floxuridine; cathepsin D as potential targeted enzyme. Molecules 2012, 17, 3672–3689. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Chubb, S.; Hertel, L.W.; Grindey, G.B.; Plunkett, W. Action of 2′,2′-difluorodeoxycytidine on DNA synthesis. Cancer Res. 1991, 51, 6110–6117. [Google Scholar] [PubMed]
- Tsume, Y.; Incecayir, T.; Song, X.; Hilfinger, S.M.; Amidon, G.L. The development of orally administrable gemcitabine prodrugs with D-enantiomer amino acid: Enhanced membrane permeability and enzymatic stability. Eur. J. Pharm. Sci. 2014, 86, 514–523. [Google Scholar] [CrossRef] [PubMed]
- Vale, N.; Ferreira, A.; Fernandes, I.; Alves, C.; Araújo, M.J.; Mateus, N.; Gomes, P. Gemcitabine anti-proliferative activity significantly enhanced upon conjugation with cell-penetrating peptides. Bioorg. Med. Chem. Lett. 2017, 27, 2898–2901. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Padilla, I.; Siu, L.L. Brivanib alaninate for cancer. Exp. Opin. Investig. Drugs 2011, 20, 577–586. [Google Scholar] [CrossRef] [PubMed]
- Huynth, H.; Fargnoli, J. Brivanib alaninate: VEGFR/FGFR inhibitor oncolytic. Drug Fut. 2009, 34, 881–895. [Google Scholar] [CrossRef]
- Marathe, P.H.; Kamath, A.V.; Zhang, Y.; D’Arienzo, C.; Bhide, R.; Fargnoli, J. Preclinical pharmacokinetics and in vitro metabolism of brivanib (BMS-540215) a potent VEGFR2 inhibitor and its alanine ester prodrug brivanib alaninate. Cancer Chemother. Pharmacol. 2009, 65, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Gan, J.; Caceres-Cortes, J.; Christopher, L.J.; Arora, V.; Masson, E.; Williams, D.; Pursley, J.; Allentoff, A.; Lago, M.; et al. Metabolism and disposition of [14C] brivanib alaninate after oral administration to rats, monkeys, and humans. Drug Metab. Dispos. 2011, 39, 891–903. [Google Scholar] [CrossRef] [PubMed]
- Vale, N.; Correia-Branco, A.; Patrício, B.; Duarte, D.; Martel, F. In vitro studies on the inhibition of colon cancer by amino acid derivatives of bromothiazole. Bioorg. Med. Chem. Lett. 2017, 27, 3507–3510. [Google Scholar] [CrossRef] [PubMed]
- Senkowski, W.; Zhang, X.; Olofsson, M.H.; Isacson, R.; Höglund, U.; Gustafsson, M.; Nygren, P.; Linder, S.; Larsson, R.; Frynknäs, M. Three-dimensional cell culture-based screening identifies the anthelminthic drug nitazoxanide as a candidate for treatment of colorectal cancer. Mol. Cancer Ther. 2015, 14, 1504–1516. [Google Scholar] [CrossRef] [PubMed]
- Stockis, A.; Deroubaix, X.; Lins, R.; Jeanbaptiste, B.; Calderon, P.; Rossignol, J. Pharmacokinetics of nitazoxanide after single oral dose administration in 6 healthy volunteers. Int. J. Clin. Pharmacol. Ther. 1996, 34, 349–351. [Google Scholar] [PubMed]
- Borrego, S.L.; Fahrmann, J.; Datta, R.; Stringari, C.; Grapov, D.; Zeller, M.; Chen, Y.; Wang, P.; Baldi, P.; Gratton, E.; et al. Metabolic changes associated with methionine stress sensitivity in MDA-MB-468 breast cancer cells. Cancer Metabol. 2016, 4, 9. [Google Scholar] [CrossRef] [PubMed]
- Jones-Bolin, S.; Zhao, H.; Hunter, K.; Klein-Szanto, A.; Ruggeri, B. The effect of the oral, pan-VEGFR kinase inhibitor CEP-7055 and chemotherapy in orthopedic models of glioblastoma and colon carcinoma in mice. Mol. Cancer Ther. 2006, 5, 1744–1753. [Google Scholar] [CrossRef] [PubMed]
- Chan, O.H.; Schmid, H.L.; Stilgenbauer, L.A.; Howson, W.; Horwell, D.C.; Stewart, S.B.H. Evaluation of a targeted prodrug strategy of enhanced oral absorption of poorly-water-soluble compounds. Pharm. Res. 1998, 15, 1012–1018. [Google Scholar] [CrossRef] [PubMed]
- Ruggeri, B.; Singh, J.; Gingrich, D.; Angeles, T.; Albom, M.; Yang, S.; Chang, H.; Robinson, C.; Hunter, K.; Dobrzanski, P.; et al. CEP-7055, a novel orally active pan inhibitor of vascular endothelial growth factor receptor tyrosine kinase with potent antiangiogenic activity and antitumor efficacy in preclinical models. Cancer Res. 2003, 63, 5978–5991. [Google Scholar] [PubMed]
- Barth, R.F.; Yang, W.; Al-Madhoun, A.S.; Johnsamuel, J.; Byun, Y.; Chandra, S.; Smith, D.R.; Tjarks, W.; Eriksson, S. Boron-containing nucleosides as potential delivery agents for neutron capture therapy of brain tumors. Cancer Res. 2004, 64, 6287–6295. [Google Scholar] [CrossRef] [PubMed]
- Barth, R.F.; Coderre, J.A.; Vicente, M.; Blue, T.E. Boron neutron capture therapy of cancer: Current status and future prospects. Clin. Cancer Res. 2005, 11, 3987–4002. [Google Scholar] [CrossRef] [PubMed]
- Hasabelnaby, S.; Goudah, A.; Agarwal, H.K.; abd Alla, M.S.M.; Tjarks, W. Synthesis, chemical and enzymatic hydrolysis, and aqueous solubility of amino acid ester prodrugs of 3-carboranyl thymidine analogs for boron neutron capture therapy of brain tumors. Eur. J. Med. Chem. 2012, 55, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Houchens, D.P.; Ovejera, A.A.; Riblet, S.M.; Slagel, D.E. Human brain tumor xenografts in nude mice as a chemotherapy model. Eur. J. Cancer Clin. Oncol. 1983, 19, 799–805. [Google Scholar] [CrossRef]
- Supko, J.G.; Malspeis, L. Dose-dependent pharmacokinetics of rapamycin-28-N,N-dimethylglycinate in the mouse. Cancer Chemother. Pharmacol. 1994, 33, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Spataro, A.; Kessel, D. Studies on camptothecin-induced degradation and apparent reaggregation of DNA from L1210 cells. Biochem. Biophys. Res. Commun. 1972, 48, 643–648. [Google Scholar] [CrossRef]
- Yurkovetskiy, A.V.; Fram, R.J. XMT-1001, a novel polymeric camptothecin pro-drug in clinical development for patients with advanced cancer. Adv. Drug Deliv. Rev. 2009, 61, 1193–1202. [Google Scholar] [CrossRef] [PubMed]
- Pantazis, P.; Hinz, H.R.; Mendoza, J.T.; Kozielski, A.J.; Williams, L.J., Jr.; Stehlin, J.S., Jr.; Giovanella, B.C. Complete inhibition of growth followed by death of human malignant melanoma cells in vitro and regression of human melanoma xenografts in immunodeficient mice induced by camptothecins. Cancer Res. 1992, 52, 3980–3987. [Google Scholar] [PubMed]
- Deshmukh, M.; Chao, P.; Kutscher, H.L.; Gao, D.; Sinko, P.J. A series of α-amino acid ester prodrugs of camptothecin: In vitro hydrolysis and A549 human lung carcinoma cell cytotoxicity. J. Med. Chem. 2010, 53, 1038–1047. [Google Scholar] [CrossRef] [PubMed]
- Chua, M.-S.; Kashiyama, E.; Bradshaw, T.D.; Stinson, S.F.; Brantley, E.; Sausville, E.A.; Stevens, M.F.G. Role of CYP1A1 in modulation of antitumor properties of the novel agent 2-(4-amino-3-methylphenyl)-benzothiazole (DF 203, NSC 674495) in human breast cancer cells. Cancer Res. 2000, 60, 5196–5203. [Google Scholar] [PubMed]
- Hose, C.; Riviera, M.; Sausville, E.; Monks, A. Induction of cytochrome P450 CYP1A1 and cytochrome P450 CYP1B1 by 2-(4-amino-3-methylphenyl)benzothiazole (BZ) in 60 human tumor cell lines: Correlation with BZ toxicity. Proc. Am. Assoc. Cancer Res. 2001, 42, 511. [Google Scholar]
- Bradshaw, T.D.; Bibby, M.C.; Double, J.A.; Fichtner, I.; Cooper, P.A.; Alley, M.C.; Donohue, S.; Stinson, S.F.; Tomaszewjsti, J.E.; Sausville, E.A.; et al. Preclinical evaluation of amino acid prodrugs of a novel antitumor 2-(4-amino-3-methylphenyl)benzothiazoles. Mol. Cancer Ther. 2002, 1, 239–246. [Google Scholar] [PubMed]
- Farmer, P.B. Metabolism and reactions of alkylating agents. Pharmacol. Ther. 1987, 35, 301–358. [Google Scholar] [CrossRef]
- Chrzanowski, K.; Bielawska, A.; Palka, J. Proline analogue of melphalan as a prodrug susceptible to the action of prolidase in breast cancer MDA-MB 231. Il Farmaco 2003, 58, 1113–1119. [Google Scholar] [CrossRef]
- Wu, Z.; Shah, A.; Patel, N.; Yuan, X. Development of methotrexate proline prodrug to overcome resistance by MDA-MB-231 cells. Bioorg. Med. Chem. Lett. 2010, 20, 5108–5112. [Google Scholar] [CrossRef] [PubMed]
- Mittal, S.; Song, X.; Vig, B.S.; Landowski, C.P.; Kim, I.; Hilfinger, J.M.; Amidon, G. Prolidase, a potential enzyme targeted for melanoma: Design of proline-containing dipeptide-linker prodrugs. Mol. Pharm. 2005, 2, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Nam, N.H.; Kim, Y.; You, Y.J.; Hong, D.H.; Kim, H.M.; Ahn, B.Z. Synthesis and anti-tumor activity of a novel combretastatins: Combretocyclopentenones and related analogues. Bioorg. Med. Chem. Lett. 2002, 12, 1955–1958. [Google Scholar] [CrossRef]
- Nam, N.H.; Kim, Y.; You, Y.J.; Hong, D.H.; Kim, H.M.; Ahn, B.Z. Water soluble prodrugs of the antitumor agent 3-[(3-amino-4-methoxy)phenyl]-2-(3,4,5-trimethoxyphenyl)cyclopent-2-ene-1-one. Bioorg. Med. Chem. 2003, 11, 1021–1029. [Google Scholar] [CrossRef]
- Hirpara, K.; Aggarwal, P.; Mukherjee, A.K.; Joshi, N.; Burman, A.C. Quercetin and its derivatives: Synthesis, pharmacological uses with special emphasis on anti-tumor properties and prodrug with enhanced bio-availability. Anti-Cancer Agent Med. Chem. 2009, 9, 138–161. [Google Scholar] [CrossRef]
- Mulholand, P.J.; Ferry, D.R.; Anderson, D.; Hussain, S.A.; Young, A.M.; Cook, J.E.; Hodgkin, E.; Seymour, L.W.; Kerr, D.J. Pre-clinical and clinical study of QC-12, a water-soluble prodrug of quercetin. Ann. Oncol. 2001, 12, 245–248. [Google Scholar] [CrossRef]
- Kyoung, M.K.; Oh, Y.M.; Park, K.-S.; Chong, Y. A novel prodrug of quercetin, 3-N,N-dimethyl carbamoyl quercetin (DCQ), with improved stability against hydrolysis in cell culture medium. Bull. Korean Chem. Soc. 2009, 30, 2114–2116. [Google Scholar]
- Vissiennon, C.; Nieber, H.; Kelber, O.; Butterweck, V. Route of administration determines the anxiolytic activity of the flavonols kampferol, quercetin and myricetin-are they prodrugs? J. Nutr. Biochem. 2012, 23, 733–740. [Google Scholar] [CrossRef] [PubMed]
- Mu, L.; Feng, S.S. A novel controlled release formulation for the anticancer drug paclitaxel (Taxol®): PLGA nanoparticles containing vitamin E TPGS. J. Control. Release 2003, 86, 33–48. [Google Scholar] [CrossRef]
- Lundberg, B.B.; Risovic, V.; Ramaswamy, M.; Wasan, K.M. A lipophilic paclitaxel derivative incorporated in a lipid emulsion for parenteral administration. J. Control. Release 2003, 86, 93–100. [Google Scholar] [CrossRef]
- Ma, H.; Chen, G.; Wang, T.; Li, Q.; Liu, Y. Design, synthesis, and biological evaluation of a novel water-soluble prodrug of docetaxel with amino acid as a linker. Chem. Biol. Drug Des. 2016, 88, 363–369. [Google Scholar] [CrossRef] [PubMed]
- McKerrow, M.H.; Caffrey, C.; Kelly, B.; Loke, P.; Sajid, M. Proteases in parasitic diseases. Annu. Rev. Pathol. Mech. Dis. 2006, 1, 497–536. [Google Scholar] [CrossRef] [PubMed]
- Molyneux, D.H. Control of human parasitic diseases: Context and overview. Adv. Parasitol. 2006, 61, 1–45. [Google Scholar] [PubMed]
- Renslo, A.R.; McKerrow, J.H. Drug discovery and development for neglected parasitic diseases. Nat. Chem. Biol. 2006, 2, 701–710. [Google Scholar] [CrossRef] [PubMed]
- Chung, M.C.; Gonçalves, M.F.; Colli, N.; Ferreira, E.I.; Miranda, M.T.M. Synthesis and in vitro evaluation of potential antichagasic dipeptide prodrugs of primaquine. J. Pharm. Sci. 1997, 86, 1127–1131. [Google Scholar] [CrossRef] [PubMed]
- Howard, J.E.; Rios, C.; Ebensperger, I.; Olivos, P. Congenital Chagas disease. Bol. Chil. Parasitol. 1957, 12, 42–45. [Google Scholar] [PubMed]
- Denise, H.; Barrett, M.P. Uptake and mode of action of drugs used against sleeping sickness. Biochem. Pharmacol. 2001, 61, 1–5. [Google Scholar] [CrossRef]
- Sands, M.; Kron, M.A.; Brown, R.B. Pentamidine, a review. Rev. Infect. Dis. 1985, 7, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Kotthaus, J.; Hungeling, H.; Reeh, C.; Kotthaus, J.; Schade, D.; Wein, S.; Wolffram, S.; Clement, B. Synthesis and biological evaluation of L-valine-amidoxiesters as double prodrugs of amidines. Bioorg. Med. Chem. 2011, 19, 1907–1914. [Google Scholar] [CrossRef] [PubMed]
- Hanboonkunupakarn, B.; White, N.J. The threat of antimalarial resistance. Trop. Dis. Travel Med. Vaccines 2016, 2, 10. [Google Scholar] [CrossRef] [PubMed]
- Baker, J.K.; Yarber, R.H.; Nanayakkara, N.P.D.; McChesney, J.D.; Homo, F.; Landau, I. Effect of aliphatic side-chain substituents on the antimalarial activity and on the metabolism of primaquine studied using mitochondria and microsome preparations. Pharm. Res. 1990, 7, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Umbreit, J. Methemoglobin-It’s not just blue: A concise review. Am. J. Hematol. 2007, 82, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Araújo, M.J.; Bom, J.; Capela, R.; Casimiro, C.; Chambel, P.; Gomes, P.; Iley, J.; Lopes, F.; Morais, J.; Moreira, R.; et al. Imidizolidin-4-one derivatives of primaquine as novel transmission-blocking antimalarials. J. Med. Chem. 2005, 48, 888–892. [Google Scholar] [CrossRef] [PubMed]
- Vagapandu, S.; Sachdeva, S.; Jain, M.; Singh, S.; Singh, P.P.; Kaul, C.L.; Jain, R. 8-aminoquinoamides conjugates with amino acids are exhibiting potent blood-schizontocidal antimalarial activities. Bioorg. Med. Chem. 2004, 12, 239–247. [Google Scholar] [CrossRef]
- Vale, N.; Matos, J.; Gut, J.; Nogueira, F.; do Rosário, V.; Rosenthal, P.J.; Moreira, R.; Gomes, P. Imidazolidin-4-one peptidomimetic derivatives of primaquine: Synthesis and antimalarial activity. Bioorg. Med. Chem. Lett. 2009, 18, 4150–4153. [Google Scholar] [CrossRef] [PubMed]
- Srinivasarao, K.; Argawal, P.; Srivastava, K.; Haq, W.; Puri, S.K.; Katti, S.B. Design, synthesis and in vitro antiplasmodial activity of 4-aminoquinolines containing modified amino acid conjugates. Med. Chem. Res. 2016, 25, 1148–1162. [Google Scholar] [CrossRef]
- World Health Organization (WHO). Control of the Leishmaniasis; Technical Report Series; WHO: Geneva, Switzerland, 2010; pp. 1–186. [Google Scholar]
- Chappuis, F.; Sundar, S.; Hailu, A.; Ghalib, H.; Rijal, S.; Peeling, R.W.; Alvar, J.; Boelaert, M. Visceral leishmaniasis: What are the needs for diagnosis, treatment, and control? Nat. Rev. Microbiol. 2007, 5, 873–882. [Google Scholar] [CrossRef] [PubMed]
- Maltezou, H.C. Drug resistance in visceral leishmaniasis. J. Biomed. Biotechnol. 2010, 2010, 617521. [Google Scholar] [CrossRef] [PubMed]
- Vale-Costa, S.; Vale, N.; Matos, J.; Tomás, A.; Moreira, R.; Gomes, P.; Gomes, M.S. Peptidomimetic and organometallic derivatives of primaquine active against Leishmania infantum. Antimicrob. Agent Chemother. 2012, 56, 5774–5781. [Google Scholar] [CrossRef] [PubMed]
- Goodman, D.A.; Huskins, W.C. Control of nosocomial antimicrobial-resistant bacteria: A strategic priority for hospitals worldwide. Clin. Infect. Dis. 1997, 24, S139–S145. [Google Scholar] [CrossRef]
- Ibrahim, M.A.; Panda, S.S.; Birds, A.S.; Serrano, J.C.; Gonzalez, C.F.; Alamry, K.A.; Katrizky, A.R. Synthesis and antibacterial evaluation of amino-acid antibiotic conjugates. Bioorg. Med. Chem. 2014, 24, 1856–1861. [Google Scholar] [CrossRef] [PubMed]
- De Sarro, A.; De Sarro, G. Adverse reactions to fluoroquinolones. An overview on mechanistic aspects. Curr. Med. Chem. 2001, 8, 371–384. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Dixit, P. Multidrug-resistant to extensively drug resistant tuberculosis: What is next? J. Biosci. 2008, 33, 605–616. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization (WHO). Anti-Tuberculosis Drug Resistance in the World: 4th Global Report; WHO: Geneva, Switzerland, 2008. [Google Scholar]
- Krátký, M.; Vinšová, J.; Butcha, V.; Horvati, K.; Bösze, S.; Stolaříková, J. New amino acid esters of salicylanilides active against MDR-TB and other microbes. Eur. J. Med. Chem. 2008, 45, 6106–6113. [Google Scholar] [CrossRef] [PubMed]
- Pochopin, N.L.; Charman, W.N.; Stella, V.J. Aminoacid derivatives of dapsone as water-soluble prodrugs. Int. J. Pharm. 1995, 121, 157–167. [Google Scholar] [CrossRef]
- Pardridge, W.M. Brain Drug Targeting: The Future of Brain Development; Cambridge University Press: Cambridge, UK, 2001; Volume 1, pp. 1–12. [Google Scholar]
- Gynther, M.; Laine, K.; Ropponen, J.; Leppänen, J.; Mannila, A.; Nevalainen, T.; Savolainen, J.; Järvinen, T.; Rautio, J. Large neutral amino acid transporter enables brain drug delivery via prodrugs. J. Med. Chem. 2008, 51, 932–936. [Google Scholar] [CrossRef] [PubMed]
- Gynther, M.; Ropponen, J.; Laine, K.; Leppänen, J.; Haapakoski, P.; Peura, L.; Järvinen, T.; Rautio, J. Glucose promoiety enables glucose transporter mediated brain uptake of ketoprofen and imdomethacin prodrugs in rats. J. Med. Chem. 2009, 52, 3348–3353. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.S.; Acevedo, C.; Arzimanoglou, A.; Bogacz, A.; Cross, J.H.; Elger, C.E.; Engel, J., Jr.; Forsgren, L.; French, J.A.; Glynn, M.; et al. A practical clinical definition of epilepsy. Epilepsia 2014, 55, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Moshé, S.L.; Perucca, E.; Ryvlin, P.; Tomson, T. Epilepsy: New advances. Lancet 2015, 385, 884–898. [Google Scholar] [CrossRef]
- Bialer, M.; Johannessen, S.I.; Levy, R.H.; Perucca, E.; Tomson, T.; White, S.H. Progress report on new antiepileptic drugs: A summary of the Eight Eilat Conference (EILT XI). Epilepsy Res. 2013, 103, 2–30. [Google Scholar] [CrossRef] [PubMed]
- French, J.A. Refractory epilepsy: Clinical overview. Epilepsia 2007, 48 (Suppl. 1), 3–7. [Google Scholar] [CrossRef] [PubMed]
- Villalba, M.L.; Enrique, A.V.; Higgs, J.; Castaño, R.A.; Goicoechea, S.; Taborda, F.D.; Gavernet, L.; Lick, I.D.; Marder, M.; Blanch, L.E.B. Novel sulfamides and sulfamates derived from amino esters: Synthetic studies and anticonvulsant activity. Eur. J. Pharmacol. 2016, 774, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Barré, J.; Chamouard, J.M.; Houin, G.; Tillement, J.P. Equilibrium dialysis, ultrafiltration, and ultracentrifugation compared for determining the plasma-protein-binding characteristics of valproic acid. Clin. Chem. 1985, 31, 60–64. [Google Scholar]
- Svennebring, A.M. Investigation of the prerequisites for the optimization of specific plasma protein binding as a strategy for the reduction of first-pass hepatic metabolism. Xenobiotica 2015, 45, 286–301. [Google Scholar] [CrossRef] [PubMed]
- Cornford, E.M.; Diep, C.P.; Pardridge, W.M. Blood-brain barrier transport of valproic acid. J. Neurochem. 1985, 44, 1541–1550. [Google Scholar] [CrossRef] [PubMed]
- Peura, L.; Malmioja, K.; Laine, K.; Leppänen, J.; Gynther, M.; Isotalo, A.; Rautio, J. Large amino acid transporter 1 (LAT1) prodrugs of valproic acid: New prodrug design ideas for central nervous system delivery. Mol. Pharm. 2011, 8, 1857–1866. [Google Scholar] [CrossRef] [PubMed]
- Gynther, M.; Peura, L.; Vernerová, M.; Leppänen, J.; Kärkkäinen, J.; Lehtonen, M.; Rautio, J.; Huttunen, K.M. Amino acid promoieties alter valproic acid pharmacokinetics and enable extended brain exposure. Neurochem. Res. 2016, 41, 2797–2809. [Google Scholar] [CrossRef] [PubMed]
- McLean, M.J. Gabapentin in the management of convulsive disorders. Epilepsia 1999, 40, S39–S50. [Google Scholar] [CrossRef] [PubMed]
- Rice, A.S.; Maton, S.; Postherpetic Neuralgia Study Group. Gabapentin in postherpetic neuralgia: A randomized double-blind placebo controlled study. Pain 2001, 94, 215–224. [Google Scholar] [CrossRef]
- Pollack, M.H.; Matthews, J.; Scott, E.L. Gabapentin as a potential treatment for anxiety disorders. Am. J. Phsychiatry 1998, 155, 992–993. [Google Scholar] [CrossRef] [PubMed]
- Backonja, M.; Beydoun, A.; Edwards, K.R.; Schwartz, S.L.; Fonseca, V.; Hes, M.; LaMoreaux, L.; Garofalo, E. Gabapentin for the symptomatic treatment of painful neuropathy in patients with diabetes mellitus: A randomized controlled trial. JAMA 1998, 280, 1831–1836. [Google Scholar] [CrossRef] [PubMed]
- Uchino, H.; Kanai, Y.; Kim, K.; Wempe, M.F.; Chairoungdua, A.; Morimoto, E.; Anders, M.W.; Endou, E. Transport of amino acid-related compound mediate to L-type amino acid transporter 1 (LAT-1): Insights into mechanisms of substrate recognition. Mol. Pharm. 2002, 61, 729–737. [Google Scholar] [CrossRef]
- Richter, A.; Anton, S.E.; Koch, P.; Dennett, S.L. The impact of reducing dose frequency on health outcomes. Clin. Ther. 2003, 25, 2307–2335. [Google Scholar] [CrossRef]
- Kriel, R.L.; Birnbaum, A.K.; Cloyd, J.C.; Ricker, B.J.; Jones Saete, C.; Caruso, K.J. Failure of absorption of gabapentin after rectal administration. Epilepsia 1997, 38, 1242–1244. [Google Scholar] [CrossRef] [PubMed]
- Cundy, C.K.; Branch, R.; Chernov-Rogan, T.; Dias, T.; Estrada, T.; Hold, K.; Koller, K.; Liu, X.; Mann, A.; Panuwat, M.; et al. XP13515 [(±)-1([α-isobutanoyloxyethoxylcarbonyl]aminomethyl)-1-cyclohexane acetic acid], a novel gabapentin prodrug: I. Design, synthesis, enzymatic conversion to gabapentin, an a intestinal transport by intestinal solute transporters. J. Pharm. Ther. 2004, 311, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Wuis, E.W.; Dirkis, M.J.; Termond, E.F.; Vree, T.B.; Van der Kleijn, E. Plasma and urinary excretion kinetics of oral baclofen in healthy subjects. Eur. J. Clin. Pharmacol. 1989, 37, 181–184. [Google Scholar] [CrossRef] [PubMed]
- Lal, R.; Sukbuntherng, J.; Tai, E.H.L.; Upadhay, S.; Yao, F.; Warren, M.S.; Luo, W.; Bu, L.; Nguyen, S.; Zamora, J.; et al. Arbaclofen placarbil, a novel R-baclofen prodrug: Improved absorption, distribution, metabolism, and elimination properties compared with R-baclofen. J. Pharm. Exp. Ther. 2009, 330, 911–921. [Google Scholar] [CrossRef] [PubMed]
- Loiseau, P.; Duche, B.; Pedespan, J.M. Absence epilepsies. Epilepsia 1985, 36, 1182–1186. [Google Scholar] [CrossRef]
- Chen, X.-Q.; Venkatesh, S. Miniature device for aqueous and non-aqueous solubility measurements during drug discovery. Pharm. Res. 2004, 21, 1758–1761. [Google Scholar] [CrossRef] [PubMed]
- Hemenway, J.N.; Jarho, P.; Henri, J.T.; Nair, S.K.; VanderVelde, D.; Georg, G.I.; Stella, V.J. Preparation and physicochemical characterization of a novel water-soluble prodrug of carbamazepine. J. Pharm. Sci. 2010, 99, 1810–1825. [Google Scholar] [CrossRef] [PubMed]
- Hemenway, J.N.; Stella, V.J. In vitro and in vivo evaluation of a novel water-soluble N-glycyl prodrug (N-Gly-CBZ) of carbamazepine. J. Pharm. Sci. 2010, 99, 4565–4575. [Google Scholar] [CrossRef] [PubMed]
- Hemenway, J.N.; Nti-Addae, K.; Guarino, V.R.; Stella, V.J. Preparation, characterization and in vivo conversion of new water-soluble sulfenamide prodrugs of carbamazepine. Bioorg. Med. Chem. Lett. 2007, 17, 6629–6632. [Google Scholar] [CrossRef] [PubMed]
- Dunayevich, E.; Erickson, J.; Levine, L.; Landbloom, R.; Schoepp, D.D.; Tollefson, G.D. Efficacy and tolerability of an mGlu2/3 agonist in the treatment of generalized anxiety disorder. Neuropsychopharmacology 2008, 33, 1603–1610. [Google Scholar] [CrossRef] [PubMed]
- Rorick-Kehn, L.M.; Perkins, E.J.; Knitowski, K.M.; Hart, J.C.; Johnson, B.G.; Schoepp, D.D.; McKinzie, D. Improved bioavailability of the mGlu213 receptor agonist LY354740 using a prodrug strategy: In vivo pharmacology of LY544344. J. Pharm. Exp. Ther. 2005, 316, 905–913. [Google Scholar] [CrossRef] [PubMed]
- Varma, M.V.; Eriksson, A.H.; Sawada, G.; Pak, Y.A.; Perkins, E.J.; Zimmerman, C.L. Transepithelial transport of the group II metabotropic glutamate 2/3 receptor agonist (1S,2S,5R,6S)-2-aminobicyclo[3.1.0]hexane-2,6-dicarboxylate (LY354740) and its prodrug (1S,2S,5R,6S)-2-[(2′S)-(2′-amino)propionyl]aminobicyclo[3.1.0]hexane-2,6-dicarboxylate (LY544344). Drug Metabol. Dis. 2009, 37, 211–220. [Google Scholar]
- Goodman, D.W. Lisdexamfetamine dimesylate (Vyvanase), a prodrug stimulant or attention deficit/hyperactivity disorder. Pharm. Ther. 2010, 35, 273–387. [Google Scholar]
- Pennick, M. Absorption of lisdexamphetamine dismesylate and its enzymatic conversion to d-amphetamine. Neuropsychiatr. Dis. Treat. 2010, 6, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Cowles, B.J. Lisdexamfetamine for treatment of attention-deficit/hyperactivity disorder. Ann. Pharmacother. 2009, 43, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Fearnley, J.M.; Lees, A.J. Ageing and Parkinson´s disease: Substantia nigra regional selectivity. Brain 1991, 114, 2283–2301. [Google Scholar] [CrossRef] [PubMed]
- Di Stefano, A.; Sazio, P.; Cuasa, L.S. Antiparkison prodrug. Molecules 2008, 13, 46–58. [Google Scholar] [CrossRef] [PubMed]
- Felix, A.M.; Winter, D.P.; Wang, S.S.; Kulesha, I.D.; Pool, W.R.; Hane, D.L.; Sheppard, H. Synthesis and antineserpine activity of peptides of L-dopa. J. Med. Chem. 1974, 17, 422–426. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Lee, J.; Tsai, M.; Lu, H.; Hsu, W. Synthesis and pharmacological activities of a novel tripeptide mimetic dopamine prodrug. Bioorg. Med. Chem. Lett. 1995, 5, 2195–2198. [Google Scholar] [CrossRef]
- Peura, L.; Malmioja, K.; Huttunen, K.; Leppänen, J.; Hämäläinen, M.; Forsberg, M.M.; Gynther, M.; Rautio, J.; Laine, K. Design, synthesis and brain uptake of LAT1-targeted aminoacid prodrugs of dopamine. Pharm. Res. 2013, 30, 2523–2537. [Google Scholar] [CrossRef] [PubMed]
- Zeilhofer, H.U.; Wildner, H.; Yévenws, G.E. Fast synaptic inhibition in spinal sensory processing and pain control. Physiol. Rev. 2012, 92, 193–235. [Google Scholar] [CrossRef] [PubMed]
- Sherman, S.E.; Loomis, C.W. Morphine insensitive allodynia is produced by intrathecal strychnine in the lightly anesthetized rat. Pain 1994, 56, 17–26. [Google Scholar] [CrossRef]
- Yamamoto, T.; Yaksh, T.L. Effects of intrathecal strychnine and bicuculine on nerve compression-induced thermal hyperalgesia and selective antagonism by MK-801. Pain 1993, 54, 79–84. [Google Scholar] [CrossRef]
- Haranishi, Y.; Hara, K.; Terada, T.; Nakamura, S.; Sata, T. The antinociceptive effect of intrathecal administration of glycine transporter-2 inhibition ALX 1393 in rat acute pain model. Anesth. Anal. 2010, 110, 615–621. [Google Scholar] [CrossRef] [PubMed]
- Seta, K.; Sershen, H.; Lajtha, A. Cerebral amino acid uptake in vivo in newborn mice. Brain Res. 1972, 47, 415–425. [Google Scholar] [CrossRef]
- Toth, E.; Lajtha, A. Elevation of cerebral levels of non-essential amino acids in vivo by administration of large doses. Neurochem. Res. 1981, 6, 1309–1317. [Google Scholar] [CrossRef] [PubMed]
- Doheny, M.; Nagaki, S.; Patsalos, P.N. A microdialysis study of glycinamide, glycine and other amino acid neurotransmitters in rat frontal cortex and hippocampus after the administration of milacemide, a glycine prodrug. Naunyn Schmiedebergs Arch. Pharmacol. 1996, 354, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Beyer, C.; Komisaruk, B.K.; González-Flores, O.; Gómora-Arrati, P. Glycinamide a glycine prodrug, induces antinociception by intraperitoneal or oral ingestion in ovariectomized rats. Life Sci. 2013, 92, 576–581. [Google Scholar] [CrossRef] [PubMed]
- Gynther, M.; Jalkanen, A.; Lethonen, M.; Forsberg, M.; Laine, K.; Ropponen, J.; Leppänen, J.; Knuuti, J.; Rautio, J. Brain uptake of ketoprofen-lysine prodrug in rats. Int. J. Pharm. 2010, 399, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Bennet, S.N.; McNeil, M.M.; Bland, L.A.; Arduino, M.J.; Villarino, M.E.; Perrotta, D.M.; Burwen, D.R.; Welbel, S.F.; Pegues, D.A.; Stroud, L.; et al. Post-operative infections traced to contamination of an intravenous anesthetic, Propofol. N. Engl. J. Med. 1995, 33, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Trapani, G.; Latrofa, A.; Franco, M.; Lopedota, A.; Maciocco, E.; Liso, G. Water-soluble salts of amino acid esters of the anesthetic agent Propofol. Int. J. Pharm. 1998, 175, 195–204. [Google Scholar] [CrossRef]
- Trapani, G.; Altomare, C.; Sanna, E.; Biggio, G.; Liso, G. Propofol in anaesthesic. Mechanism of action, structure-activity relationships and drug delivery. Curr. Med. Chem. 2000, 7, 249–271. [Google Scholar] [CrossRef] [PubMed]
- Altomare, C.; Trapani, G.; Latrofa, A.; Serra, M.; Sanna, E.; Biggio, G.; Liso, G. Highly water-soluble derivatives of the anesthetic agent propofol: In vitro and in vivo evaluation of cyclic amino acid esters. Eur. J. Pharm. Sci. 2003, 20, 17–26. [Google Scholar] [CrossRef]
- Zhou, Y.; Yang, J.; Liu, J.; Wang, Y.; Zhang, W.S. Efficacy comparison of the novel water-soluble propofol prodrug HX0969w and fosfopropofol in mice and rats. Br. J. Anaesth. 2013, 111, 825–832. [Google Scholar] [CrossRef] [PubMed]
- Lang, B.C.; Yang, J.; Wang, Y.; Luo, Y.; Kang, Y.; Liu, J.; Zhang, W.S. An improved design of water-soluble propofol prodrugs characterized by rapid onset of action. Anesth. Anal. 2014, 118, 745–754. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |



































































© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vale, N.; Ferreira, A.; Matos, J.; Fresco, P.; Gouveia, M.J. Amino Acids in the Development of Prodrugs. Molecules 2018, 23, 2318. https://doi.org/10.3390/molecules23092318
Vale N, Ferreira A, Matos J, Fresco P, Gouveia MJ. Amino Acids in the Development of Prodrugs. Molecules. 2018; 23(9):2318. https://doi.org/10.3390/molecules23092318
Chicago/Turabian StyleVale, Nuno, Abigail Ferreira, Joana Matos, Paula Fresco, and Maria João Gouveia. 2018. "Amino Acids in the Development of Prodrugs" Molecules 23, no. 9: 2318. https://doi.org/10.3390/molecules23092318
APA StyleVale, N., Ferreira, A., Matos, J., Fresco, P., & Gouveia, M. J. (2018). Amino Acids in the Development of Prodrugs. Molecules, 23(9), 2318. https://doi.org/10.3390/molecules23092318